
Adaptive Evolution of New Variants of Dengue Virus Serotype 1 Genotype V Circulating in the Brazilian Amazon

 ,
,  , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. DENV Samples
2.2. Complete Genomes Sequencing
2.3. Dataset Construction
2.4. Phylogenetic, Molecular Clock, and Phylogeographic Analysis
2.5. Selection Pressure Analysis
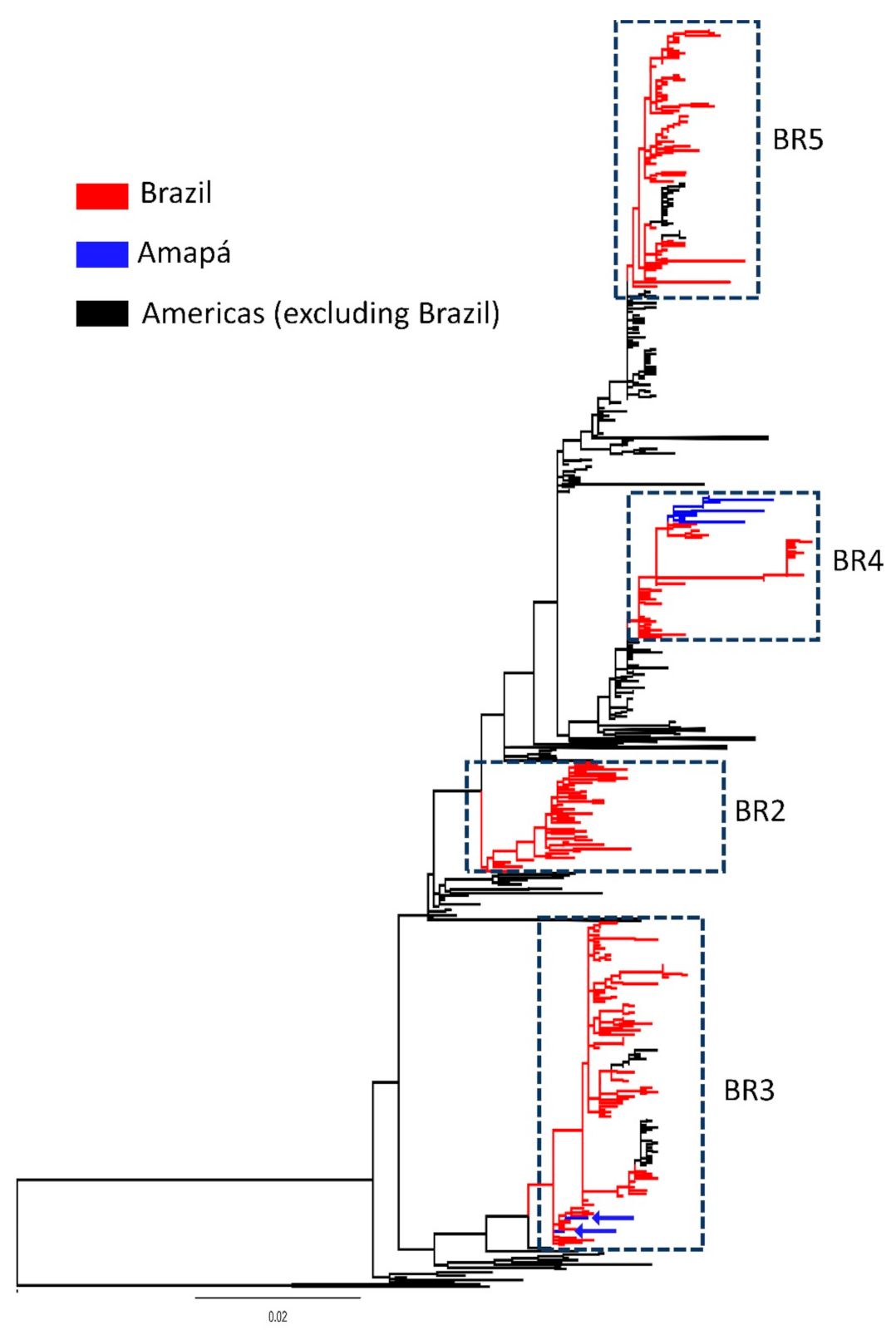
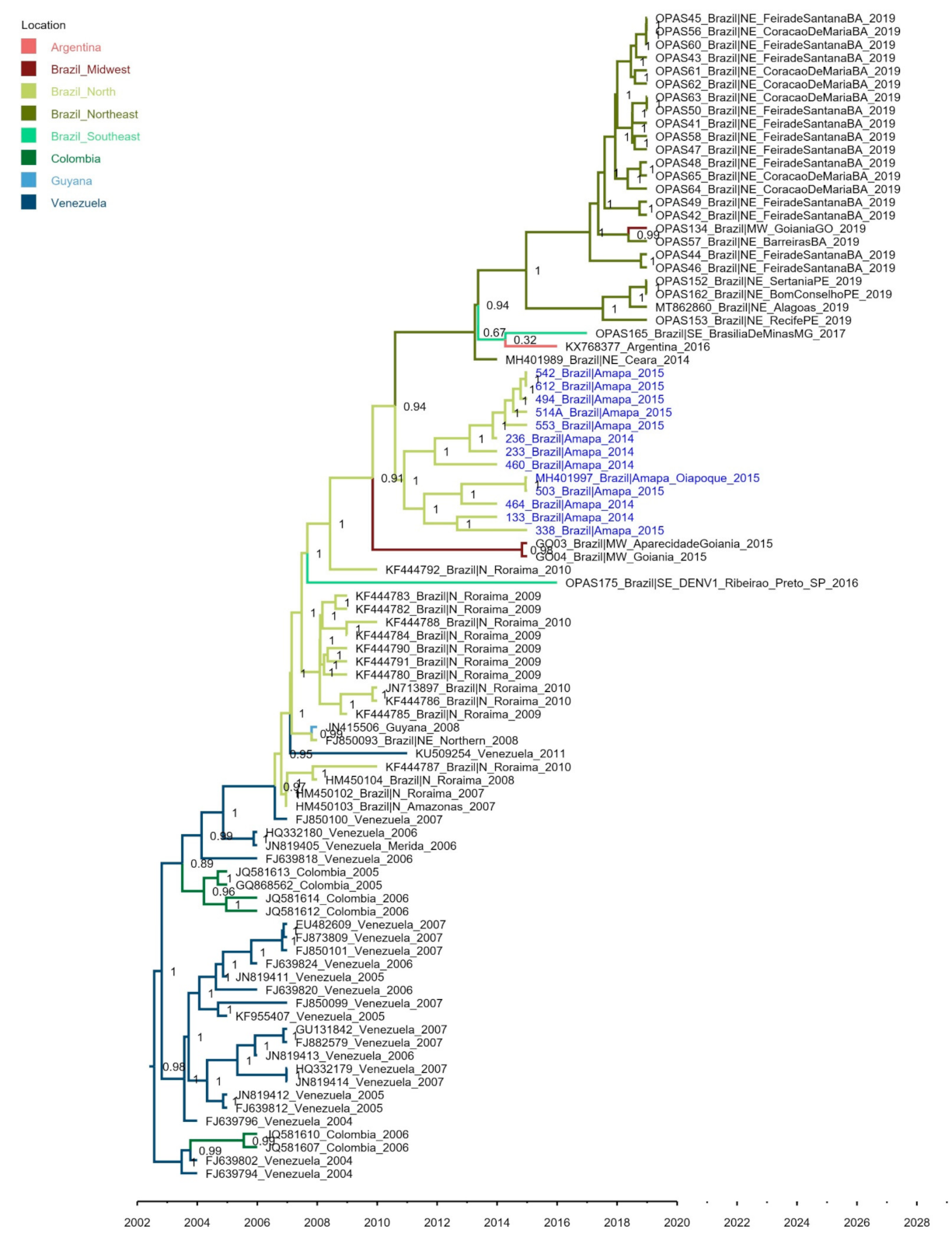
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jenkins, G.M.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of Molecular Evolution in RNA Viruses: A Quantitative Phylogenetic Analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nat. Cell Biol. 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Allicock, O.M.; Lemey, P.; Tatem, A.J.; Pybus, O.G.; Bennett, S.N.; Mueller, B.A.; Suchard, M.A.; Foster, J.E.; Rambaut, A.; Carrington, C.V.F. Phylogeography and Population Dynamics of Dengue Viruses in the Americas. Mol. Biol. Evol. 2012, 29, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrington, C.V.F.; Foster, J.E.; Pybus, O.G.; Bennett, S.N.; Holmes, E.C. Invasion and Maintenance of Dengue Virus Type 2 and Type 4 in the Americas. J. Virol. 2005, 79, 14680–14687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughlin, C.A.; Morens, D.M.; Cassetti, M.C.; Denis, A.C.-S.; Martin, J.-L.S.; Whitehead, S.S.; Fauci, A.S. Dengue Research Opportunities in the Americas. J. Infect. Dis. 2012, 206, 1121–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, M.R.T.; Palacios, G.; Faria, N.R.; Sousa, E.C., Jr.; Pantoja, J.A.; Rodrigues, S.G.; Carvalho, V.L.; Medeiros, D.B.A.; Savji, N.; Baele, G.; et al. Air Travel Is Associated with Intracontinental Spread of Dengue Virus Serotypes 1–3 in Brazil. PLoS Negl. Trop. Dis. 2014, 8, e2769. [Google Scholar] [CrossRef] [PubMed]
- Villabona-Arenas, C.J.; Zanotto, P.M.D.A. Worldwide Spread of Dengue Virus Type 1. PLoS ONE 2013, 8, e62649. [Google Scholar] [CrossRef] [Green Version]
