
Molecular Mechanisms during Hepatitis B Infection and the Effects of the Virus Variability

 and
and
Abstract
:1. Introduction
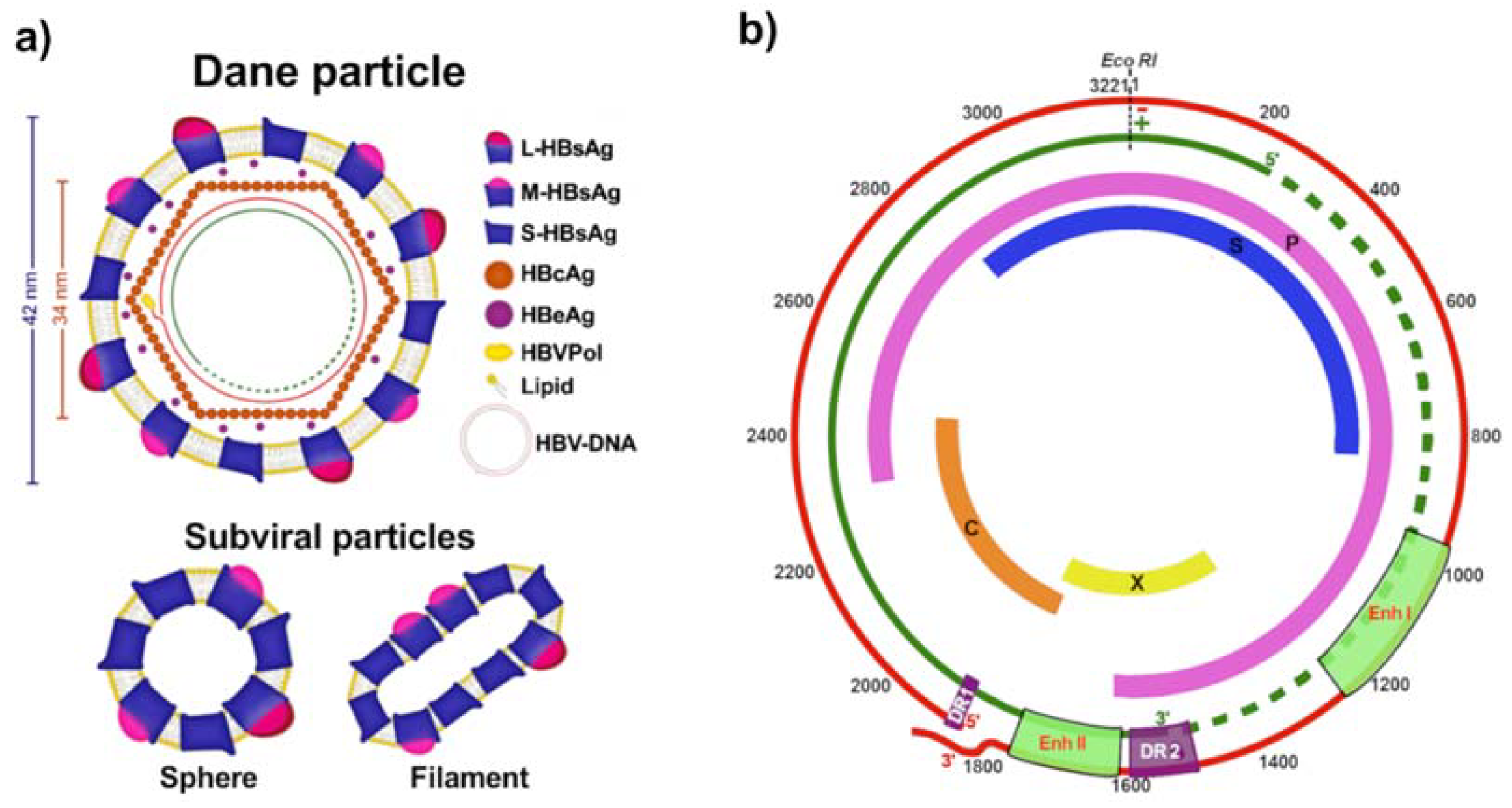
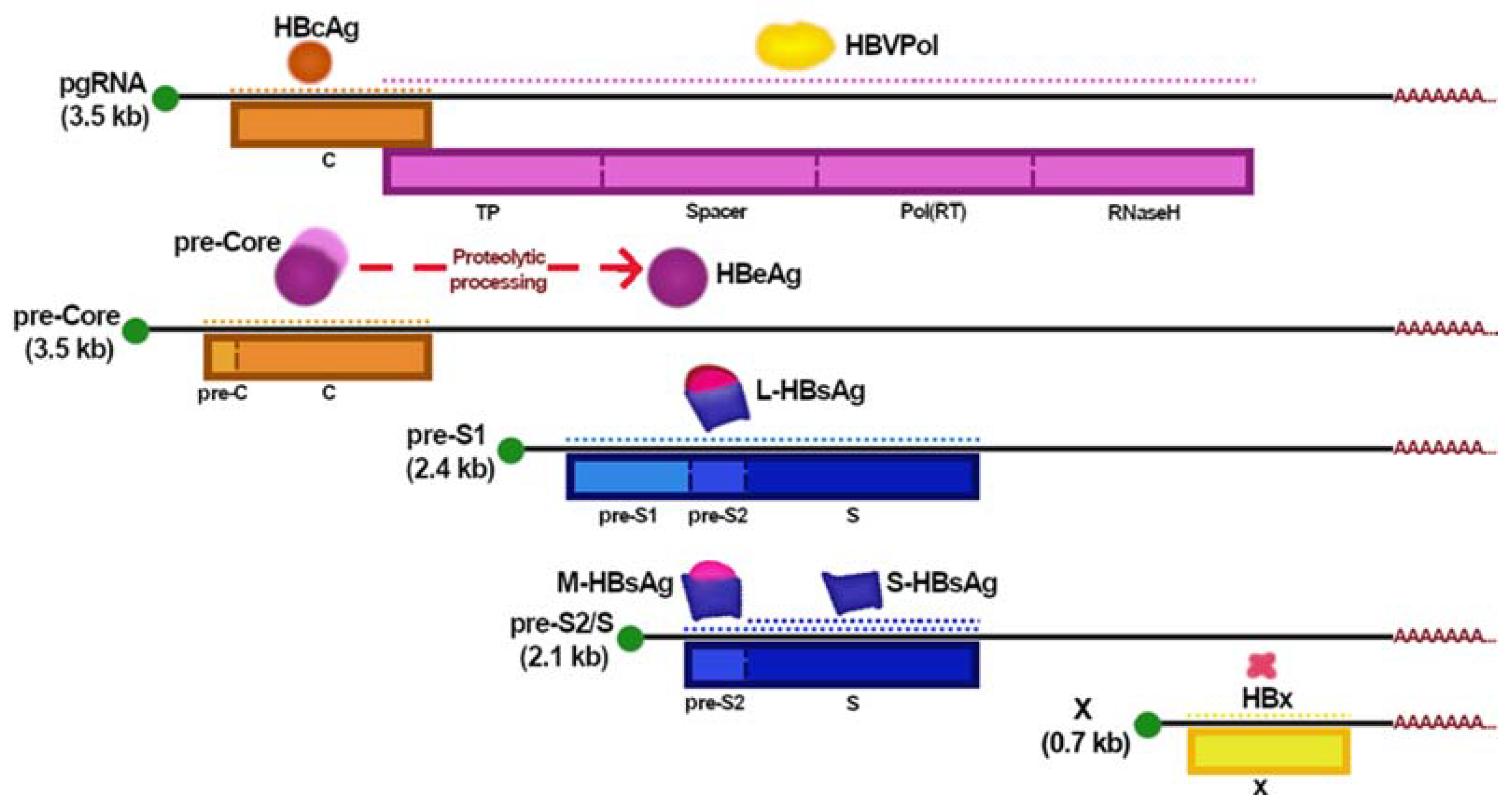
