
Heparan Sulfate Is a Cellular Receptor for Enteric Human Adenoviruses

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses and Antibodies
2.2. Recombinant Fiber Knobs, Enzymes, Metabolic Inhibitors, Soluble GAGs and Cholera Toxin
2.3. Fiber Knob Binding Assay
2.4. Infection Assay
2.5. 35S-Labeled Virion Binding Assay
2.6. HAdV Uptake Assay
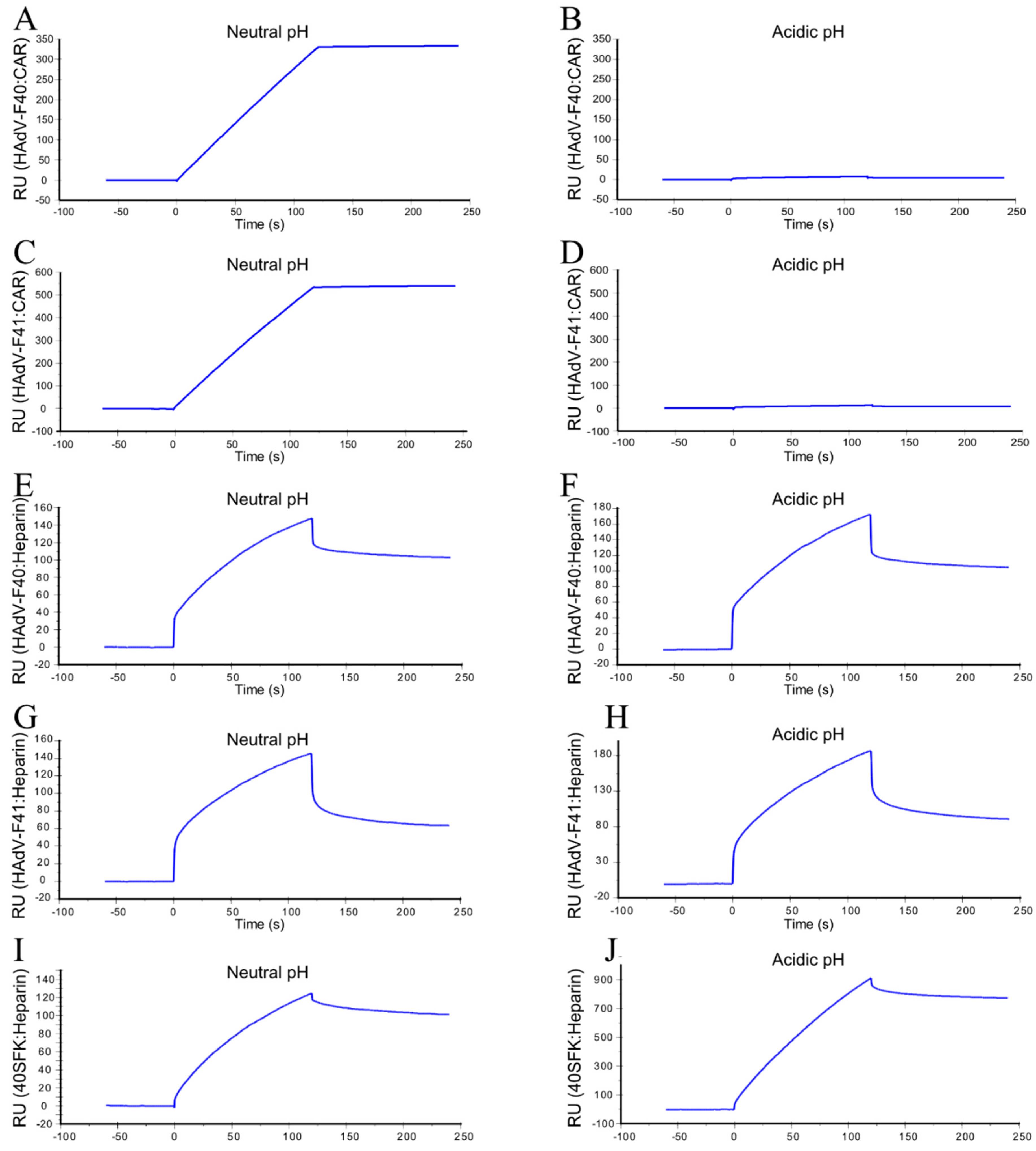
2.7. Surface Plasmon Resonance
2.8. Statistical Analysis
3. Results
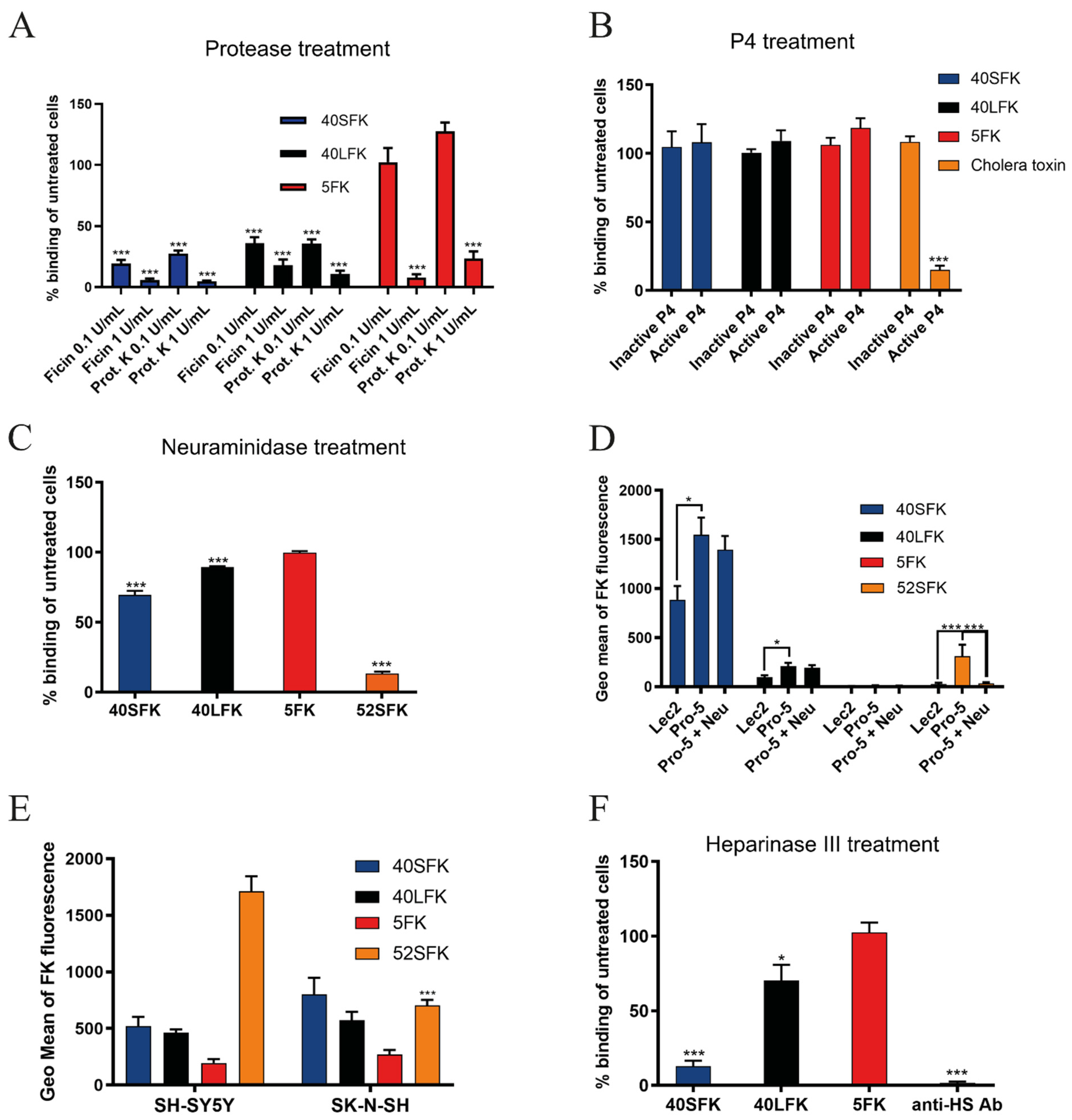
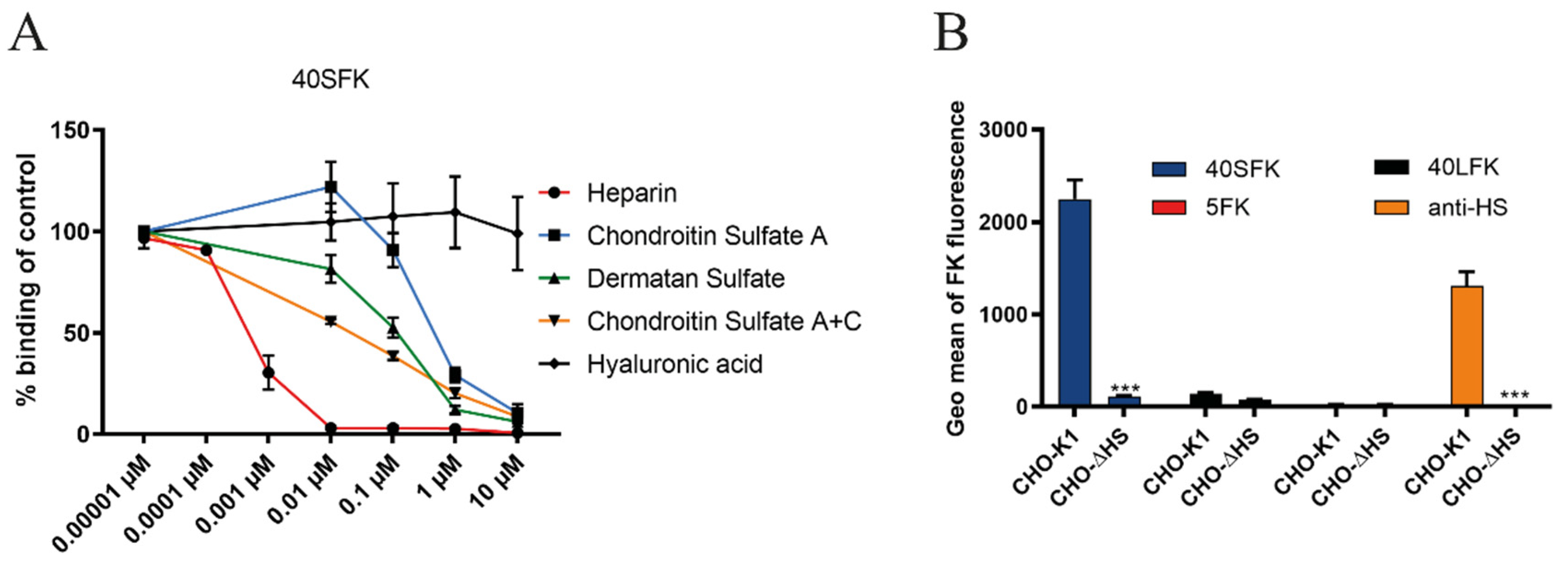
3.1. HAdV-F40 Short Fiber Knob Binding to Cells Requires Heparan Sulfate-Containing Proteins
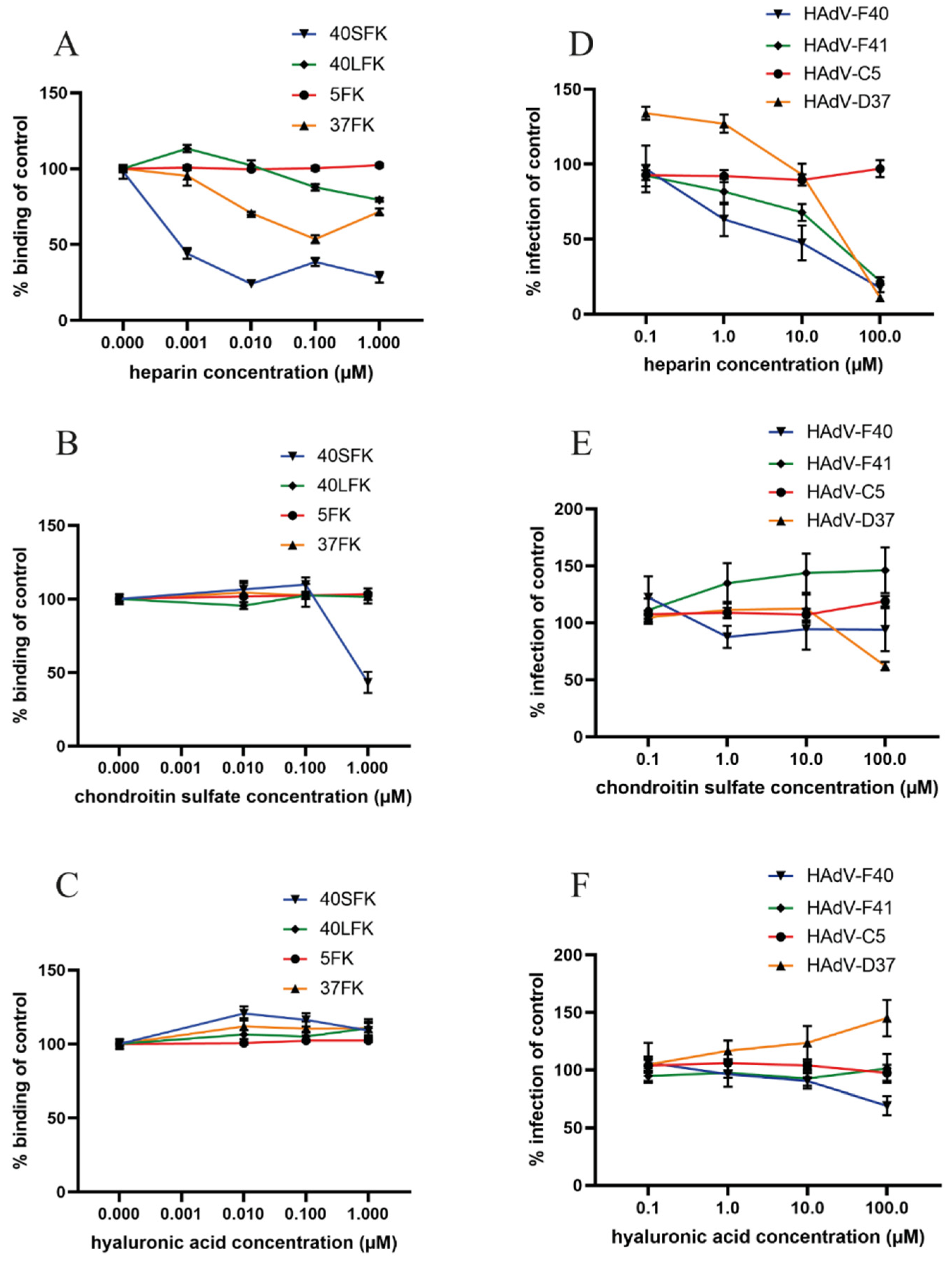