- Walimbe, A.M.; Lotankar, M.; Cecilia, D.; Cherian, S.S. Global phylogeography of Dengue type 1 and 2 viruses reveals the role of India. Infect. Genet. Evol. 2014, 22, 30–39. [Google Scholar] [CrossRef]
- De Bruycker-Nogueira, F.; Mir, D.; Dos Santos, F.B.; Bello, G. Evolutionary history and spatiotemporal dynamics of DENV-1 genotype V in the Americas. Infect. Genet. Evol. 2016, 45, 454–460. [Google Scholar] [CrossRef]
- da Cunha, A.C. Tempo, Clima e Recursos Hídricos: Resultados do Projeto REMETAP no Estado do Amapá; IEPA: Macapá, Brazil, 2010; ISBN 8587794159. [Google Scholar]
- Monteiro, F.J.C.; Mourão, F.R.P.; Ribeiro, E.S.D.; Rêgo, M.O.D.S.; Frances, P.A.D.C.; Souto, R.N.P.; Façanha, M.D.S.; Tahmasebi, R.; Da Costa, A.C. Prevalence of dengue, Zika and chikungunya viruses in Aedes (Stegomyia) aegypti (Diptera: Culicidae) in a medium-sized city, Amazon, Brazil. Rev. Inst. Med. Trop. Sao Paulo 2020, 62, e10. [Google Scholar] [CrossRef] [Green Version]
- Cunha, M.S.; Da Costa, A.C.; Fernandes, N.C.C.D.A.; Guerra, J.M.; Dos Santos, F.C.P.; Nogueira, J.S.; D’Agostino, L.G.; Komninakis, S.V.; Witkin, S.S.; Ressio, R.A.; et al. Epizootics due to Yellow Fever Virus in São Paulo State, Brazil: Viral dissemination to new areas (2016–2017). Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Faria, N.R.; Kraemer, M.U.G.; Hill, S.C.; De Jesus, J.G.; Aguiar, R.S.; Iani, F.C.M.; Xavier, J.; Quick, J.; Du Plessis, L.; Dellicour, S.; et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018, 361, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Nunes, M.R.T.; Faria, N.R.; De Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.G.; De Oliveira, L.F.; Azevedo, R.D.S.D.S.; Da Silva, D.E.A.; Da Silva, E.V.P.; Da Silva, S.P.; et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Da Costa, A.C.; Lourenço, J.; Loureiro, P.; Lopes, M.E.; Ribeiro, R.; Alencar, C.S.; Kraemer, M.U.G.; Villabona-Arenas, C.J.; Wu, C.