
Synergistic Chromatin-Modifying Treatments Reactivate Latent HIV and Decrease Migration of Multiple Host-Cell Types

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of Ld2G THP-1 Library
2.3. Establishment of a Monocytic Latency Model (TLat)
2.4. Detection of Human Immunodeficiency Virus (HIV) DNA by Polymerase Chain Reaction (PCR) and Agarose Gel Electrophoresis
2.5. Drug Treatments
2.6. CXCR4 Antibody Staining
2.7. Migration Assay
2.8. Flow Cytometry Analyses and Gating Strategy
2.9. Gene Expression Noise Calculations
2.10. Bliss Independence Score Calculation
2.11. Two-State Model Simulations
2.12. Statistical Methods
3. Results
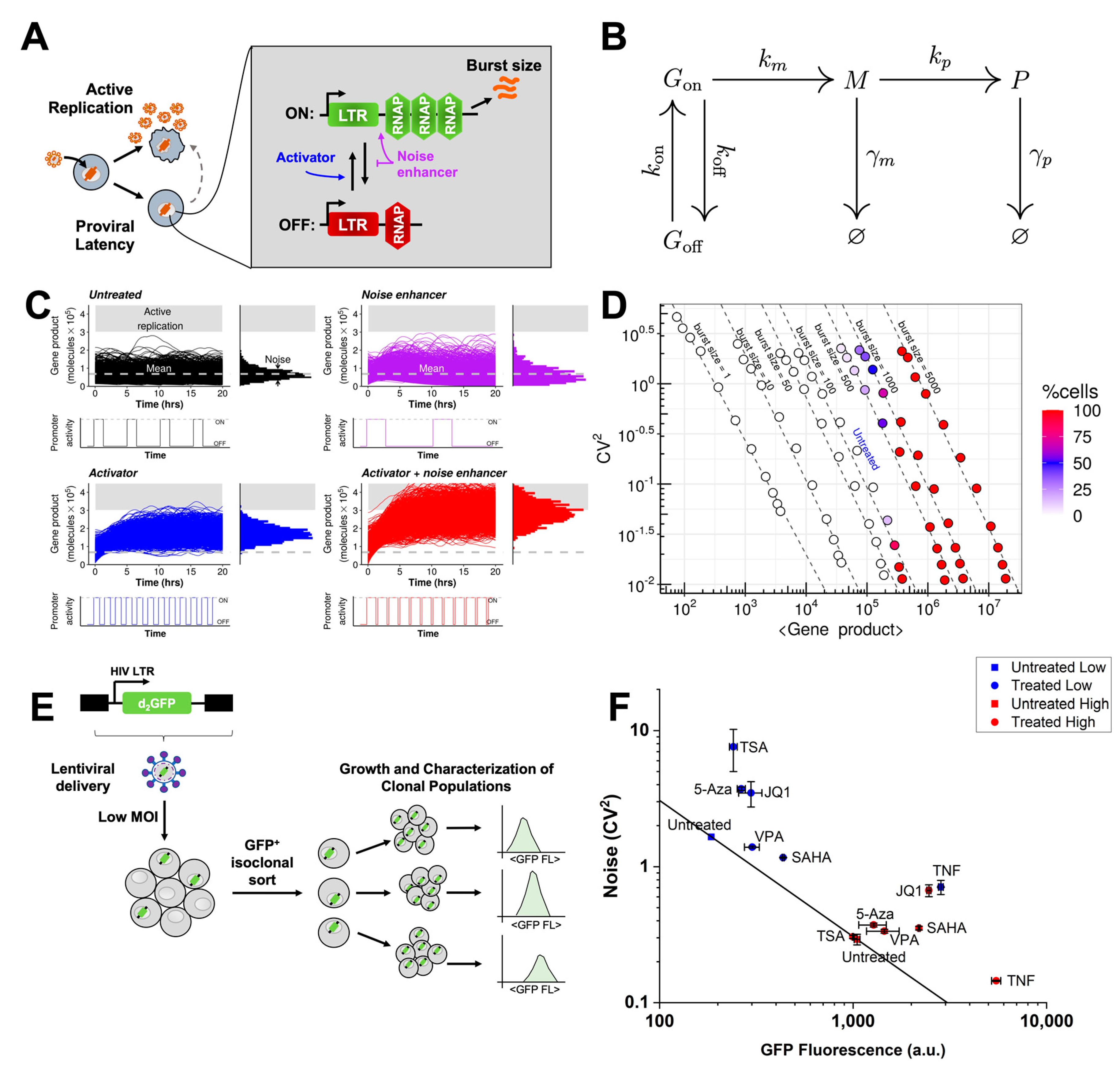
3.1. Stochastic Simulations Reveal an Over-Expression Strategy with Noise Enhancement
3.2. Generation of Single-Integration Clones to Study HIV Promoter Expression in Monocytes
