
Molecular Characterization of Closely Related H6N2 Avian Influenza Viruses Isolated from Turkey, Egypt, and Uganda

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Legal Permits
2.2. Viruses
2.3. Whole Genome Sequencing
2.4. Nucleic Acid and Amino Acid Identities of Virus Isolates
2.5. Sequence Analyses
2.6. Phylogenetic Tree Construction
2.7. Sequence Accession Numbers
3. Results
3.1. Genetic Relatedness of H6N2 Isolates from Turkey, Egypt, and Uganda
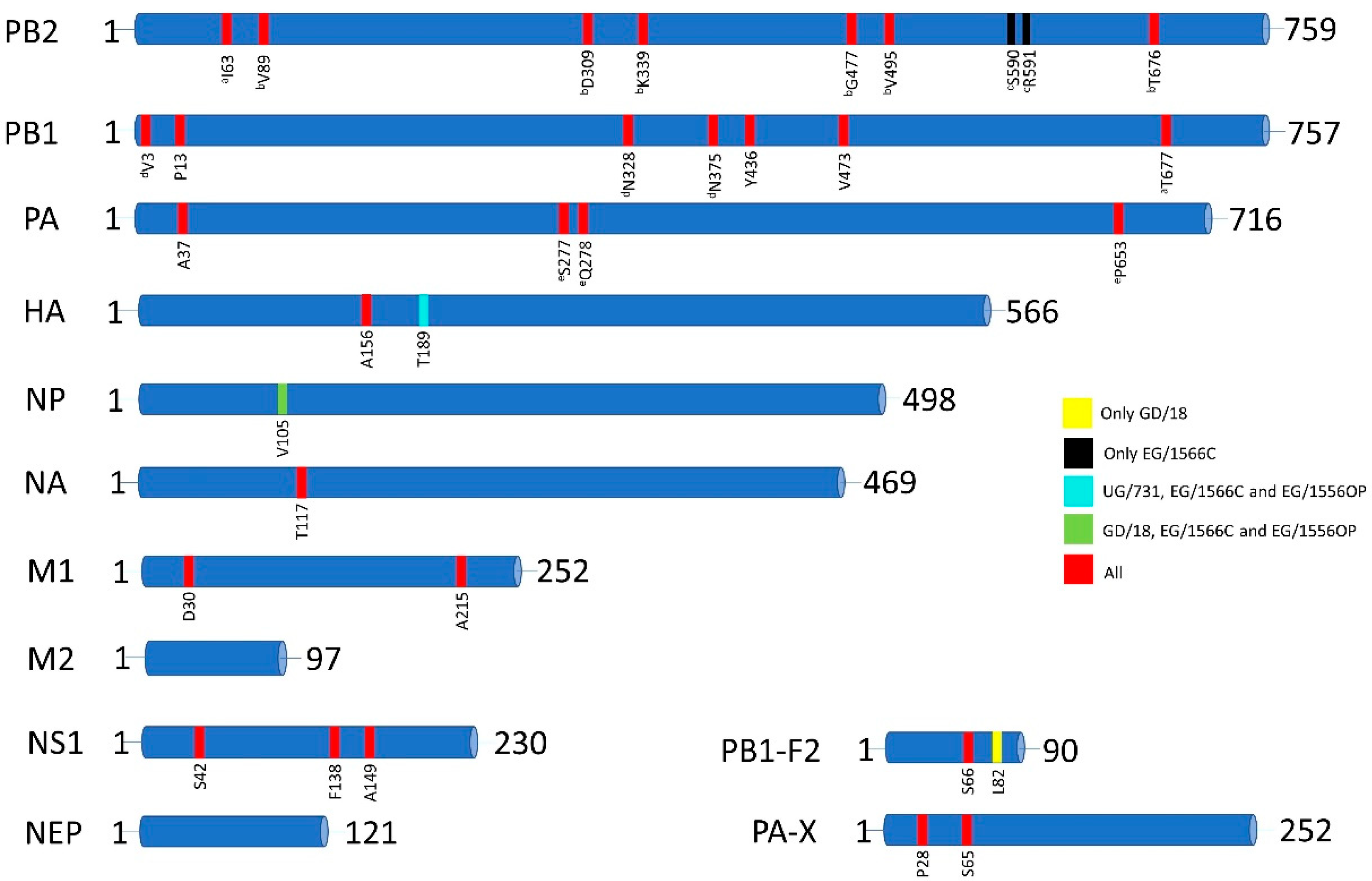
3.2. Amino Acid Markers of Pathogenicity, Mammalian Adaptation and Antiviral Resistance
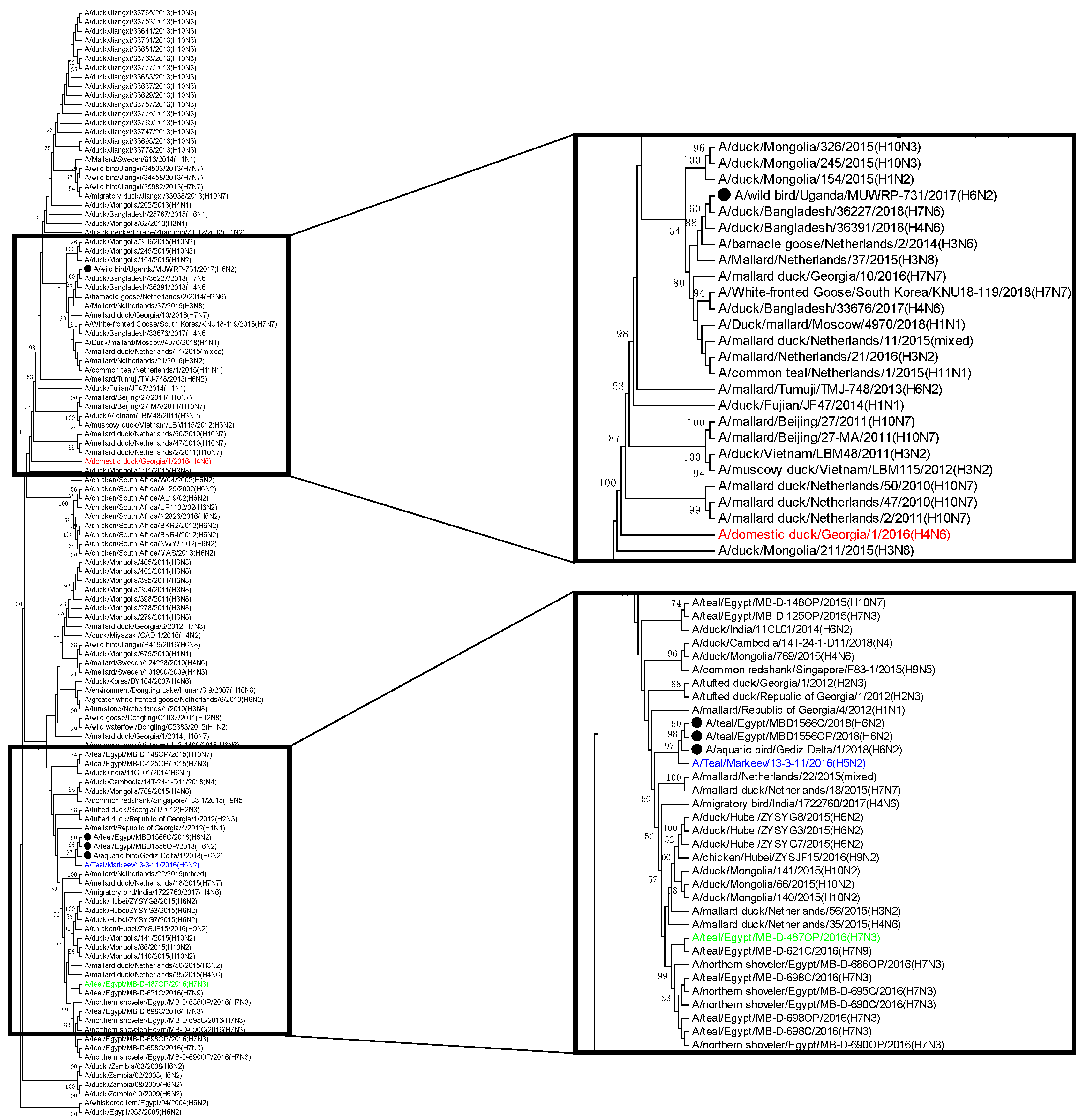
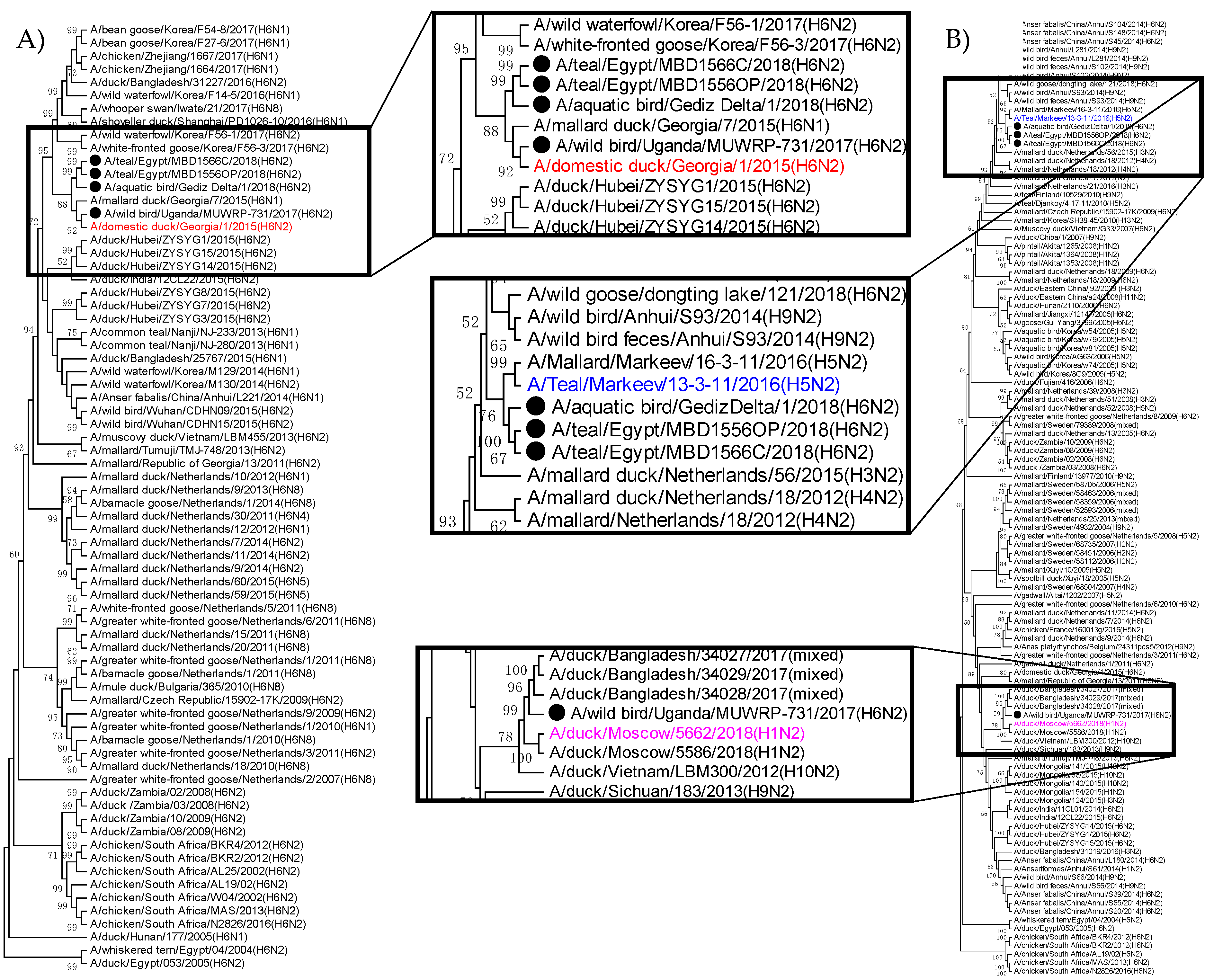
3.3. Phylogenetic Relatedness of H6N2 Isolates from Turkey, Egypt, and Uganda
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scholtissek, C.; Rohde, W.V.; Von Hoyningen, V.; Rott, R. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 1978, 87, 13–20. [Google Scholar] [CrossRef]
- Kawaoka, Y.; Krauss, S.; Webster, R.G. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J. Virol. 1989, 63, 4603–4608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The mother of all pandemics. Emerg. Infect. Dis. 2006, 17, 69–79. [Google Scholar] [CrossRef]
- Smith, G.J.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumming, G.S.; Caron, A.; Abolnik, C.; Cattoli, G.; Bruinzeel, L.W.; Burger, C.E.; Cecchettin, K.; Chiweshe, N.; Mochotlhoane, B.; Mutumi, G.L.; et al. The ecology of influenza A viruses in wild birds in southern Africa. EcoHealth 2011, 8, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Zhu, H.; Fan, X.; Wang, J.; Cheung, C.L.; Duan, L.; Hong, W.; Liu, Y.; Li, L.; Smith, D.K.; et al. Establishment and lineage replacement of H6 influenza viruses in domestic ducks in southern China. J. Virol. 2012, 86, 6075–6083. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Nagarajan, S.; Murugkar, H.V.; Saikia, B.; Singh, B.; Mishra, A.; Tripathi, S.K.; Agarwal, S.; Shukla, S.; Kulkarni, D.D.; et al. Emergence of novel reassortant H6N2 avian influenza viruses in ducks in India. Infect. Genet. Evol. 2018, 61, 20–23. [Google Scholar] [CrossRef]