3.2. HAdV-F40 SFK Binding to A549 Cells Requires Sulfated GAGs
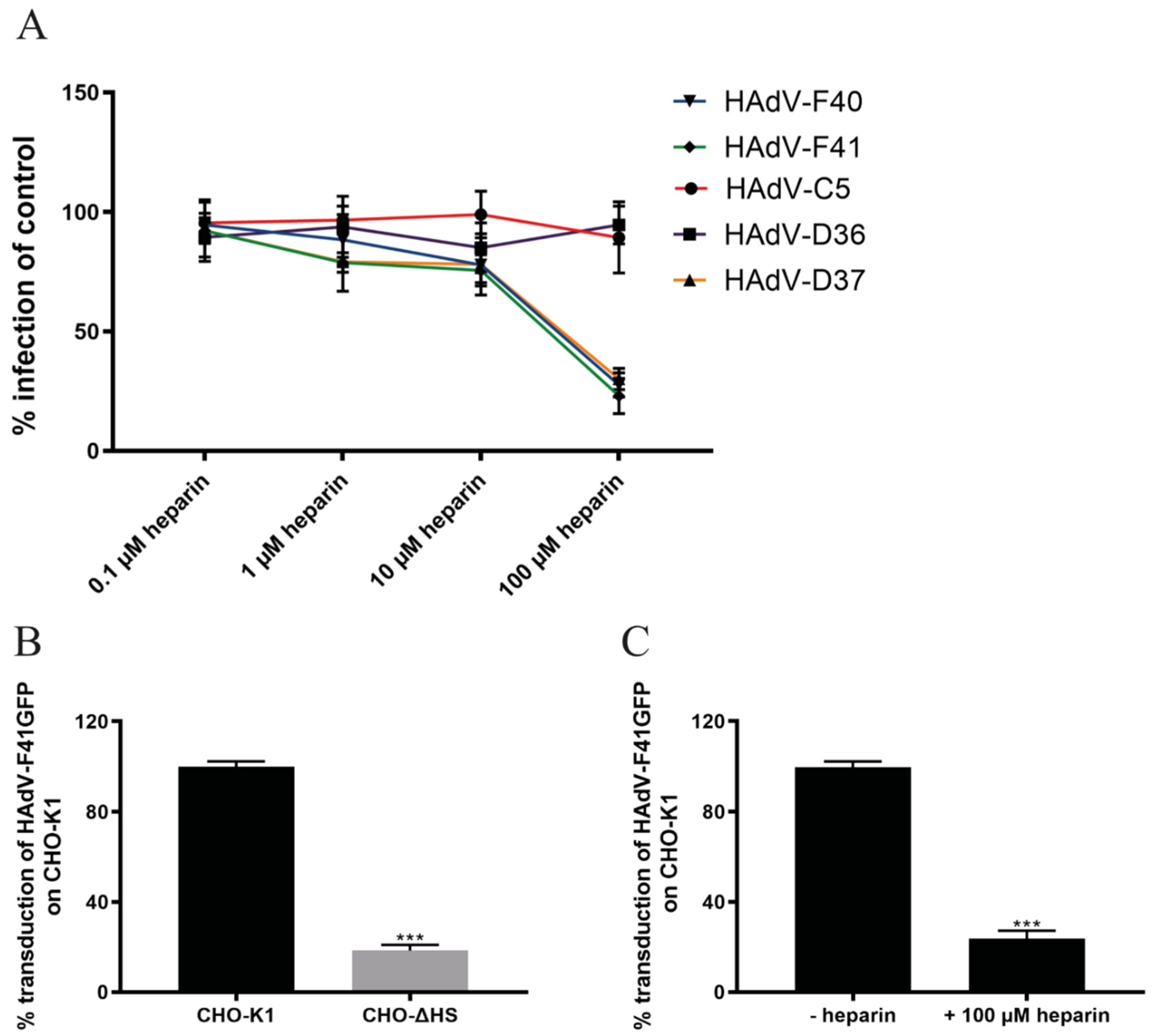
3.3. Sulfated GAGs Play an Important Role in HAdV-F40 and -F41 Infection of A549 Cells
3.4. HAdV-F40 and -F41 Binding to and Uptake in A549 Cells Is Not Affected by Heparin
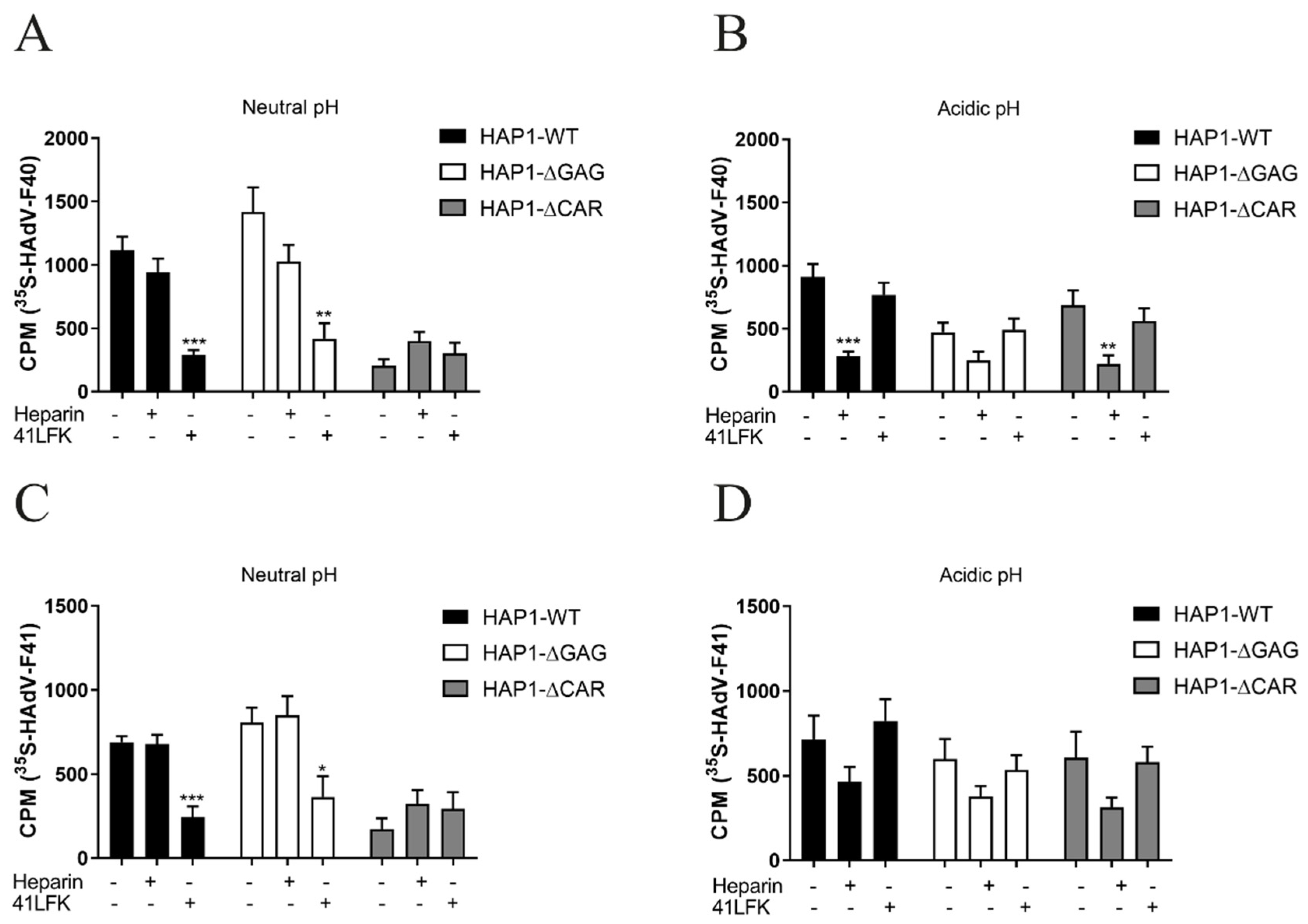
