
An Intracellular Model of Hepatitis B Viral Infection: An In Silico Platform for Comparing Therapeutic Strategies

 , , , , and
, , , , and
Abstract
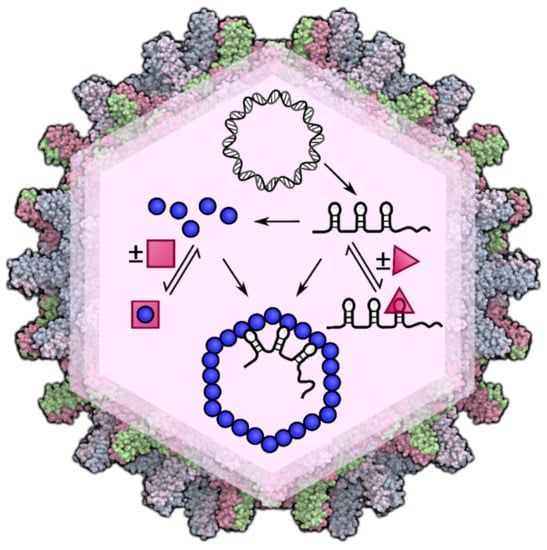
:
{kind=link}
{kind=link}
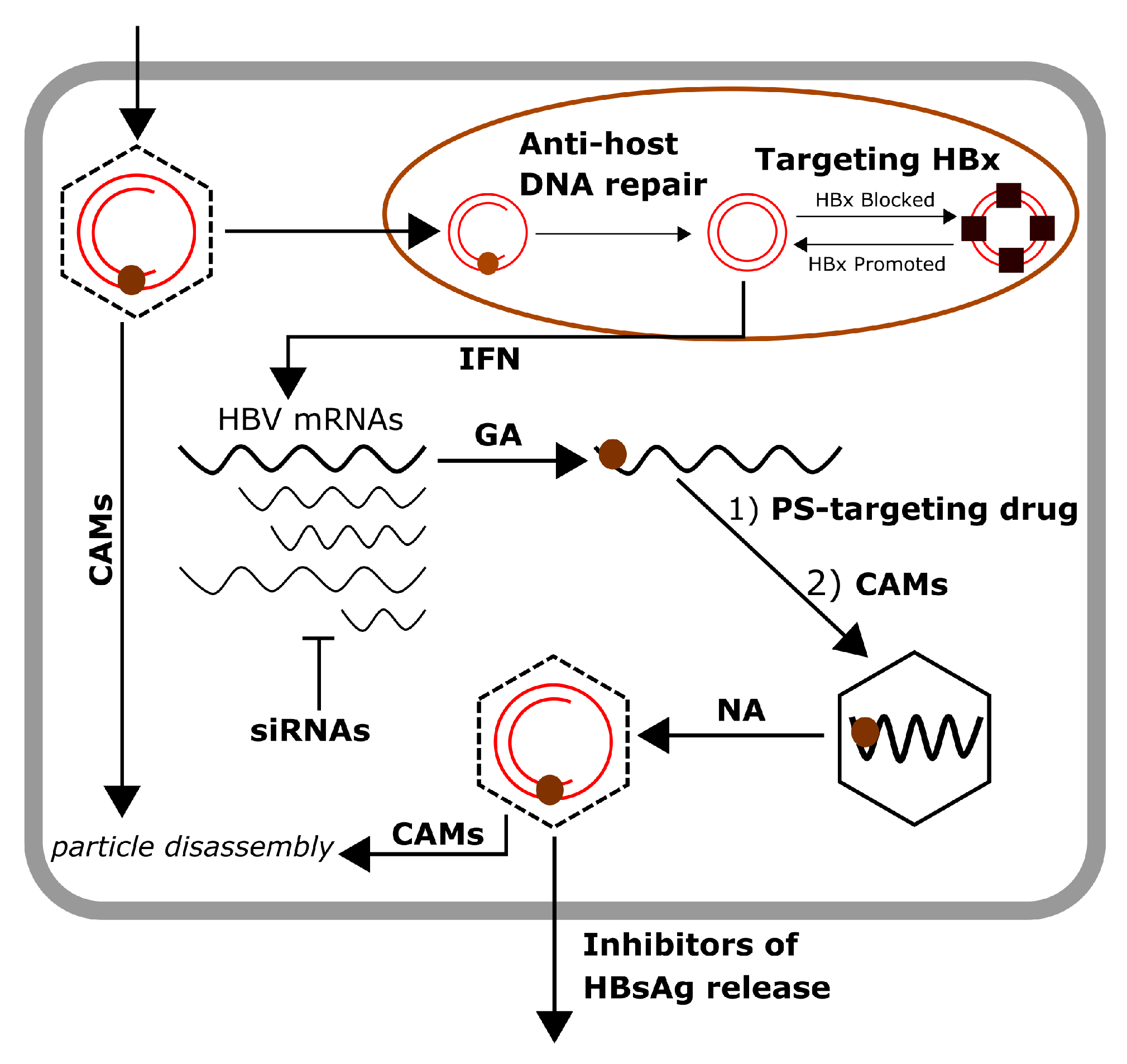{kind=link}
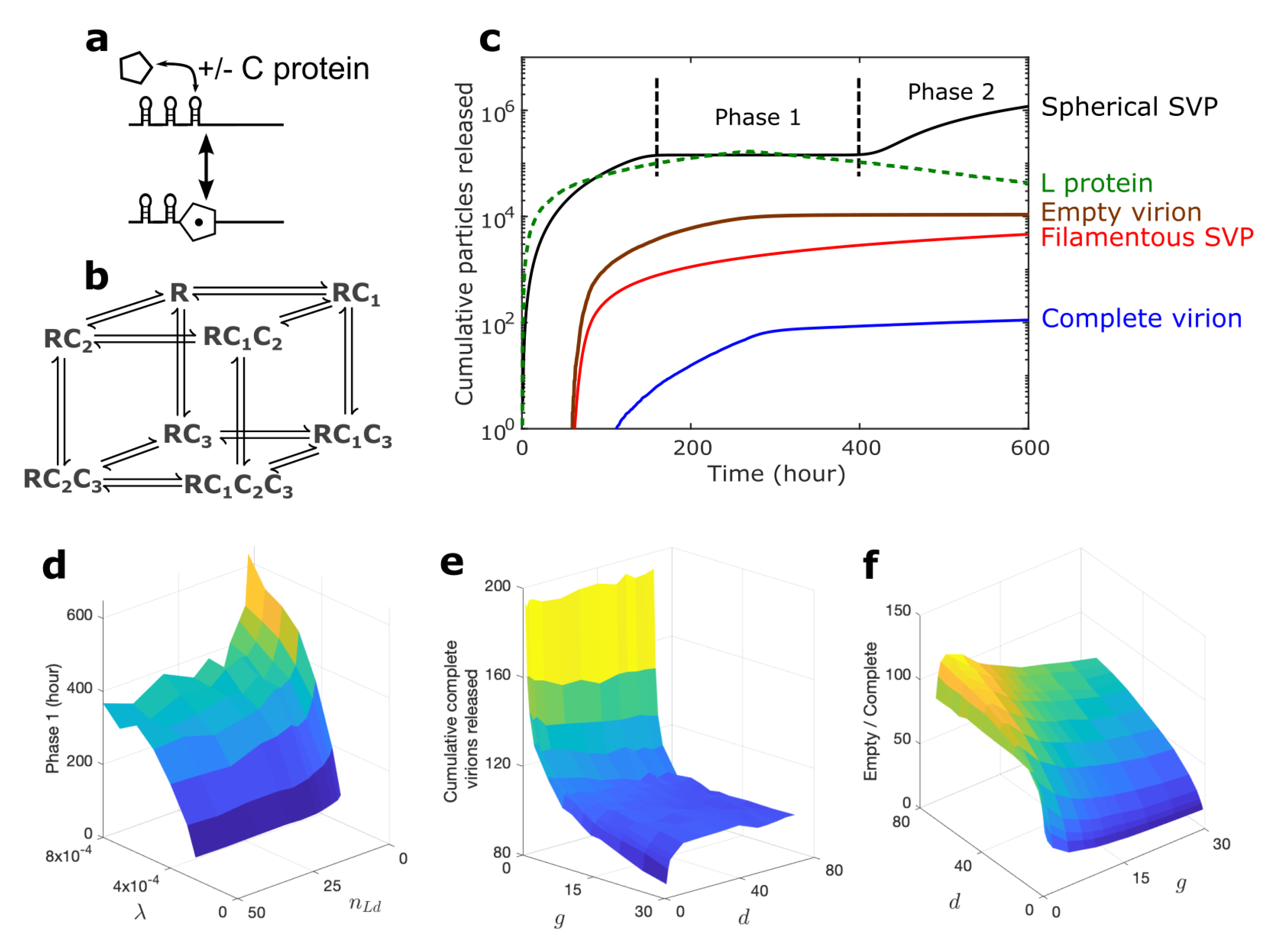{kind=link}
{kind=link}
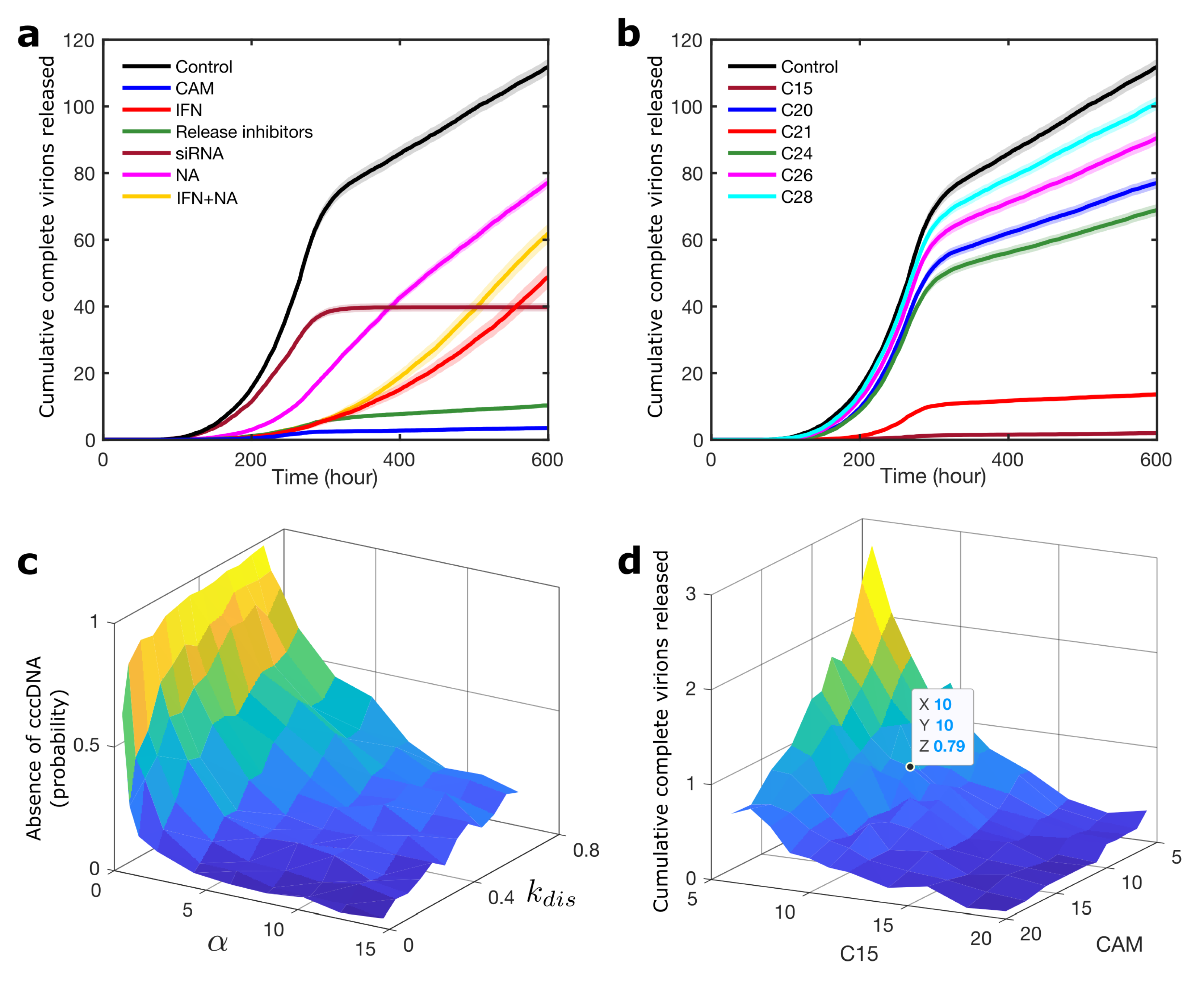{kind=link}
1. Introduction
2. Materials and Methods
2.1. An In Silico Model of Intracellular HBV Infection Dynamics
2.2. Modelling of Different Drug Treatments
3. Results
3.1. Novel Insights into Infection Kinetics in the Drug-Free Situation
3.2. Modelling Synergies between Antiviral Strategies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Hepatitis Report; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Lok, A.S.; Zoulim, F.; Dusheiko, G.; Ghany, M.G. Hepatitis B cure: From discovery to regulatory approval. J. Hepatol. 2017, 67, 847–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar]
- Nakabayashi, J.; Sasaki, A. A mathematical model of the intracellular replication and within host evolution of hepatitis type B virus: Understanding the long time course of chronic hepatitis. J. Theor. Biol. 2011, 269, 318–329. [Google Scholar] [CrossRef]
- Nakabayashi, J. The intracellular dynamics of hepatitis B virus (HBV) replication with reproduced virion “re-cycling”. J. Theor. Biol. 2016, 396, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Goyal, A. In silico single cell dynamics of hepatitis B virus infection and clearance. J. Theor. Biol. 2015, 366, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; White, S.J.; Thompson, R.F.; Bingham, R.; Weiß, E.U.; Maskell, D.P.; Zlotnick, A.; Dykeman, E.C.; Tuma, R.; Twarock, R.; et al. HBV RNA pre-genome encodes specific motifs that mediate interactions with the viral core protein that promote nucleocapsid assembly. Nat. Microbiol. 2017, 2, 17098. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Seeger, C. Hepadnavirus genome replication and persistence. Cold Spring Harb. Perspect. Med. 2015, 5, a021386. [Google Scholar] [CrossRef] [Green Version]