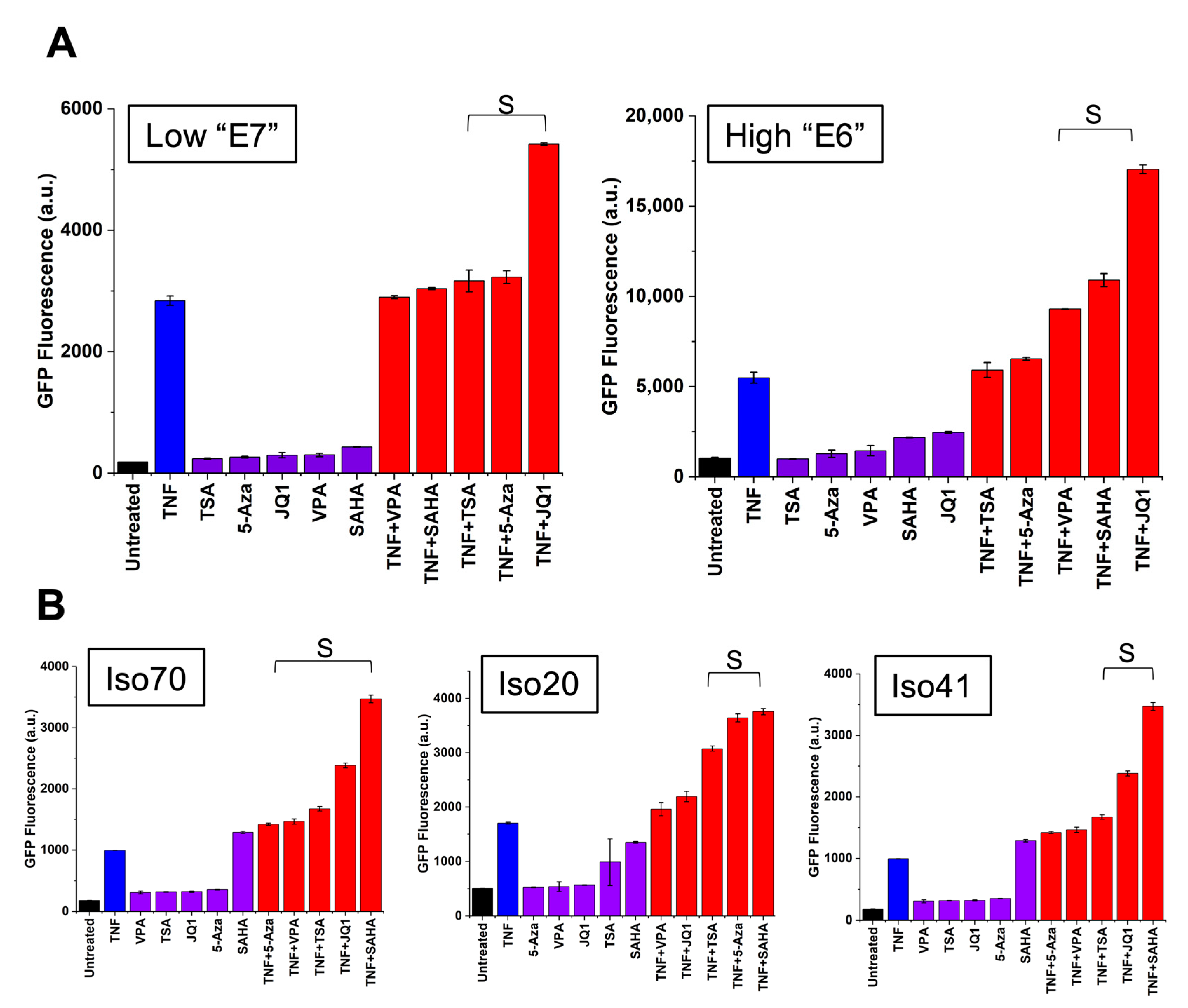
3.3. Chromatin Modifiers Combined with Activators Enhance HIV Promoter Expression in Both Monocytes and T-Cells
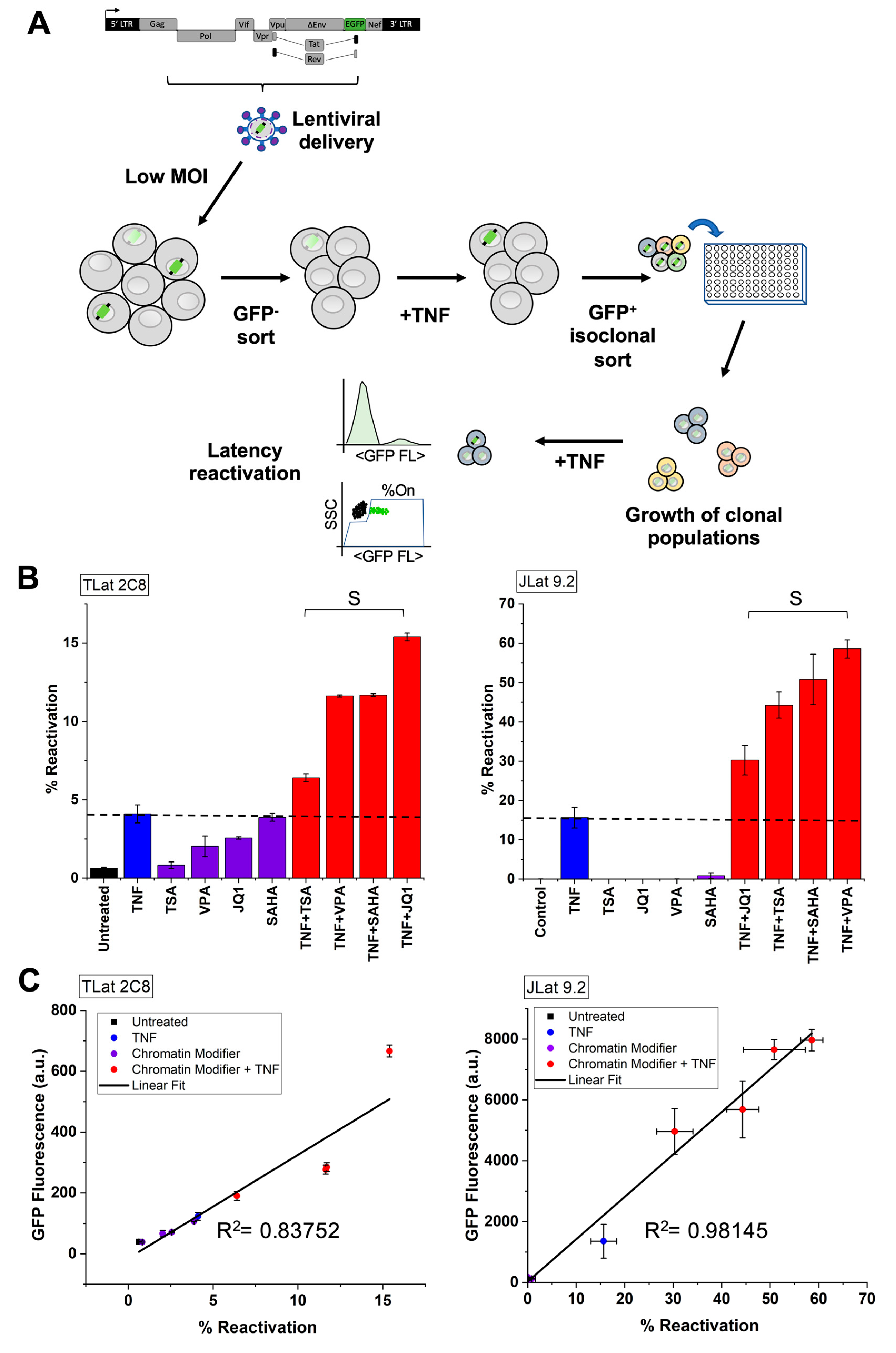
3.4. Establishment of an In Vitro Model of HIV-1 Latency in Monocytes (TLat)
3.5. Synergistic Cocktails Enhance Reactivation from Latency
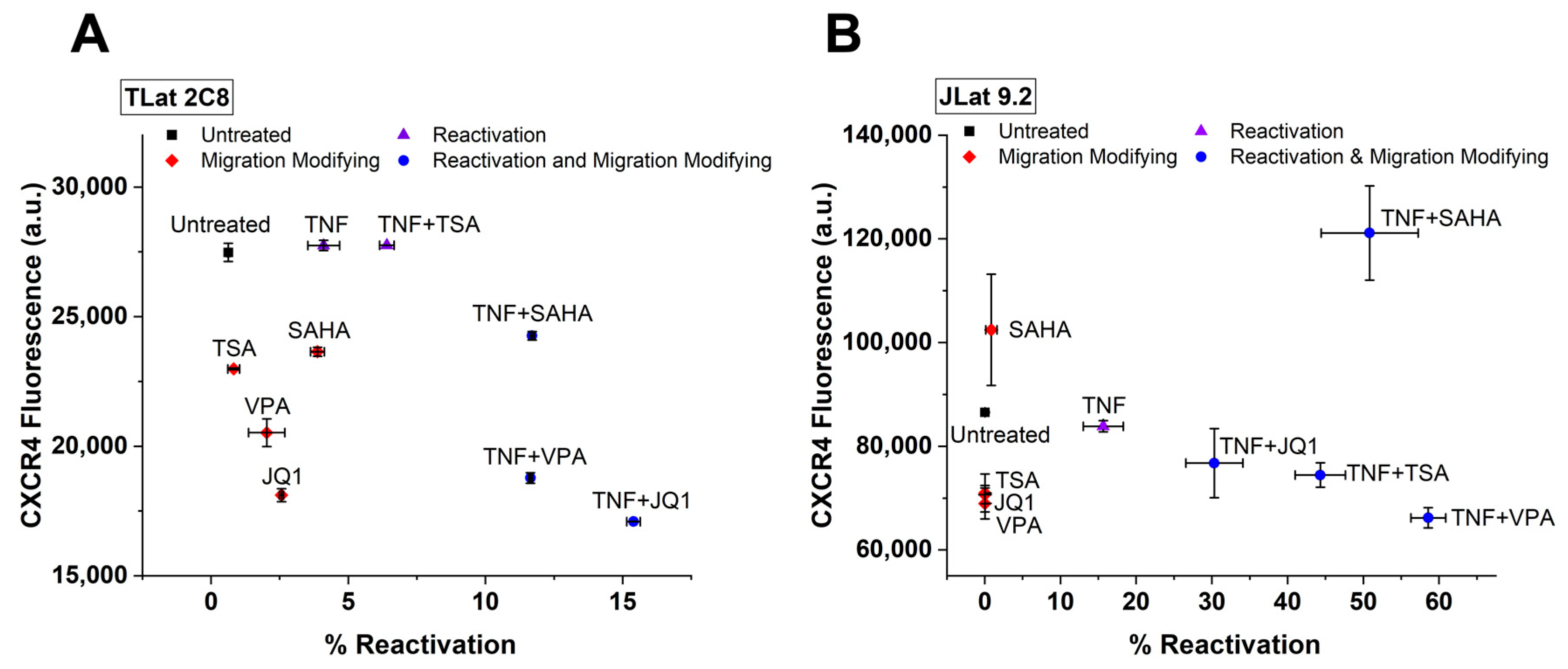
3.6. Leading Drug Cocktails Affect Host Cell CXCR4 Surface Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, S.G. Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.-C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L. In vivo suppression of HIV rebound by didehydro-cortistatin A, a “block-and-lock” strategy for HIV-1 treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Bohn-Wippert, K.; Pazerunas, P.J.; Moy, J.M.; Singh, H.; Dar, R.D. Screening for gene expression fluctuations reveals latency-promoting agents of HIV. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Moranguinho, I.; Valente, S.T. Block-And-Lock: New Horizons for a Cure for HIV-1. Viruses 2020, 12, 1443. [Google Scholar] [CrossRef]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Dar, R.D.; Hosmane, N.N.; Arkin, M.R.; Siliciano, R.F.; Weinberger, L.S. Screening for noise in gene expression identifies drug synergies. Science 2014, 344, 1392–1396. [Google Scholar] [CrossRef] [Green Version]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.-C.; Planelles, V.; Bradner, J.E. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, L.S.; Dar, R.D.; Simpson, M.L. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat. Genet. 2008, 40, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Dar, R.D.; Razooky, B.S.; Singh, A.; Trimeloni, T.V.; McCollum, J.M.; Cox, C.D.; Simpson, M.L.; Weinberger, L.S. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc. Natl. Acad. Sci. 2012, 109, 17454–17459. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Razooky, B.; Cox, C.D.; Simpson, M.L.; Weinberger, L.S. Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression. Biophys. J. 2010, 98, L32–L34. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Coleman, C.M.; Wu, L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Khanal, S.; Schank, M.; El Gazzar, M.; Moorman, J.P.; Yao, Z.Q. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells 2021, 10, 475. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2020, 10, 3060. [Google Scholar] [CrossRef] [Green Version]
- Zaikos, T.D.; Collins, K.L. Long-lived reservoirs of HIV-1. Trends Microbiol 2014, 22, 173–175. [Google Scholar] [CrossRef] [Green Version]
- Bohn-Wippert, K.; Tevonian, E.N.; Megaridis, M.R.; Dar, R.D. Similarity in viral and host promoters couples viral reactivation with host cell migration. Nat. Commun. 2017, 8, 15006. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.R.S.; Ghaemmaghami, S.; Ihmels, J.; Breslow, D.K.; Noble, M.; DeRisi, J.L.; Weissman, J.S. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 2006, 441, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Bliss, C.I. The toxicity of poisons applied jointly 1. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Cannoodt, R.; Saelens, W. GillespieSSA2: Gillespie’s Stochastic Simulation Algorithm for Impatient People; 0.2.7; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://github.com/rcannood/GillespieSSA2 (accessed on 27 March 2021).