- Zanaty, A.M.; Erfan, A.M.; Mady, W.H.; Amer, F.; Nour, A.A.; Rabie, N.; Samy, M.; Selim, A.A.; Hassan, W.M.; Naguib, M.M. Avian influenza virus surveillance in migratory birds in Egypt revealed a novel reassortant H6N2 subtype. Avian Res. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gillim-Ross, L.; Santos, C.; Chen, Z.; Aspelund, A.; Yang, C.F.; Ye, D.; Jin, H.; Kemble, G.; Subbarao, K. Avian influenza H6 viruses productively infect and cause illness in mice and ferrets. J. Virol. 2008, 82, 10854–10863. [Google Scholar] [CrossRef] [Green Version]
- Koçer, Z.A.; Krauss, S.; Stallknecht, D.E.; Rehg, J.E.; Webster, R.G. The potential of avian H1N1 influenza A viruses to replicate and cause disease in mammalian models. PLoS ONE 2012, 7, e41609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Z.; Ma, S.; Kong, H.; Deng, G.; Shi, J.; Liu, L.; Suzuki, Y.; Chen, H. Identification of a key amino acid in hemagglutinin that increases human-type receptor binding and transmission of an H6N2 avian influenza virus. Microb. Infect. 2017, 19, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.P.; Setterquist, S.F.; Capuano, A.W.; Gray, G.C. Infection due to 3 avian influenza subtypes in United States veterinarians. Clin. Infect. Dis. 2007, 45, 4–9. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, L.; Kan, X.; Jiang, L.; Yang, J.; Guo, Z.; Ren, Q. Origin and molecular characteristics of a novel 2013 avian influenza A (H6N1) virus causing human infection in Taiwan. Clin. Infect. Dis. 2013, 57, 1367–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, L.; Bai, T.; Zhou, J.F.; Chen, Y.K.; Li, X.D.; Zhu, W.F.; Li, Y.; Tang, J.; Chen, T.; Qin, K.; et al. Seropositivity for avian influenza H6 virus among humans, China. Emerg. Infect. Dis. 2015, 21, 1267. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, J. The Flyway Approach to Conserving Migratory Birds. Its Necessity and Value; UNEP/CMS Secretariat: Bonn, Germany, 2009. [Google Scholar]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef]