- Ciupe, S.M.; Ribeiro, R.M.; Perelson, A.S. Antibody responses during hepatitis B viral infection. PLoS Comput. Biol. 2014, 10, e1003730. [Google Scholar] [CrossRef]
- Aunins, T.R.; Marsh, K.A.; Subramanya, G.; Uprichard, S.L.; Perelson, A.S.; Chatterjee, A. Intracellular hepatitis C virus modeling predicts infection dynamics and viral protein mechanisms. J. Virol. 2018, 92, e02098-17. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Hu, J. Hepatitis B virus virology and replication. In Hepatitis B Virus in Human Diseases; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–34. [Google Scholar]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhao, Q.; Sheraz, M.; Cheng, J.; Qi, Y.; Su, Q.; Cuconati, A.; Wei, L.; Du, Y.; Li, W.; et al. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog. 2017, 13, e1006658. [Google Scholar] [CrossRef]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Li, S.; Shen, F.; Li, H.; Qian, S.; Lee, D.H.S.; Wu, J.Z.; Yang, W. Characterization of nucleosome positioning in hepadnaviral covalently closed circular DNA minichromosomes. J. Virol. 2012, 86, 10059–10069. [Google Scholar] [CrossRef] [Green Version]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [Green Version]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Investig. 2012, 122, 529–537. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Campagna, M.; Qi, Y.; Zhao, X.; Guo, F.; Xu, C.; Li, S.; Li, W.; Block, T.M.; Chang, J.; et al. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog. 2013, 9, e1003613. [Google Scholar] [CrossRef] [Green Version]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Jansen, M.; Pfaffelhuber, P. Stochastic gene expression with delay. J. Theor. Biol. 2015, 364, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Suk-Fong Lok, A. Hepatitis B treatment: What we know now and what remains to be researched. Hepatol. Commun. 