3.5. LFK:CAR-Dependent Cell Attachment of Enteric HAdVs Is Switched to SFK:HS-Dependent Cell Attachment after Virion Exposure to Acidic pH
3.6. Acidic pH-Exposed HAdV-F40 and -F41 Virions Lose CAR-Binding Capacity But Retain HS-Binding Capacity
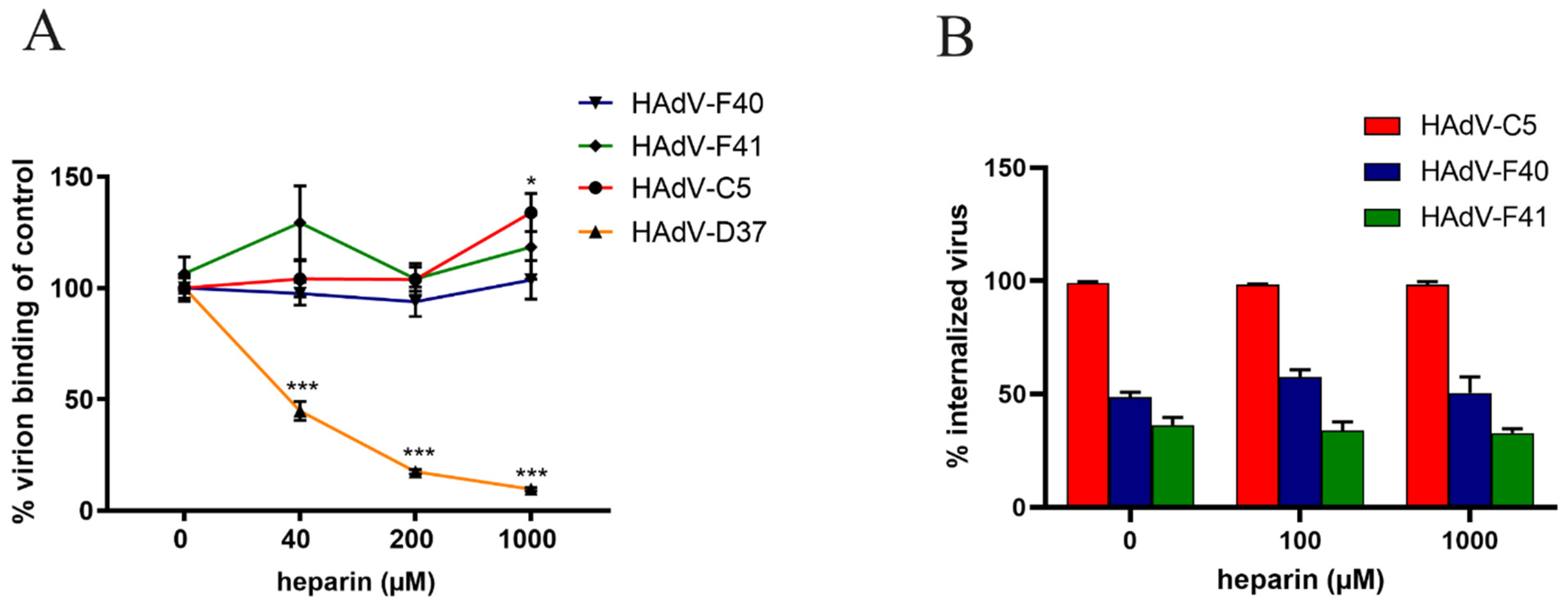
3.7. Heparin Specifically Reduces Infection and Short Fiber Knob Binding on a Small Intestinal Cell Line
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brister, J.R.; Chodosh, J.; Curiel, D.T.; Heim, A.; Jones, M.S.; Kajon, A.; Lion, T.; Seto, D.; Zhang, Q. HAdV Working Group. Available online: http://hadvwg.gmu.edu/ (accessed on 31 December 2019).