- Kepler, T.B.; Elston, T.C. Stochasticity in transcriptional regulation: Origins, consequences, and mathematical representations. Biophys. J. 2001, 81, 3116–3136. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.L.; Cox, C.D.; Sayler, G.S. Frequency domain chemical Langevin analysis of stochasticity in gene transcriptional regulation. J. Theor. Biol. 2004, 229, 383–394. [Google Scholar] [CrossRef]
- Bohn-Wippert, K.; Tevonian, E.N.; Lu, Y.; Huang, M.Y.; Megaridis, M.R.; Dar, R.D. Cell Size-Based Decision-Making of a Viral Gene Circuit. Cell Rep 2018, 25, 3844–3857 e5. [Google Scholar] [CrossRef]
- Dar, R.D.; Shaffer, S.M.; Singh, A.; Razooky, B.S.; Simpson, M.L.; Raj, A.; Weinberger, L.S. Transcriptional Bursting Explains the Noise-Versus-Mean Relationship in mRNA and Protein Levels. PLoS ONE 2016, 11, e0158298. [Google Scholar] [CrossRef] [Green Version]
- Megaridis, M.R.; Lu, Y.; Tevonian, E.N.; Junger, K.M.; Moy, J.M.; Bohn-Wippert, K.; Dar, R.D. Fine-tuning of noise in gene expression with nucleosome remodeling. APL Bioeng. 2018, 2, 026106. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.C.; Bass, V.L.; Bullock, M.E.; Chavali, A.K.; Lee, R.E.C.; Mothes, W.; Gaudet, S.; Miller-Jensen, K. NF-kappaB-Chromatin Interactions Drive Diverse Phenotypes by Modulating Transcriptional Noise. Cell Rep 2018, 22, 585–599. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.D.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef]
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.-S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS ONE 2009, 4, e6093. [Google Scholar] [CrossRef]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2′-deoxycytidine and deacetylase inhibitors reactivates HIV -1. EMBO Mol. Med. 2016, 8, 117–138. [Google Scholar] [CrossRef]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog 2009, 5, e1000495. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An In-Depth Comparison of Latent HIV-1 Reactivation in Multiple Cell Model Systems and Resting CD4+ T Cells from Aviremic Patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Humphreys, T.D.; Kremer, K.N.; Bramati, P.S.; Bradfield, L.; Edgar, C.E.; Hedin, K.E. CXCR4 Physically Associates with the T Cell Receptor to Signal in T Cells. Immunity 2006, 25, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Ottoson, N.C.; Pribila, J.T.; Chan, A.S.H.; Shimizu, Y. Cutting Edge: T Cell Migration Regulated by CXCR4 Chemokine Receptor Signaling to ZAP-70 Tyrosine Kinase. J. Immunol. 2001, 167, 1857–1861. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 Entry Cofactor: Functional cDNA Cloning of a Seven-Transmembrane, G Protein-Coupled Receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Stellbrink, H.-J.; van Lunzen, J.; Westby, M.; O’Sullivan, E.; Schneider, C.; Adam, A.; Weitner, L.; Kuhlmann, B.; Hoffmann, C.; Fenske, S.; et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial). AIDS 2002, 16, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Estes, J.D.; Kityo, C.; Ssali, F.; Swainson, L.; Makamdop, K.N.; Del Prete, G.Q.; Deeks, S.G.; Luciw, P.A.; Chipman, J.G.; Beilman, G.J. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017, 23, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, G.; Graziosi, C.; Demarest, J.F.; Butini, L.; Montroni, M.; Fox, C.H.; Orenstein, J.M.; Kotler, D.P.; Fauci, A.S. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 1993, 362, 355–358. [Google Scholar] [CrossRef]
- Henderson, L.J.; Reoma, L.B.; Kovacs, J.A.; Nath, A. Advances toward Curing HIV-1 Infection in Tissue Reservoirs. J. Virol. 2020, 94, e00375-19. [Google Scholar] [CrossRef]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 Latency in Monocytes/Macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [Green Version]