- Barman, S.; Turner, J.C.; Hasan, M.K.; Akhtar, S.; El-Shesheny, R.; Franks, J.; Walker, D.; Seiler, P.; Friedman, K.; Kercher, L.; et al. Continuing evolution of highly pathogenic H5N1 viruses in Bangladeshi live poultry markets. Emerg. Microbes. Infect. 2019, 8, 650–661. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT; Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Hasegawa, M.; Kishino, H.; Yano, T.A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, E.; Dugan, V.G.; Stockwell, T.B.; Lindsay, L.L.; Plancarte, M.; Boyce, W.M. Evaluation of phenotypic markers in full genome sequences of avian influenza isolates from California. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 521–536. [Google Scholar] [CrossRef]
- Pu, Z.; Xiang, D.; Li, X.; Luo, T.; Shen, X.; Murphy, R.W.; Liao, M.; Shen, Y. Potential pandemic of H7N9 avian influenza A virus in human. Front. Cell. Infect. Microbiol. 2018, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Suttie, A.; Deng, Y.M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of molecular markers affecting biological characteristics of avian influenza A viruses. Virus Genes 2019, 55, 739–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ishaq, M.; Prudence, M.; Xi, X.; Hu, T.; Liu, Q.; Guo, D. Single mutation at the amino acid position 627 of PB2 that leads to increased virulence of an H5N1 avian influenza virus during adaptation in mice can be compensated by multiple mutations at other sites of PB2. Virus Res. 2009, 144, 123–129. [Google Scholar] [CrossRef]
- Mehle, A.; Doudna, J.A. Adaptive strategies of the influenza virus polymerase for replication in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 21312–21316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, Y.; Hu, Y.; Chang, G.; Sun, W.; Yang, Y.; Kang, X.; Wu, X.; Zhu, Q. PB1-mediated virulence attenuation of H5N1 influenza virus in mice is associated with PB2. J. Gen. Virol. 