-H.; et al. Genomic and epidemiological characterisation of a dengue virus outbreak among blood donors in Brazil. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Nunes, M.R.T.; Faria, N.R.; Vasconcelos, H.B.; Medeiros, D.B.D.A.; De Lima, C.P.S.; Carvalho, V.L.; Da Silva, E.V.P.; Cardoso, J.F.; Sousa, E.C.; Nunes, K.N.B.; et al. Phylogeography of Dengue Virus Serotype 4, Brazil, 2010–2011. Emerg. Infect. Dis. 2012, 18, 1858–1864. [Google Scholar] [CrossRef] [Green Version]
- Lowe, R.; Lee, S.; Lana, R.M.; Codeço, C.T.; Castro, M.C.; Pascual, M. Emerging arboviruses in the urbanized Amazon rainforest. BMJ 2020, 371, m4385. [Google Scholar] [CrossRef]
- Johnson, N.; Wakeley, P.R.; Mansfield, K.L.; McCracken, F.; Haxton, B.; Phipps, L.P.; Fooks, A.R. Assessment of a Novel Real-Time Pan-Flavivirus RT-Polymerase Chain Reaction. Vector-Borne Zoonotic Dis. 2010, 10, 665–671. [Google Scholar] [CrossRef]
- Scaramozzino, N.N.; Crance, J.-M.; Jouan, A.; DeBriel, D.A.; Stoll, F.; Garin, D. Comparison of Flavivirus Universal Primer Pairs and Development of a Rapid, Highly Sensitive Heminested Reverse Transcription-PCR Assay for Detection of Flaviviruses Targeted to a Conserved Region of the NS5 Gene Sequences. J. Clin. Microbiol. 2001, 39, 1922–1927. [Google Scholar] [CrossRef] [Green Version]
- Chien, L.-J.; Liao, T.-L.; Shu, P.-Y.; Huang, J.-H.; Gubler, D.J.; Chang, G.-J.J. Development of Real-Time Reverse Transcriptase PCR Assays To Detect and Serotype Dengue Viruses. J. Clin. Microbiol. 2006, 44, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Landt, O.; Kaiser, M.; Faye, O.; Koppe, T.; Lass, U.; Sall, A.; Niedrig, M. Development of one-step quantitative reverse transcription PCR for the rapid detection of flaviviruses. Virol. J. 2013, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, V.; Libin, P.J.K.; Theys, K.; Faria, N.R.; Nunes, M.R.T.; Restovic, M.I.; Freire, M.; Giovanetti, M.; Cuypers, L.; Nowé, A.; et al. A computational method for the identification of Dengue, Zika and Chikungunya virus species and genotypes. PLoS Negl. Trop. Dis. 2019, 13, e0007231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.L.K.