- Wold, W.S.M.; Ison, M.G. Adenoviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1732–1767. [Google Scholar]
- Kidd, A.H.; Banatvala, J.E.; de Jong, J.C. Antibodies to fastidious faecal adenoviruses (species 40 and 41) in sera from children. J. Med. Virol. 1983, 11, 333–341. [Google Scholar] [CrossRef]
- Tiemessen, C.T.; Kidd, A.H. The subgroup F adenoviruses. J. Gen. Virol. 1995, 76, 481–497. [Google Scholar] [CrossRef]
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000-15: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- GBD 2016 Diarrhoeal Disease Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef] [Green Version]
- Roelvink, P.W.; Lizonova, A.; Lee, J.G.; Li, Y.; Bergelson, J.M.; Finberg, R.W.; Brough, D.E.; Kovesdi, I.; Wickham, T.J. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J. Virol. 1998, 72, 7909–7915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenman, A.; Liaci, A.M.; Liu, Y.; Ardahl, C.; Rajan, A.; Nilsson, E.; Bradford, W.; Kaeshammer, L.; Jones, M.S.; Frangsmyr, L.; et al. Human adenovirus 52 uses sialic acid-containing glycoproteins and the coxsackie and adenovirus receptor for binding to target cells. PLoS Pathog. 2015, 11, e1004657. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Filardo, E.J.; Cheresh, D.A.; Nemerow, G.R. Integrin alpha v beta 5 selectively promotes adenovirus mediated cell membrane permeabilization. J. Cell. Biol. 1994, 127, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Rajan, A.; Persson, B.D.; Frangsmyr, L.; Olofsson, A.; Sandblad, L.; Heino, J.; Takada, Y.; Mould, A.P.; Schnapp, L.M.; Gall, J.; et al. Enteric Species F Human Adenoviruses use Laminin-Binding Integrins as Co-Receptors for Infection of Ht-29 Cells. Sci. Rep. 2018, 8, 10019. [Google Scholar] [CrossRef]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Persson, B.D.; Reiter, D.M.; Marttila, M.; Mei, Y.F.; Casasnovas, J.M.; Arnberg, N.; Stehle, T. Adenovirus type 11 binding alters the conformation of its receptor CD46. Nat. Struct. Mol. Biol. 2007, 14, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Marttila, M.; Persson, D.; Gustafsson, D.; Liszewski, M.K.; Atkinson, J.P.; Wadell, G.; Arnberg, N. CD46 is a cellular receptor for all species B adenoviruses except types 3 and 7. J. Virol. 2005, 79, 14429–14436. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, Z.Y.; Liu, Y.; Persson, J.; Beyer, I.; Moller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.C.; Storm, R.J.; Bauer, J.; Johansson, S.M.; Lookene, A.; Angstrom, J.; Hedenstrom, M.; Eriksson, T.L.; Frangsmyr, L.; Rinaldi, S.; et al. The GD1a glycan is a cellular receptor for adenoviruses causing epidemic keratoconjunctivitis. Nat. Med. 2011, 17, 105–109. [Google Scholar] [CrossRef]
- Lenman, A.; Liaci, A.M.; Liu, Y.; Frangsmyr, L.; Frank, M.; Blaum, B.S.; Chai, W.; Podgorski, I.I.; Harrach, B.; Benko, M.; et al. Polysialic acid is a cellular receptor for human adenovirus 52. Proc. Natl. Acad. Sci. USA 2018, 115, E4264–E4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechecchi, M.C.; Melotti, P.; Bonizzato, A.; Santacatterina, M.; Chilosi, M.; Cabrini, G. Heparan sulfate glycosaminoglycans are receptors sufficient to mediate the initial binding of adenovirus types 2 and 5. J. Virol. 2001, 75, 8772–8780. [Google Scholar] [CrossRef] [Green Version]
- Dechecchi, M.C.; Tamanini, A.; Bonizzato, A.; Cabrini, G. Heparan sulfate glycosaminoglycans are involved in adenovirus type 5 and 2-host cell interactions. Virology 2000, 268, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Tuve, S.; Wang, H.; Jacobs, J.D.; Yumul, R.C.; Smith, D.F.; Lieber, A. Role of cellular heparan sulfate proteoglycans in infection of human adenovirus serotype 3 and 35. PLoS Pathog. 2008, 4, e1000189. [Google Scholar] [CrossRef] [Green Version]
- Chandra, N.; Liu, Y.; Liu, J.X.; Frangsmyr, L.; Wu, N.; Silva, L.M.; Lindstrom, M.; Chai, W.; Pedrosa Domellof, F.; Feizi, T.; et al. Sulfated Glycosaminoglycans as Viral Decoy Receptors for Human Adenovirus Type 37. Viruses 2019, 11, 247. [Google Scholar] [CrossRef] [Green Version]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; The Consortium of Glycobiology Editors: La Jolla, CA, USA, 2009. [Google Scholar]