- Redel, L.; Le Douce, V.; Cherrier, T.; Marban, C.; Janossy, A.; Aunis, D.; Van Lint, C.; Rohr, O.; Schwartz, C. HIV-1 regulation of latency in the monocyte-macrophage lineage and in CD4+ T lymphocytes. J. Leukoc. Biol. 2010, 87, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Kim, K.-C.; Son, J.; Shin, Y.; Yoon, C.-H.; Kang, C.; Choi, B.-S. Synergistic reactivation of latent HIV-1 provirus by PKA activator dibutyryl-cAMP in combination with an HDAC inhibitor. Virus Res. 2017, 227, 1–5. [Google Scholar] [CrossRef]
- Adam, E.; Quivy, V.; Bex, F.; Chariot, A.; Collette, Y.; Vanhulle, C.; Schoonbroodt, S.; Goffin, V.; Nguyên, T.L.-A.; Gloire, G.; et al. Potentiation of Tumor Necrosis Factor-Induced NF-κB Activation by Deacetylase Inhibitors Is Associated with a Delayed Cytoplasmic Reappearance of IκBα. Mol. Cell. Biol. 2003, 23, 6200–6209. [Google Scholar] [CrossRef] [Green Version]
- Quivy, V.; Adam, E.; Collette, Y.; Demonte, D.; Chariot, A.; Vanhulle, C.; Berkhout, B.; Castellano, R.M.; De Launoit, Y.; Burny, A.N.; et al. Synergistic Activation of Human Immunodeficiency Virus Type 1 Promoter Activity by NF-κB and Inhibitors of Deacetylases: Potential Perspectives for the Development of Therapeutic Strategies. J. Virol. 2002, 76, 11091–11103. [Google Scholar] [CrossRef] [Green Version]
- Kula, A.; Delacourt, N.; Bouchat, S.; Darcis, G.; Avettand-Fenoel, V.; Verdikt, R.; Corazza, F.; Necsoi, C.; Vanhulle, C.; Bendoumou, M.; et al. Heterogeneous HIV-1 Reactivation Patterns of Disulfiram and Combined Disulfiram+Romidepsin Treatments. JAIDS J. Acquir. Immune Defic. Syndr. 2019, 80, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Kutsch, O.; Benveniste, E.N.; Shaw, G.M.; Levy, D.N. Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J. Virol. 2002, 76, 8776–8786. [Google Scholar] [CrossRef] [Green Version]
- Cassol, E.; Alfano, M.; Biswas, P.; Poli, G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J. Leukoc. Biol. 2006, 80, 1018–1030. [Google Scholar] [CrossRef]
- Folks, T.M.; Justement, J.; Kinter, A.; Schnittman, S.; Orenstein, J.; Poli, G.; Fauci, A.S. Characterization of a promonocyte clone chronically infected with HIV and inducible by 13-phorbol-12-myristate acetate. J. Immunol. 1988, 140, 1117–1122. [Google Scholar]
- Han, Y.; Wind-Rotolo, M.; Yang, H.-C.; Siliciano, J.D.; Siliciano, R.F. Experimental approaches to the study of HIV-1 latency. Nat. Rev. Microbiol. 2007, 5, 95–106. [Google Scholar] [CrossRef]
- Poli, G.; Kinter, A.L.; Justement, J.S.; Biswas, P.; Weissman, D.; Fox, L.M.; Goletti, D.; Bressler, P.; Stanley, S.K.; Fauci, A.S. The Chronically Infected Promonocytic Cell Line U1: A Model of HIV Expression Regulated by Cytokines. ImmunoMethods 1993, 3, 50–55. [Google Scholar] [CrossRef]
- Pomerantz, R.J.; Feinberg, M.B.; Trono, D.; Baltimore, D. Lipopolysaccharide is a potent monocyte/macrophage-specific stimulator of human immunodeficiency virus type 1 expression. J. Exp. Med. 1990, 172, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, C.; Alfano, M.; Bugatti, A.; Camozzi, M.; Poli, G.; Rusnati, M. Inhibition of intra- and extra-cellular Tat function and HIV expression by pertussis toxin B-oligomer. Eur. J. Immunol. 2004, 34, 530–536. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+ JQ1 and ingenol-B+ JQ1 to potently reactivate viral gene expression. PLoS Pathog 2015, 11, e1005063. [Google Scholar] [CrossRef]
- Nekhai, S.; Jeang, K.-T. Transcriptional and post-transcriptional regulation of HIV-1 gene expression: Role of cellular factors for Tat and Rev. Future Microbiol. 2006, 1, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Kwas, C.; Wu, L. Transcriptional restriction of human immunodeficiency virus type 1 gene expression in undifferentiated primary monocytes. J. Virol. 2009, 83, 3518–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, M.E.; Mayall, T.P.; Suess, E.M.; Meisenhelder, J.; Thompson, N.E.; Jones, K.A. CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat–P-TEFb complex to TAR RNA. Mol. Cell. Biol. 2000, 20, 6958–6969. [Google Scholar] [CrossRef] [Green Version]