2019, 3, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, R.; Salahub, D. Delay stochastic simulation of single-gene expression reveals a detailed relationship between protein noise and mean abundance. FEBS Lett. 2008, 582, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Toft, D.O.; Seeger, C. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 1997, 16, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef]
- Hu, J.; Lin, L. RNA-protein interactions in hepadnavirus reverse transcription. Front. Biosci. 2009, 14, 1606. [Google Scholar] [CrossRef] [Green Version]
- Junker-Niepmann, M.; Bartenschlager, R.; Schaller, H. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990, 9, 3389–3396. [Google Scholar] [CrossRef]
- Hirsch, R.C.; Loeb, D.D.; Pollack, J.R.; Ganem, D. cis-acting sequences required for encapsidation of duck hepatitis B virus pregenomic RNA. J. Virol. 1991, 65, 3309–3316. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.J.C.; Beales, L.; Blond, D.; Simon, M.N.; Lin, B.Y.; Chisari, F.V.; Stuart, D.I.; Rowlands, D.J. Hepatitis B small surface antigen particles are octahedral. Proc. Natl. Acad. Sci. USA 2005, 102, 14783–14788. [Google Scholar] [CrossRef] [Green Version]
- Prange, R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 2012, 201, 449–461. [Google Scholar] [CrossRef]
- Schreiner, S.; Nassal, M. A role for the host DNA damage response in hepatitis B virus cccDNA formation—and beyond? Viruses 2017, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Nishi, R.; Wijnhoven, P.W.G.; Kimura, Y.; Matsui, M.; Konietzny, R.; Wu, Q.; Nakamura, K.; Blundell, T.L.; Kessler, B.M. The deubiquitylating enzyme UCHL3 regulates Ku80 retention at sites of DNA damage. Sci. Rep. 2018, 8, 17891. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.; Shackel, N.; Urban, S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Döring, T.; Prange, R. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and γ2-adaptin. J. Virol. 2007, 81, 9050–9060. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.; Smith, P.M.; Horwich, A.L. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 1990, 64, 2819–2824. [Google Scholar] [CrossRef] [Green Version]