2011, 92, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Govorkova, E.A.; Rehg, J.E.; Krauss, S.; Yen, H.L.; Guan, Y.; Peiris, M.; Nguyen, T.D.; Hanh, T.H.; Puthavathana, P.; Long, H.T.; et al. Lethality to ferrets of H5N1 influenza viruses isolated from humans and poultry in 2004. J. Virol. 2005, 79, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Hulse-Post, D.J.; Franks, J.; Boyd, K.; Salomon, R.; Hoffmann, E.; Yen, H.L.; Webby, R.J.; Walker, D.; Nguyen, T.D.; Webster, R.G. Molecular changes in the polymerase genes (PA and PB1) associated with high pathogenicity of H5N1 influenza virus in mallard ducks. J. Virol. 2007, 81, 8515–8524. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.D.; Stech, J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590–18595. [Google Scholar] [CrossRef] [Green Version]
- Salomon, R.; Franks, J.; Govorkova, E.A.; Ilyushina, N.A.; Yen, H.L.; Hulse-Post, D.J.; Humberd, J.; Trichet, M.; Rehg, J.E.; Webby, R.J.; et al. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. JEM 2006, 203, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Hu, W.B.; Xu, K.; He, Y.X.; Wang, T.Y.; Chen, Z.; Li, T.X.; Liu, J.H.; Buchy, P.; Sun, B. Amino acids 473V and 598P of PB1 from an avian-origin influenza A virus contribute to polymerase activity, especially in mammalian cells. J. Gen. Virol. 2012, 93, 531–540. [Google Scholar] [CrossRef]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007, 2, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Conenello, G.M.; Tisoncik, J.R.; Rosenzweig, E.; Varga, Z.T.; Palese, P.; Katze, M.G. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 2011, 85, 652–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alymova, I.V.; Green, A.M.; van de Velde, N.; McAuley, J.L.; Boyd, K.L.; Ghoneim, H.E.; McCullers, J.A. Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 2011, 85, 12324–12333. [Google Scholar] [CrossRef] [Green Version]