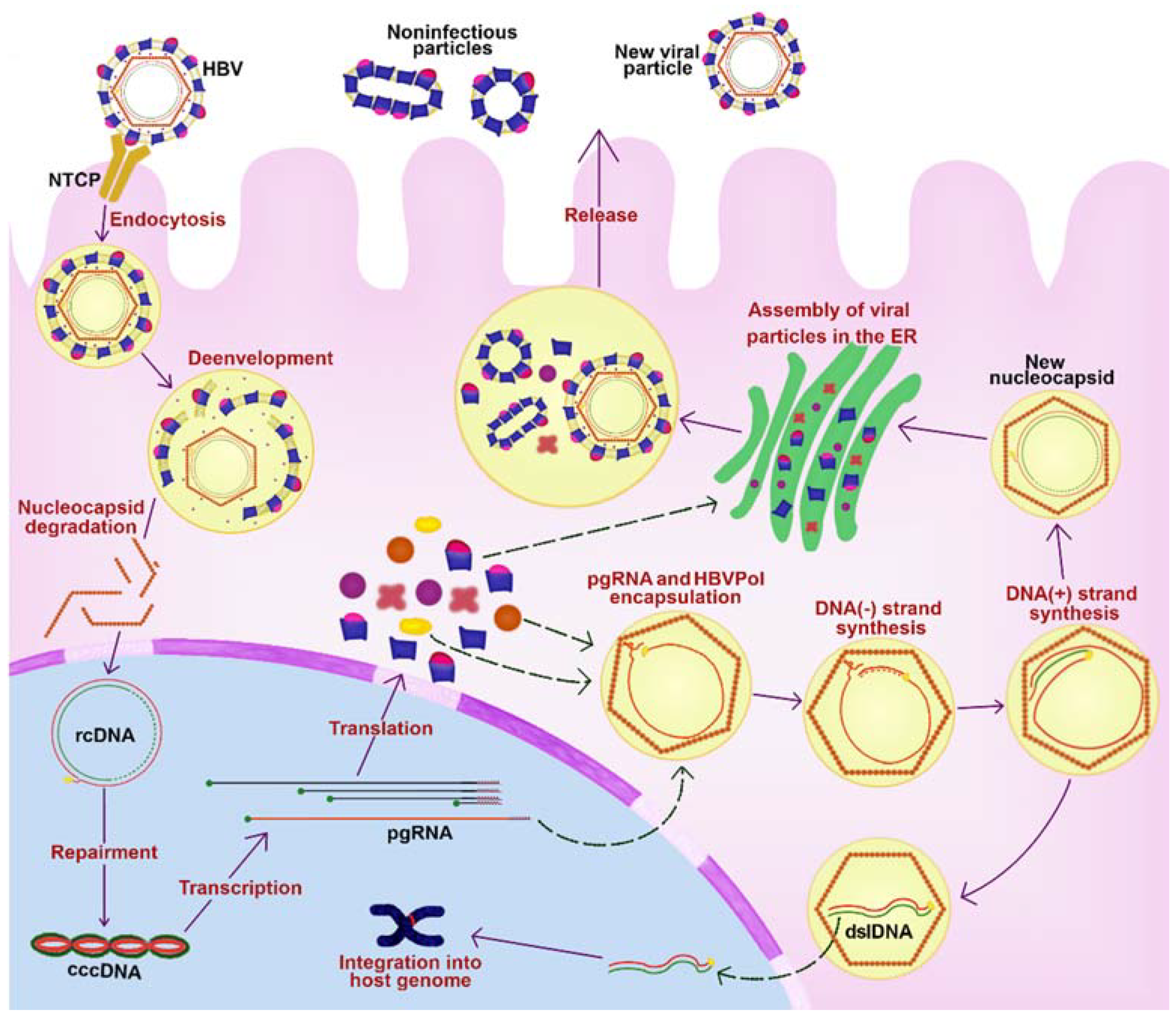
2. HBV Replicative Cycle
3. HBV Genotypes and Variants
3.1. Clinical Relevance of HBV Diversity during Infection
- (a)
- The association of genotype A with durable remission after HBeAg seroconversion. Those patients infected with genotypes A and B have higher rates of HBsAg seroclearance than genotypes D and C, respectively [27].