- Short, J.M.; Chen, S.; Roseman, A.M.; Butler, P.J.G.; Crowther, R.A. Structure of hepatitis B surface antigen from subviral tubes determined by electron cryomicroscopy. J. Mol. Biol. 2009, 390, 135–141. [Google Scholar] [CrossRef]
- Roingeard, P.; Sureau, C. Ultrastructural analysis of hepatitis B virus in HepG2-transfected cells with special emphasis on subviral filament morphogenesis. Hepatology 1998, 28, 1128–1133. [Google Scholar] [CrossRef]
- Dryden, K.A.; Wieland, S.F.; Whitten-Bauer, C.; Gerin, J.L.; Chisari, F.V.; Yeager, M. Native hepatitis B virions and capsids visualized by electron cryomicroscopy. Mol. Cell 2006, 22, 843–850. [Google Scholar] [CrossRef]
- Patient, R.; Hourioux, C.; Roingeard, P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 2009, 11, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Van Bömmel, F.; Bartens, A.; Mysickova, A.; Hofmann, J.; Krüger, D.H.; Berg, T.; Edelmann, A. Serum hepatitis B virus RNA levels as an early predictor of hepatitis B envelope antigen seroconversion during treatment with polymerase inhibitors. Hepatology 2015, 61, 66–76. [Google Scholar] [CrossRef]
- Rokuhara, A.; Matsumoto, A.; Tanaka, E.; Umemura, T.; Yoshizawa, K.; Kimura, T.; Maki, N.; Kiyosawa, K. Hepatitis B virus RNA is measurable in serum and can be a new marker for monitoring lamivudine therapy. J. Gastroenterol. 2006, 41, 785–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.W.; Chayama, K.; Tsuge, M.; Takahashi, S.; Hatakeyama, T.; Abe, H.; Hu, J.T.; Liu, C.J.; Lai, M.Y.; Chen, D.S.; et al. Differential effects of interferon and lamivudine on serum HBV RNA inhibition in patients with chronic hepatitis B. Antivir. Ther. 2010, 15, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.W.; Takahashi, S.; Tsuge, M.; Chen, C.L.; Wang, T.C.; Abe, H.; Hu, J.T.; Chen, D.S.; Yang, S.S.; Chayama, K.; et al. On-treatment low serum HBV RNA level