- Liou, L.-Y.; Herrmann, C.H.; Rice, A.P. Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J. Virol. 2002, 76, 10579–10587. [Google Scholar] [CrossRef] [Green Version]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR Nucleosome Positioning by the BAF Complex Is Required for HIV Latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.A.; Søgaard, O.S.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Østergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development. Hum. Vaccines Immunother. 2013, 9, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV–human protein complexes. Nature 2012, 481, 365–370. [Google Scholar] [CrossRef]
- Vérollet, C.; Souriant, S.; Bonnaud, E.; Jolicoeur, P.; Raynaud-Messina, B.; Kinnaer, C.; Fourquaux, I.; Imle, A.; Benichou, S.; Fackler, O.T.; et al. HIV-1 reprograms the migration of macrophages. Blood 2015, 125, 1611–1622. [Google Scholar] [CrossRef]
- Choe, E.Y.; Schoenberger, E.S.; Groopman, J.E.; Park, I.-W. HIV Nef Inhibits T Cell Migration. J. Biol. Chem. 2002, 277, 46079–46084. [Google Scholar] [CrossRef] [Green Version]
- Murooka, T.T.; Deruaz, M.; Marangoni, F.; Vrbanac, V.D.; Seung, E.; von Andrian, U.H.; Tager, A.M.; Luster, A.D.; Mempel, T.R. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 2012, 490, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef]
- Hidalgo-Estévez, A.M.; Punzón, C.; Sanchez-Duffhues, G.; Muñoz, E.; Fresno, M. HIV-1-Tat potentiates CXCL12/Stromal Cell-Derived Factor 1-induced downregulation of membrane CXCR4 in T lymphocytes through Protein kinase C zeta. Mol. Immunol. 2008, 46, 106–115. [Google Scholar] [CrossRef]
- Xiao, H.; Neuveut, C.; Tiffany, H.L.; Benkirane, M.; Rich, E.A.; Murphy, P.M.; Jeang, K.T. Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. 2000, 97, 11466–11471. [Google Scholar] [CrossRef] [Green Version]
- Venzke, S.; Michel, N.; Allespach, I.; Fackler, O.T.; Keppler, O.T. Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. J. Virol. 2006, 80, 11141–11152. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Rodepeter, F.R.; Rossmann, A.; Teymoortash, A.; Lee, J.-S.; Quint, K.; Di Fazio, P.; Ocker, M.; Werner, J.A.; Mandic, R. SIVmac239-Nef down-regulates cell surface expression of CXCR4 in tumor cells and inhibits proliferation, migration and angiogenesis. Anticancer Res. 2012, 32, 2759–2768. [Google Scholar]
- Ghezzi, S.; Noonan, D.M.; Aluigi, M.G.; Vallanti, G.; Cota, M.; Benelli, R.; Morini, M.; Reeves, J.D.; Vicenzi, E.; Poli, G. Inhibition of CXCR4-dependent HIV-1 infection by extracellular HIV-1 Tat. Biochem. Biophys. Res. Commun. 2000, 270, 992–996. [Google Scholar] [CrossRef]
- Castro-Gonzalez, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for HIV Cure: The Latent Reservoir. AIDS Res. Hum. Retrovir. 2018, 34, 739–759. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, A.; Mahajan, T.; Coronado, R.A.; Ma, K.; Demma, D.R.; Dar, R.D. Synergistic Chromatin-Modifying Treatments Reactivate Latent HIV and Decrease Migration of Multiple Host-Cell Types. Viruses 2021, 13, 1097. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061097
Blanco A, Mahajan T, Coronado RA, Ma K, Demma DR, Dar RD. Synergistic Chromatin-Modifying Treatments Reactivate Latent HIV and Decrease Migration of Multiple Host-Cell Types. Viruses. 2021; 13(6):1097. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061097
Chicago/Turabian StyleBlanco, Alexandra, Tarun Mahajan, Robert A. Coronado, Kelly Ma, Dominic R. Demma, and Roy D. Dar. 2021. "Synergistic Chromatin-Modifying Treatments Reactivate Latent HIV and Decrease Migration of Multiple Host-Cell Types" Viruses 13, no. 6: 1097. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061097