- Mei, K.; Liu, G.; Chen, Z.; Gao, Z.; Zhao, L.; Jin, T.; Yu, X.; Chen, Q. Deep sequencing reveals the viral adaptation process of environment-derived H10N8 in mice. Infect. Genet. Evol. 2016, 37, 8–13. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamada, S.; Fukuyama, S.; Murakami, S.; Zhao, D.; Uraki, R.; Watanabe, T.; Tomita, Y.; Macken, C.; Neumann, G.; et al. Virulence-affecting amino acid changes in the PA protein of H7N9 influenza A viruses. J. Virol. 2014, 88, 3127–3134. [Google Scholar] [CrossRef] [Green Version]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Identification of amino acid residues in influenza A virus PA-X that contribute to enhanced shutoff activity. Front. Microbiol. 2019, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhang, Y.; Shinya, K.; Deng, G.; Jiang, Y.; Li, Z.; Guan, Y.; Tian, G.; Li, Y.; Shi, J.; et al. Identification of amino acids in HA and PB2 critical for the transmission of H5N1 avian influenza viruses in a mammalian host. PLoS Pathog. 2009, 5, 1000709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Li, Q.; Gao, R.; He, D.; Xu, Y.; Xu, H.; Xu, L.; Wang, X.; Hu, J.; Liu, X.; et al. The T160A hemagglutinin substitution affects not only receptor binding property but also transmissibility of H5N1 clade 2.3. 4 avian influenza virus in guinea pigs. Vet. Res. 2017, 48, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Bouwman, K.M.; McBride, R.; Grant, O.C.; Woods, R.J.; Verheije, M.H.; Paulson, J.C.; de Vries, R.P. Enhanced human-type receptor binding by ferret-transmissible H5N1 with a K193T mutation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, T.; Suzuki, K.; Sakurai, Y.; Kubo, M.; Okada, H.; Itoh, T.; Tsukamoto, K. NP body domain and PB2 contribute to increased virulence of H5N1 highly pathogenic avian influenza viruses in chickens. J. Virol. 2011, 85, 1834–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kode, S.S.; Pawar, S.D.; Tare, D.S.; Keng, S.S.; Hurt, A.C.; Mullick, J. A novel I117T substitution in neuraminidase of highly pathogenic avian influenza H5N1 virus conferring reduced susceptibility to oseltamivir and zanamivir. Vet. Microbiol. 2019, 235, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Deng, G.; Song, J.; Tian, G.; Suo, Y.; Jiang, Y.; Guan, Y.; Bu, Z.; Kawaoka, Y.; Chen, H. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology 2009, 384, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Macken, C.A.; Li, C.; Ozawa, M.; Goto, H.; Iswahyudi, N.F.N.; Nidom, C.A.; Chen, H.; Neumann, G.; Kawaoka, Y. Synergistic effect of the PDZ and p85β-binding domains of the NS1 protein on virulence of an avian H5N1 influenza A virus. J. Virol. 2013, 87, 4861–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Jiang, Y.; Jiao, P.; Wang, A.; Zhao, F.; Tian, G.; Wang, X.; Yu, K.; Bu, Z.; Chen, H. The NS1 gene contributes to the virulence of H5N1 avian influenza viruses. J. Virol. 2006, 80, 11115–11123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, E.; Stech, J.; Leneva, I.; Krauss, S.; Scholtissek, C.; San Chin, P.; Peiris, M.; Shortridge, K.F.; Webster, R.G. Characterization of the influenza A virus gene pool in avian species in southern China: Was H6N1 a derivative or a precursor of H5N1? J. Virol. 2000, 74, 6309–6315. [Google Scholar] [CrossRef] [Green Version]
- Driskell, E.A.; Pickens, J.A.; Humberd-Smith, J.; Gordy, J.T.; Bradley, K.C.; Steinhauer, D.A.; Berghaus, R.D.; Stallknecht, D.E.; Howerth, E.W.; Tompkins, S.M. Low pathogenic avian influenza isolates from wild birds replicate and transmit via contact in ferrets without prior adaptation. PLoS ONE 2012, 7, 38067. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.H.; Yang, J.R.; Wu, H.S.; Chang, M.C.; Lin, J.S.; Lin, C.Y.; Liu, Y.L.; Lo, Y.C.; Yang, C.H.; Chuang, J.H.; et al. Human infection with avian influenza A H6N1 virus: An epidemiological analysis. Lancet Respir. Med. 2013, 1, 771–778. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Protein | GD/18 | UG/731 | EG/1566C | EG/1556OP |
|---|---|---|---|---|
| PB2 | 7 | 7 | 8 | 7 |
| PB1 | 7 | 7 | 7 | 7 |
| PB1-F2 | 2 | 1 | 1 | 1 |
| PA | 4 | 4 | 4 | 4 |
| PA-X | 2 | 2 | 2 | 2 |
| HA | 1 | 2 | 2 | 2 |
| NP | 1 | 0 | 1 | 1 |
| NA | 1 | 1 | 1 | 1 |
| M1 | 2 | 2 | 2 | 2 |
| M2 | 0 | 0 | 0 | 0 |
| NS1 | 3 | 3 | 3 | 3 |
| NEP | 0 | 0 | 0 | 0 |