predicts initial virological response in chronic hepatitis B patients receiving nucleoside analogue therapy. Antivir. Ther. 2015, 20, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, D.; Zlotnick, A. Model-based analysis of assembly kinetics for virus capsids or other spherical polymers. Biophys. J. 2002, 83, 1217–1230. [Google Scholar] [CrossRef] [Green Version]
- Porterfield, J.Z.; Zlotnick, A. An overview of capsid assembly kinetics. In Emerging Topics in Physical Virology; Imperial College Press: London, UK, 2010; pp. 131–158. [Google Scholar]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral hepatitis B virus filaments, like infectious viral particles, are released via multivesicular bodies. J. Virol. 2016, 90, 3330–3341. [Google Scholar] [CrossRef] [Green Version]
- Patient, R.; Hourioux, C.; Sizaret, P.Y.; Trassard, S.; Sureau, C.; Roingeard, P. Hepatitis B virus subviral envelope particle morphogenesis and intracellular trafficking. J. Virol. 2007, 81, 3842–3851. [Google Scholar] [CrossRef] [Green Version]
- Molnar-Kimber, K.L.; Jarocki-Witek, V.; Dheer, S.K.; Vernon, S.K.; Conley, A.J.; Davis, A.R.; Hung, P.P. Distinctive properties of the hepatitis B virus envelope proteins. J. Virol. 1988, 62, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Fatehi Chenar, F.; Kyrychko, Y.N.; Blyuss, K.B. Mathematical model of immune response to hepatitis B. J. Theor. Biol. 2018, 447, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Micco, L.; Peppa, D.; Loggi, E.; Schurich, A.; Jefferson, L.; Cursaro, C.; Panno, A.M.; Bernardi, M.; Brander, C.; Bihl, F.; et al. Differential boosting of innate and adaptive antiviral responses during pegylated-interferon-alpha therapy of chronic hepatitis B. J. Hepatol. 2013, 58, 225–233. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Thimme, R.; Dandri, M. Dissecting the divergent effects of interferon-alpha on immune cells: Time to rethink combination therapy in chronic hepatitis B? J. Hepatol. 