- Kidd, A.H.; Chroboczek, J.; Cusack, S.; Ruigrok, R.W. Adenovirus type 40 virions contain two distinct fibers. Virology 1993, 192, 73–84. [Google Scholar] [CrossRef]
- Song, J.D.; Liu, X.L.; Chen, D.L.; Zou, X.H.; Wang, M.; Qu, J.G.; Lu, Z.Z.; Hung, T. Human adenovirus type 41 possesses different amount of short and long fibers in the virion. Virology 2012, 432, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.S., 2nd; Harrach, B.; Ganac, R.D.; Gozum, M.M.; Dela Cruz, W.P.; Riedel, B.; Pan, C.; Delwart, E.L.; Schnurr, D.P. New adenovirus species found in a patient presenting with gastroenteritis. J. Virol. 2007, 81, 5978–5984. [Google Scholar] [CrossRef] [Green Version]
- Westerberg, S.; Hagbom, M.; Rajan, A.; Loitto, V.; Persson, B.D.; Allard, A.; Nordgren, J.; Sharma, S.; Magnusson, K.E.; Arnberg, N.; et al. Interaction of Human Enterochromaffin Cells with Human Enteric Adenovirus 41 Leads to Serotonin Release and Subsequent Activation of Enteric Glia Cells. J. Virol. 2018, 92, e00026-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafie, K.; Lenman, A.; Fuchs, J.; Rajan, A.; Arnberg, N.; Carlson, L.-A. The structure of enteric human adenovirus 41—A leading cause of diarrhea in children. Sci. Adv. 2021, 7, eabe0974. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.; Romero, C.; Rio, A.; Miralles, M.; Raventos, A.; Planells, L.; Burgueno, J.F.; Hamada, H.; Perales, J.C.; Bosch, A.; et al. Short-fiber protein of ad40 confers enteric tropism and protection against acidic gastrointestinal conditions. Hum. Gene Ther. Methods 2013, 24, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Favier, A.L.; Burmeister, W.P.; Chroboczek, J. Unique physicochemical properties of human enteric Ad41 responsible for its survival and replication in the gastrointestinal tract. Virology 2004, 322, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Lidholt, K.; Weinke, J.L.; Kiser, C.S.; Lugemwa, F.N.; Bame, K.J.; Cheifetz, S.; Massague, J.; Lindahl, U.; Esko, J.D. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 2267–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutscher, S.L.; Nuwayhid, N.; Stanley, P.; Briles, E.I.; Hirschberg, C.B. Translocation across Golgi vesicle membranes: A CHO glycosylation mutant deficient in CMP-sialic acid transport. Cell 1984, 39, 295–299. [Google Scholar] [CrossRef]
- Stanley, P.; Caillibot, V.; Siminovitch, L. Selection and characterization of eight phenotypically distinct lines of lectin-resistant Chinese hamster ovary cell. Cell 1975, 6, 121–128. [Google Scholar] [CrossRef]
- Johansson, S.M.; Arnberg, N.; Elofsson, M.; Wadell, G.; Kihlberg, J. Multivalent HSA conjugates of 3′-sialyllactose are potent inhibitors of adenoviral cell attachment and infection. Chembiochem 2005, 6, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Lemiale, F.; Haddada, H.; Nabel, G.J.; Brough, D.E.; King, C.R.; Gall, J.G. Novel adenovirus vaccine vectors based on the enteric-tropic serotype 41. Vaccine 2007, 25, 2074–2084. [Google Scholar] [CrossRef] [Green Version]
- Arnberg, N.; Kidd, A.H.; Edlund, K.; Nilsson, J.; Pring-Akerblom, P.; Wadell, G. Adenovirus type 37 binds to cell surface sialic acid through a charge-dependent interaction. Virology 2002, 302, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Mistry, N.; Inoue, H.; Jamshidi, F.; Storm, R.J.; Oberste, M.S.; Arnberg, N. Coxsackievirus A24 variant uses sialic acid-containing O-linked glycoconjugates as cellular receptors on human ocular cells. J. Virol. 2011, 85, 11283–11290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Witt, D.J.; Bousquet, E.B. Comparison of Enteric Adenovirus Infection in Various Human Cell-Lines. J. Virol. Methods 1988, 20, 295–308. [Google Scholar] [CrossRef]
- Arnberg, N.; Kidd, A.H.; Edlund, K.; Olfat, F.; Wadell, G. Initial interactions of subgenus D adenoviruses with A549 cellular receptors: Sialic acid versus alpha(v) integrins. J. Virol. 2000, 74, 7691–7693. [Google Scholar] [CrossRef] [Green Version]