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Pond, S.L.K. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Dutra, K.R.; Drumond, B.P.; De Rezende, I.M.; Nogueira, M.L.; Lopes, D.D.O.; Silva, C.E.C.; Ferreira, J.M.S.; Dos Santos, L.L. Molecular surveillance of dengue in Minas Gerais provides insights on dengue virus 1 and 4 circulation in Brazil. J. Med Virol. 2017, 89, 966–973. [Google Scholar] [CrossRef]
- Adelino, T.E.R.; Giovanetti, M.; Fonseca, V.; Xavier, J.; Salgado, A.; Nascimento, V.; Demarchi, L.H.; Oliveira, M.; Silva, V.; Mello, A. Portable sequencing in the field and the classroom: A retrospective examination of the circulation of DENV1 and DENV2 in Brazil. medRxiv 2020. [Google Scholar] [CrossRef]
- De Bruycker-Nogueira, F.; Souza, T.M.A.; Chouin-Carneiro, T.; Faria, N.R.D.C.; Santos, J.B.; Torres, M.C.; Ramalho, I.L.C.; De Aguiar, S.F.; Nogueira, R.M.R.; De Filippis, A.M.B.; et al. DENV-1 Genotype V in Brazil: Spatiotemporal dispersion pattern reveals continuous co-circulation of distinct lineages until 2016. Sci. Rep. 2018, 8, 17160. [Google Scholar] [CrossRef]
- Salles, T.S.; Sá-Guimarães, T.D.E.; De Alvarenga, E.S.L.; Guimarães-Ribeiro, V.; De Meneses, M.D.F.; De Castro-Salles, P.F.; Dos Santos, C.R.; Melo, A.C.D.A.; Soares, M.R.; Ferreira, D.F.; et al. History, epidemiology and diagnostics of dengue in the American and Brazilian contexts: A review. Parasites Vectors 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Amapa Epidemiological Surveillance Agency Análise Epidemiológica Situacional da Dengue, Chikungunya e Febre pelo Vírus Zika no Estado do Amapá no Período de 2011 a 2015. Available online: https://editor.amapa.gov.br/arquivos_portais/publicacoes/SVS_926dca2c36aa88e0e3f2cba4e2f17cb9.pdf (accessed on 10 February 2021).