2013, 58, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locarnini, S.; Hatzakis, A.; Chen, D.S.; Lok, A. Strategies to control hepatitis B: Public policy, epidemiology, vaccine and drugs. J. Hepatol. 2015, 62, S76–S86. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Durantel, D. Antiviral therapies and prospects for a cure of chronic hepatitis B. Cold Spring Harb. Perspect. Med. 2015, 5, a021501. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Toft, D.O. Binding of p23 and hsp90 during assembly with the progesterone receptor. Mol. Endocrinol. 1995, 9, 670–678. [Google Scholar]
- Smith, D.F.; Whitesell, L.; Nair, S.C.; Chen, S.; Prapapanich, V.; Rimerman, R.A. Progesterone receptor structure and function altered by geldanamycin, an hsp90-binding agent. Mol. Cell. Biol. 1995, 15, 6804–6812. [Google Scholar] [CrossRef] [Green Version]
- Zoulim, F.; Yogaratnam, J.Z.; Vandenbossche, J.J.; Lenz, O.; Talloen, W.; Vistuer, C.; Moscalu, I.; Streinu-Cercel, A.; Bourgeois, S.; Buti, M.; et al. Safety, Tolerability, Pharmacokinetics and Antiviral Activity of JNJ-56136379, a Novel HBV Capsid Assembly Modulator, in Non-cirrhotic, Treatment-naive Subjects with Chronic Hepatitis B. Hepatology 2017, 66, 1263A–1264A. [Google Scholar]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Van Damme, E.; Mostmans, W.; Vandyck, K.; Pauwels, F. Capsid assembly modulators have a dual mechanism of action in primary human hepatocytes infected with hepatitis B virus. Antimicrob. Agents Chemother. 2017, 61, e00560-17. [Google Scholar] [CrossRef] [Green Version]
- Schlicksup, C.J.; Wang, J.C.Y.; Francis, S.; Venkatakrishnan, B.; Turner, W.W.; VanNieuwenhze, M.; Zlotnick, A. Hepatitis B virus core protein allosteric modulators can distort and disrupt intact capsids. Elife 2018, 7, e31473. [Google Scholar] [CrossRef]