- (b)
- Infection with genotype A is an independent risk factor for progression to chronic hepatitis B (CHB), and the treatment with nucleoside/nucleotide analogues (NAs) does not prevent progression to chronicity [28].
- (c)
- Genotype C is more prone to cause CHB compared to genotype B [29].
- (d)
- Genotype D appears to be more prevalent in patients with HBV-related acute liver failure [30].
- (e)
- Genotype C and D are associated with a higher viral load compared to genotypes B and A, respectively. Infection with genotype H is associated with a low viral load; nevertheless, it is difficult to elucidate their association with liver diseases since patients infected with this genotype usually have additional risk factors, such as alcohol consumption, co-infection with hepatitis C virus, and obesity [31,32].
3.2. Drug-Resistant Variants
3.3. HBV-Related HCC
4. Natural History of the HBV Infection
4.1. Acute Hepatitis B
4.2. Chronic Hepatitis B
5. Immunopathogenesis of HBV Infection
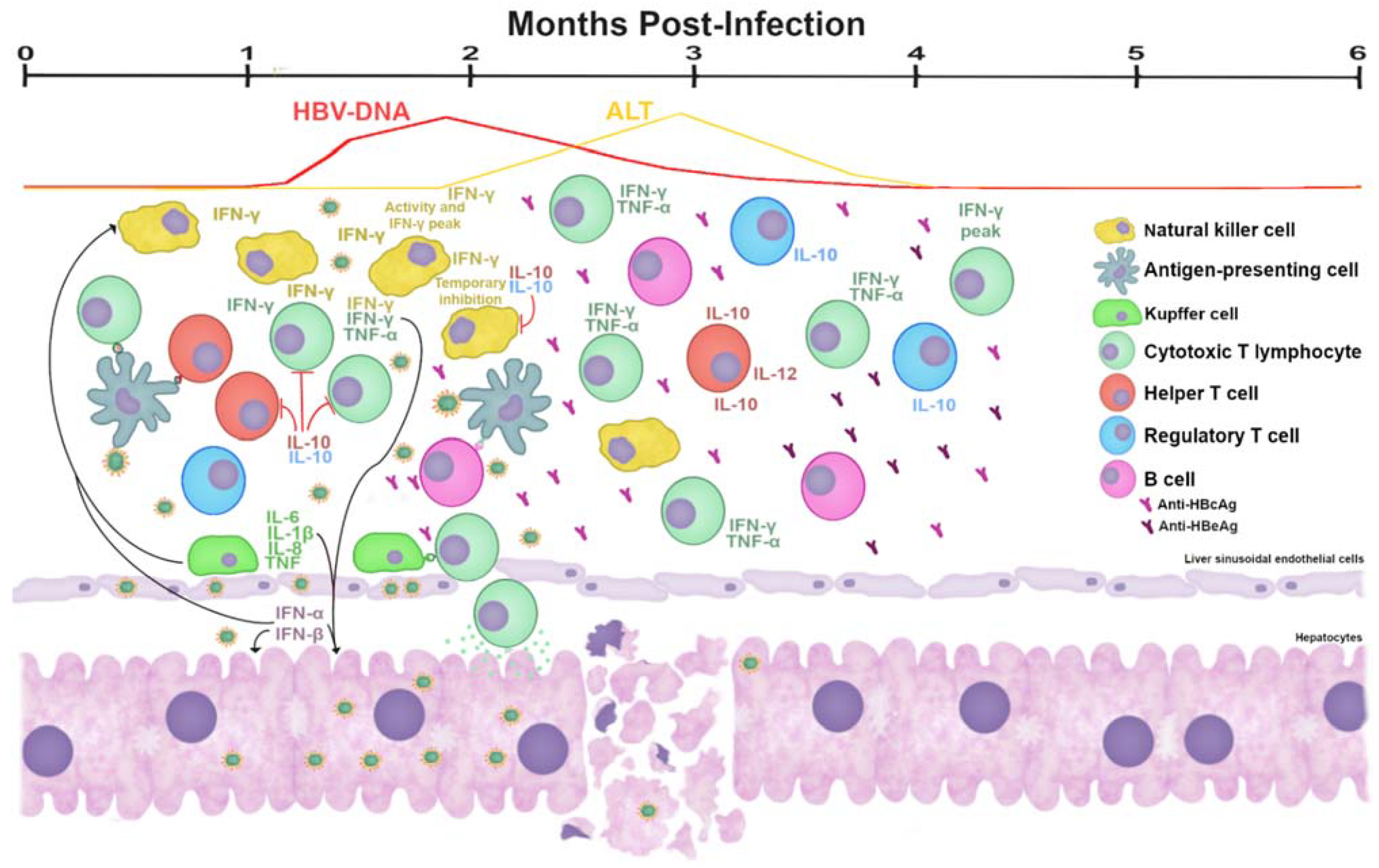
5.1. Immune Response to HBV in the Evolution of the Infection
5.2. Response in AHB Infection
5.3. Response in CHB Infection
6. Acute Liver Failure in HBV Infection
6.1. Liver Failure in Acute Infection
6.2. Acute-on-Chronic Hepatitis B Liver Failure
6.3. Viral Factors in ACLF
7. Sensing and Response to HBV
- Hepatocytes: Primary human hepatocytes (PHHs) and differentiated HepaRG (dHepaRG) cells express pattern recognition receptors (PRRs) such as retinoic acid-inducible gene-I (RIGI), melanoma differentiation-associated protein 5 (MDA5), and most Toll-like receptors (TLRs). Both RIGI and MDA5 are important for sensing viral RNAs during viral infections [146]. After the recognition of viral RNAs, these