- Amorim, J.H.; Alves, R.P.D.S.; Boscardin, S.B.; Ferreira, L.C.D.S. The dengue virus non-structural 1 protein: Risks and benefits. Virus Res. 2014, 181, 53–60. [Google Scholar] [CrossRef]
- Chuang, Y.-C.; Wang, S.-Y.; Lin, Y.-S.; Chen, H.-R.; Yeh, T.-M. Re-evaluation of the pathogenic roles of nonstructural protein 1 and its antibodies during dengue virus infection. J. Biomed. Sci. 2013, 20, 42. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the Dengue Virus Nonstructural Glycoprotein NS1 Suggests a Role in Viral RNA Replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Nncube, N.B.; Ramharack, P.; Soliman, M.E. Using bioinformatics tools for the discovery of Dengue RNA-dependent RNA polymerase inhibitors. PeerJ 2018, 6, e5068. [Google Scholar] [CrossRef]
- Temporão, J.G.; Penna, G.O.; Carmo, E.H.; Coelho, G.E.; Azevedo, R.D.S.S.; Nunes, M.R.T.; Vasconcelos, P.F.D.C. Dengue Virus Serotype 4, Roraima State, Brazil. Emerg. Infect. Dis. 2011, 17, 938–940. [Google Scholar] [CrossRef]
- Osanai, C.; da Rosa, A.P.T.; Tang, A.T.; do Amaral, R.S.; Passos, A.D.; Tauil, P.L. Dengue outbreak in Boa Vista, Roraima [in Portuguese]. Rev. Inst. Med. Trop. Sao Paulo 1983, 25, 53–54. [Google Scholar]



{kind=link}
{kind=link}
| Codon Position | Protein | FEL 1 (p-Value < 0.1) | SLAC 2 (p-Value < 0.1) | FUBAR 3 (Probability) | MEME 4 (p-Value < 0.1) |
|---|---|---|---|---|---|
| 39 | E | Yes | Yes | 0.9793 | Yes |
| 193 | NS1 | Yes | Yes | 0.9970 | Yes |
| 1684 | NS3 | Yes | Yes | 0.9888 | Yes |
| 1752 | NS3 | Yes | Yes | 0.9668 | Yes |
| 2951 | NS5 | Yes | No | 0.9818 | Yes |
| 2913 | NS5 | Yes | No | 0.9832 | Yes |
| 2997 | NS5 | Yes | No | 0.9916 | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, G.d.O.; Gill, D.E.; Ribeiro, E.S.D.; Monteiro, F.J.C.; Morais, V.S.; Marcatti, R.; Rego, M.O.d.S.; Araújo, E.L.L.; Witkin, S.S.; Villanova, F.; et al. Adaptive Evolution of New Variants of Dengue Virus Serotype 1 Genotype V Circulating in the Brazilian Amazon. Viruses 2021, 13, 689. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040689
Ribeiro GdO, Gill DE, Ribeiro ESD, Monteiro FJC, Morais VS, Marcatti R, Rego MOdS, Araújo ELL, Witkin SS, Villanova F, et al. Adaptive Evolution of New Variants of Dengue Virus Serotype 1 Genotype V Circulating in the Brazilian Amazon. Viruses. 2021; 13(4):689. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040689
Chicago/Turabian StyleRibeiro, Geovani de Oliveira, Danielle Elise Gill, Edcelha Soares D’Athaide Ribeiro, Fred Julio Costa Monteiro, Vanessa S. Morais, Roberta Marcatti, Marlisson Octavio da S. Rego, Emerson Luiz Lima Araújo, Steven S. Witkin, Fabiola Villanova, and et al. 2021. "Adaptive Evolution of New Variants of Dengue Virus Serotype 1 Genotype V Circulating in the Brazilian Amazon" Viruses 13, no. 4: 689. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040689