- Varki, A. Sialic acids in human health and disease. Trends Mol Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [Green Version]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Parizhskaya, M.; Walpusk, J.; Mazariegos, G.; Jaffe, R. Enteric adenovirus infection in pediatric small bowel transplant recipients. Pediatr. Dev. Pathol. 2001, 4, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.W.; Freimuth, P.; Moninger, T.O.; Ganske, I.; Zabner, J.; Welsh, M.J. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 2002, 110, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Rebetz, J.; Na, M.; Su, C.; Holmqvist, B.; Edqvist, A.; Nyberg, C.; Widegren, B.; Salford, L.G.; Sjogren, H.O.; Arnberg, N.; et al. Fiber mediated receptor masking in non-infected bystander cells restricts adenovirus cell killing effect but promotes adenovirus host co-existence. PLoS ONE 2009, 4, e8484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, M.K.; Graham, D.Y.; Mason, B.B. Proteolytic enhancement of rotavirus infectivity: Molecular mechanisms. J. Virol. 1981, 39, 879–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favier, A.-L.; Grzela, R.; Tcherniuk, S.; Harsi, M.C.; Chroboczek, J. A Novel Human Membrane Protein Interacting with the Short Fiber of Enteric Adenovirus. ISRN Virol. 2013, 2013, 709734. [Google Scholar] [CrossRef] [Green Version]
- Finne, J.; Finne, U.; Deagostini-Bazin, H.; Goridis, C. Occurrence of alpha 2-8 linked polysialosyl units in a neural cell adhesion molecule. Biochem. Biophys. Res. Commun. 1983, 112, 482–487. [Google Scholar] [CrossRef]
- Galuska, S.P.; Rollenhagen, M.; Kaup, M.; Eggers, K.; Oltmann-Norden, I.; Schiff, M.; Hartmann, M.; Weinhold, B.; Hildebrandt, H.; Geyer, R.; et al. Synaptic cell adhesion molecule SynCAM 1 is a target for polysialylation in postnatal mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 10250–10255. [Google Scholar] [CrossRef] [Green Version]
- Werneburg, S.; Muhlenhoff, M.; Stangel, M.; Hildebrandt, H. Polysialic acid on SynCAM 1 in NG2 cells and on neuropilin-2 in microglia is confined to intracellular pools that are rapidly depleted upon stimulation. Glia 2015, 63, 1240–1255. [Google Scholar] [CrossRef]
- Kiermaier, E.; Moussion, C.; Veldkamp, C.T.; Gerardy-Schahn, R.; de Vries, I.; Williams, L.G.; Chaffee, G.R.; Phillips, A.J.; Freiberger, F.; Imre, R.; et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science 2016, 351, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.X.; Zou, X.H.; Jiang, S.Y.; Lu, N.N.; Han, M.; Zhao, J.H.; Guo, X.J.; Zhao, S.C.; Lu, Z.Z. Prevalence of serum neutralizing antibodies to adenovirus type 5 (Ad5) and 41 (Ad41) in children is associated with age and sanitary conditions. Vaccine 2016, 34, 5579–5586. [Google Scholar] [CrossRef]
- Shinozaki, T.; Araki, K.; Ushijima, H.; Fujii, R. Antibody response to enteric adenovirus types 40 and 41 in sera from people in various age groups. J. Clin. Microbiol. 1987, 25, 1679–1682. [Google Scholar] [CrossRef] [Green Version]
- Banyai, K.; Martella, V.; Meleg, E.; Kisfali, P.; Peterfi, Z.; Benko, M.; Melegh, B.; Szucs, G. Searching for HAdV-52, the putative gastroenteritis-associated human adenovirus serotype in Southern Hungary. New Microbiol. 2009, 32, 185–188. [Google Scholar]
- Kesisoglou, F.; Chamberlain, J.R.; Schmiedlin-Ren, P.; Kaz, A.; Fleisher, D.; Roessler, B.; Zimmermann, E.M. Chimeric Ad5 vectors expressing the short fiber of Ad41 show reduced affinity for human intestinal epithelium. Mol. Pharm. 2005, 2, 500–508. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajan, A.; Palm, E.; Trulsson, F.; Mundigl, S.; Becker, M.; Persson, B.D.; Frängsmyr, L.; Lenman, A. Heparan Sulfate Is a Cellular Receptor for Enteric Human Adenoviruses. Viruses 2021, 13, 298. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020298
Rajan A, Palm E, Trulsson F, Mundigl S, Becker M, Persson BD, Frängsmyr L, Lenman A. Heparan Sulfate Is a Cellular Receptor for Enteric Human Adenoviruses. Viruses. 2021; 13(2):298. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020298
Chicago/Turabian StyleRajan, Anandi, Elin Palm, Fredrik Trulsson, Sarah Mundigl, Miriam Becker, B. David Persson, Lars Frängsmyr, and Annasara Lenman. 2021. "Heparan Sulfate Is a Cellular Receptor for Enteric Human Adenoviruses" Viruses 13, no. 2: 298. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020298