receptors induce the stimulation of an IFN-β promoter, increasing the synthesis of this antiviral cytokine. The loss of MDA5 in Huh7 cells and in MDA5 knockout mice caused an increase in HBV replication; however, the overexpression of MDA5 did not impact IFN-β induction. In that same study, it was found that RIGI did not inhibit viral replication [147]. In contrast, the recognition of the epsilon region of the pgRNA by RIGI and how this recognition not only induced the production of type III IFNs but also counteracted the interaction of HBVPol with the pgRNA [148]. Infection of PHHs cells and dHepaRG with HBV induced a weak and transient innate response (production IL-6, IL-29 and type I IFNs) [149]. Another in vitro model of micropatterned cocultures of PHHs with stromal cells (MPCCs) incubated with HBV-infected serum stimulated the expression of interferon-stimulated genes (ISGs) (Figure 6) [150].
- Hepatic non-parenchymal cells: Liver sinusoidal endothelial cells (LSECs) play a key role in the uptake of viral particles circulating in the blood to infect adjacent hepatocytes. LSECs seem to be unable to replicate the virus, but they may serve as a reservoir of endogenous reinfection. They produce large amounts of anti-inflammatory cytokines (such as TGF-β) and constitutively express major histocompatibility complex-I restricted antigens and co-stimulatory molecules that could favor the shift of the hepatic immune balance towards tolerance [153,154].
- Dendritic cells: The exposure of BDCA1+ mDCs to HBsAg results in their strong maturation, cytokine production, and an enhanced capacity to activate antigen-specific CTLs. It was also found that CD14 and TLR4 play a crucial role in the HBsAg-mediated DCs maturation [156].