| TOTAL | 30 | 29 | 31 | 30 |
| Protein | Uganda | Egypt | Turkey |
|---|---|---|---|
| PB2 | A/domestic duck/Georgia/1/2016 (H4N6)-like | A/Teal/Markeev/13-3-11/2016 (H5N2) and A/teal/Egypt/MB-D-487OP/2016(H7N3)-like | |
| PB1 | A/teal/Egypt/MB-D-487OP/2016 (H7N3) and A/duck/Moscow/ 5662/2018 (H1N2)-like | A/domestic duck/Georgia/1/2016 (H4N6)-like | |
| PA | A/teal/Egypt/MB-D-487OP/2016 (H7N3)-like | A/Teal/Markeev/13-3-11/2016 (H5N2)-like | |
| HA | A/domestic duck/Georgia/1/2015(H6N2)-like | ||
| NP | A/teal/Egypt/MB-D-487OP/2016 (H7N3) and A/domestic duck/Georgia/1/2016 (H4N6)-like | A/Teal/Markeev/13-3-11/2016 (H5N2)-like | |
| NA | A/duck/Moscow/5662/2018 (H1N2)-like | A/Teal/Markeev/13-3-11/2016 (H5N2)-like | |
| M | A/Teal/Markeev/13-3-11/2016 (H5N2), A/domestic duck/Georgia/1/2016 (H4N6), A/duck/Moscow/5662/2018 (H1N2), and A/teal/Egypt/MB-D-487OP/2016 (H7N3)-like | ||
| NS | A/domestic duck/Georgia/1/2016 (H4N6) and A/teal/Egypt/MB-D-487OP/2016 (H7N3)-like | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercan, Y.; Atim, G.; Kayed, A.E.; Azbazdar, M.E.; Kandeil, A.; Ali, M.A.; Rubrum, A.; McKenzie, P.; Webby, R.J.; Erima, B.; et al. Molecular Characterization of Closely Related H6N2 Avian Influenza Viruses Isolated from Turkey, Egypt, and Uganda. Viruses 2021, 13, 607. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040607
Mercan Y, Atim G, Kayed AE, Azbazdar ME, Kandeil A, Ali MA, Rubrum A, McKenzie P, Webby RJ, Erima B, et al. Molecular Characterization of Closely Related H6N2 Avian Influenza Viruses Isolated from Turkey, Egypt, and Uganda. Viruses. 2021; 13(4):607. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040607
Chicago/Turabian StyleMercan, Yavuz, Gladys Atim, Ahmed E. Kayed, M. Ekin Azbazdar, Ahmed Kandeil, Mohamed A. Ali, Adam Rubrum, Pamela McKenzie, Richard J. Webby, Bernard Erima, and et al. 2021. "Molecular Characterization of Closely Related H6N2 Avian Influenza Viruses Isolated from Turkey, Egypt, and Uganda" Viruses 13, no. 4: 607. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040607