- Lahlali, T.; Berke, J.M.; Vergauwen, K.; Foca, A.; Vandyck, K.; Pauwels, F.; Zoulim, F.; Durantel, D. Novel potent capsid assembly modulators regulate multiple steps of the hepatitis B virus life cycle. Antimicrob. Agents Chemother. 2018, 62, e00835-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Y.; Wei, Z.M.; Wu, G.Y.; Wang, J.H.; Zhang, Y.J.; Li, J.; Zhang, H.H.; Xie, X.W.; Wang, X.; Wang, Z.H.; et al. In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil-resistant HBV mutations. Antivir. Ther. 2012, 17, 793–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billioud, G.; Pichoud, C.; Puerstinger, G.; Neyts, J.; Zoulim, F. The main hepatitis B virus (HBV) mutants resistant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antivir. Res. 2011, 92, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A.; Mukhopadhyay, S. Virus assembly, allostery, and antivirals. Trends Microbiol. 2011, 19, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Richvirology/HBV_Model. Available online: https://github.com/richvirology/HBV_model (accessed on 1 December 2020).
- Balagopal, A.; Grudda, T.; Ribeiro, R.M.; Saad, Y.S.; Hwang, H.S.; Quinn, J.; Murphy, M.; Ward, K.; Sterling, R.K.; Zhang, Y.; et al. Single hepatocytes show persistence and transcriptional inactivity of hepatitis B. JCI Insight 2020, 5, e140584. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatehi, F.; Bingham, R.J.; Dykeman, E.C.; Patel, N.; Stockley, P.G.; Twarock, R. An Intracellular Model of Hepatitis B Viral Infection: An In Silico Platform for Comparing Therapeutic Strategies. Viruses 2021, 13, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010011
Fatehi F, Bingham RJ, Dykeman EC, Patel N, Stockley PG, Twarock R. An Intracellular Model of Hepatitis B Viral Infection: An In Silico Platform for Comparing Therapeutic Strategies. Viruses. 2021; 13(1):11. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010011
Chicago/Turabian StyleFatehi, Farzad, Richard J. Bingham, Eric C. Dykeman, Nikesh Patel, Peter G. Stockley, and Reidun Twarock. 2021. "An Intracellular Model of Hepatitis B Viral Infection: An In Silico Platform for Comparing Therapeutic Strategies" Viruses 13, no. 1: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/v13010011