8. Viral Epitopes
8.1. HBsAg
8.2. Immune Escape Mutations and Diagnosis Failure
8.3. HBcAg and HBeAg
9. The Role of TLRs
10. Interferon Response
HBV Genotypes in the Response to IFN
11. HBV Interference against the Antiviral Activity
Interference by Viral Proteins
- (a)
- HBsAg
- (b)
- HBVPol
- (c)
- HBX
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schieck, A.; Schulze, A.; Gähler, C.; Müller, T.; Haberkorn, U.; Alexandrov, A.; Urban, S.; Mier, W. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology 2013, 58, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Patient, R.; Hourioux, C.; Roingeard, P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 2009, 11, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Pastor, F.; Herrscher, C.; Patient, R.; Eymieux, S.; Moreau, A.; Burlaud-Gaillard, J.; Seigneuret, F.; De Rocquigny, H.; Roingeard, P.; Hourioux, C. Direct interaction between the hepatitis B virus core and envelope proteins analyzed in a cellular context. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karayiannis, P. Hepatitis B virus: Virology, molecular biology, life cycle and intrahepatic spread. Hepatol. Int. 2017, 11, 500–508. [Google Scholar] [CrossRef]
- McNaughton, A.L.; D’Arienzo, V.; Ansari, M.A.; Lumley, S.; Littlejohn, M.; Revill, P.; McKeating, J.A.; Matthews, P.C. Insights From Deep Sequencing of the HBV Genome—Unique, Tiny, and Misunderstood. Gastroenterology 2019, 156, 384–399. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, Z.; Li, Y.; Ding, G. Adaptive evolution of proteins in hepatitis B virus during divergence of genotypes. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.; Lemey, P.; Hurles, M.; Moyes, C.; Horn, S.; Pryor, J.; Malani, J.; Supuri, M.; Masta, A.; Teriboriki, B.; et al. Genomic Analysis of Hepatitis B Virus Reveals Antigen State and Genotype as Sources of Evolutionary Rate Variation. Viruses 2011, 3, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, P.; Cerruti, R.; Icardi, G.; Bruzzone, B. Overview of hepatitis B virus mutations and their implications in the management of infection. World J. Gastroenterol. 2016, 22, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Araujo, N.M.; Teles, S.A.; Spitz, N. Comprehensive Analysis of Clinically Significant Hepatitis B Virus Mutations in Relation to Genotype, Subgenotype and Geographic Region. Front. Microbiol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Chen, P.; Gan, Y.; Han, N.; Fang, W.; Li, J.; Zhao, F.; Hu, K.; Rayner, S. Computational Evolutionary Analysis of the Overlapped Surface (S) and Polymerase (P) Region in Hepatitis B Virus Indicates the Spacer Domain in P Is Crucial for Survival. PLoS ONE 2013, 8, e60098. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Chen, J.; Deng, L.; Mao, Q.; Zheng, J.; Wu, J.; Zeng, C.; Li, Y. Evolutionary selection associated with the multi-function of overlapping genes in the hepatitis B virus. Infect. Genet. Evol. 2010, 10, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Casillas, R.; Tabernero, D.; Gregori, J.; Belmonte, I.; Cortese, M.F.; Gonzalez-Fernandez, C.; Riveiro-Barciela, M.; López, R.M.; Quer, J.; Esteban, R.; et al. Analysis of hepatitis B virus preS1 variability and prevalence of the rs2296651 polymorphism in a Spanish population. World J. Gastroenterol. 2018, 24, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Chen, C.-C.; Chang, W.-C.; Tao, M.-H.; Huang, C. Entry of Hepatitis B Virus into Immortalized Human Primary Hepatocytes by Clathrin-Dependent Endocytosis. J. Virol. 2012, 86, 9443–9453. [Google Scholar] [CrossRef] [Green Version]
- Mitra, B.; Thapa, R.J.; Guo, H.; Block, T.M. Host functions used by hepatitis B virus to complete its life cycle: Implications for developing host-targeting agents to treat chronic hepatitis B. Antivir. Res. 2018, 158, 185–198. [Google Scholar] [CrossRef]
- Gallucci, L.; Kann, M. Nuclear Import of Hepatitis B Virus Capsids and Genome. Viruses 2017, 9, 21. [Google Scholar] [CrossRef]
- Yuen, M.-F.; Chen, D.-S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.-L. Hepatitis B virus infection. Nat. Rev. Dis. Prim. 2018, 4, 18035. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Wu, Y.; Wen, J.; Xiao, W.; Zhang, B. Pregenomic RNA: How to assist the management of chronic hepatitis B? Rev. Med. Virol. 2019, 29, e2051. [Google Scholar] [CrossRef]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Kurbanov, F.; Van Ranst, M.; Tacke, F. Molecular identification of hepatitis B virus genotypes/subgenotypes: Revised classification hurdles and updated resolutions. World J. Gastroenterol. 2014, 20, 7152–7168. [Google Scholar] [CrossRef]
- Kramvis, A. Genotypes and Genetic Variability of Hepatitis B Virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Schaefer, S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J. Gastroenterol. 2007, 13, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velkov, S.; Protzer, U.; Michler, T. Global Occurrence of Clinically Relevant Hepatitis B Virus Variants as Found by Analysis of Publicly Available Sequencing Data. Viruses 2020, 12, 1344. [Google Scholar] [CrossRef]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-L.; Kao, J.-H. Hepatitis B Virus Genotypes and Variants. Cold Spring Harb. Perspect. Med. 2015, 5, a021436. [Google Scholar] [CrossRef] [Green Version]
- Croagh, C.M.; Desmond, P.V.; Bell, S.J. Genotypes and viral variants in chronic hepatitis B: A review of epidemiology and clinical relevance. World J. Hepatol. 2015, 7, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tapias, J.M.; Costa, J.; Mas, A.; Bruguera, M.; Rodés, J. Influence of hepatitis B virus genotype on the long-term outcome of chronic hepatitis B in western patients. Gastroenterology 2002, 123, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Yotsuyanagi, H.; Yatsuhashi, H.; Karino, Y.; Takikawa, Y.; Saito, T.; Arase, Y.; Imazeki, F.; Kurosaki, M.; Umemura, T.; et al. Risk factors for long-term persistence of serum hepatitis B surface antigen following acute hepatitis B virus infection in Japanese adults. Hepatology 2014, 59, 89–97. [Google Scholar] [CrossRef]
- Zhang, H.W.; Yin, J.H.; Li, Y.T.; Li, C.Z.; Ren, H.; Gu, C.Y.; Wu, H.Y.; Liang, X.S.; Zhang, P.; Zhao, J.F.; et al. Risk factors for acute hepatitis B and its progression to chronic hepatitis in Shanghai, China. Gut 2008, 57, 1713–1720. [Google Scholar] [CrossRef]
- Wai, C.-T.; Fontana, R.J.; Polson, J.; Hussain, M.; Shakil, A.O.; Han, S.-H.; Davern, T.J.; Lee, W.M.; Lok, A.S.-F.; The US Acute Liver Failure Study Group. Clinical outcome and virological characteristics of hepatitis B-related acute liver failure in the United States. J. Viral Hepat. 2005, 12, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Roman, S. HBV endemicity in Mexico is associated with HBV genotypes H and G. World J. Gastroenterol. 2013, 19, 5446–5453. [Google Scholar] [CrossRef]
- Panduro, A.; Maldonado-González, M.; Fierro, N.A.; Roman, S. Distribution of HBV genotypes F and H in Mexico and Central America. Antivir. Ther. 2013, 18, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Fung, J.; Lai, C.-L.; Seto, W.-K.; Yuen, M.-F. Nucleoside/nucleotide analogues in the treatment of chronic hepatitis B. J. Antimicrob. Chemother. 2011, 66, 2715–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menéndez-Arias, L.; Álvarez, M.; Pacheco, B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: Mechanism of action and resistance. Curr. Opin. Virol. 2014, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Amini-Bavil-Olyaee, S.; Herbers, U.; Sheldon, J.; Luedde, T.; Trautwein, C.; Tacke, F. The rtA194T polymerase mutation impacts viral replication and susceptibility to tenofovir in hepatitis B e antigen-positive and hepatitis B e antigen-negative hepatitis B virus strains. Hepatology 2008, 49, 1158–1165. [Google Scholar] [CrossRef]
- Thai, H.; Lara, J.; Xu, X.; Kitrinos, K.; Gaggar, A.; Chan, H.L.Y.; Xia, G.-L.; Ganova-Raeva, L.; Khudyakov, Y. Complex genetic encoding of the hepatitis B virus on-drug persistence. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Cento, V.; Mirabelli, C.; Dimonte, S.; Salpini, R.; Han, Y.; Trimoulet, P.; Bertoli, A.; Micheli, V.; Gubertini, G.; Cappiello, G.; et al. Overlapping structure of hepatitis B virus (HBV) genome and immune selection pressure are critical forces modulating HBV evolution. J. Gen. Virol. 2013, 94, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, M.; Xiang, K.; Li, Y.; Li, Y.; Deng, J.; Xu, X.; Yan, L.; Zhuang, H.; Li, T. Higher detection rates of amino acid substitutions in HBV reverse transcriptase/surface protein overlapping sequence is correlated with lower serum HBV DNA and HBsAg levels in HBeAg-positive chronic hepatitis B patients with subgenotype B2. Infect. Genet. Evol. 2016, 40, 275–281. [Google Scholar] [CrossRef]
- Monto, A.; Schooley, R.T.; Lai, J.C.; Sulkowski, M.S.; Chung, R.T.; Pawlotsky, J.-M.; McHutchison, J.G.; Jacobson, I.M. Lessons From HIV Therapy Applied to Viral Hepatitis Therapy: Summary of a Workshop. Am. J. Gastroenterol. 2010, 105, 989–1004. [Google Scholar] [CrossRef]
- Tseng, T.-C.; Liu, C.; Yang, H.; Su, T.; Wang, C.; Chen, C.; Kuo, S.F.; Liu, C.; Chen, P.; Chen, D.-S.; et al. High Levels of Hepatitis B Surface Antigen Increase Risk of Hepatocellular Carcinoma in Patients with Low HBV Load. Gastroenterology 2012, 142, 1140–1149.e3. [Google Scholar] [CrossRef] [Green Version]
- Sarma, M.P.; Bhattacharjee, M.; Kar, P.; Medhi, S. Detection of HBV Genotype C in Hepatocellular Carcinoma Patients from North East India: A Brief Report. Asian Pac. J. Cancer Prev. 2018, 19, 1741–1746. [Google Scholar]
- Asim, M.; Sarma, M.P.; Kar, P. Etiological and molecular profile of hepatocellular cancer from India. Int. J. Cancer 2013, 133, 437–445. [Google Scholar] [CrossRef]
- Datta, S.; Dasgupta, D.; Ghosh, A.; Ghosh, S.; Manna, A.; Datta, S.; Chatterjee, M.; Chowdhury, A.; Banerjee, S. Oncogenic potential of hepatitis B virus subgenotype D1 surpasses D3: Significance in the development of hepatocellular carcinoma. Carcinogenesis 2018, 39, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.-W.; Yeh, S.-H.; Chen, P.-J.; Liaw, Y.-F.; Lin, C.-L.; Liu, C.-J.; Shih, W.-L.; Kao, J.-H.; Chen, D.-S.; Chen, C.-J. Hepatitis B Virus Genotype and DNA Level and Hepatocellular Carcinoma: A Prospective Study in Men. J. Natl. Cancer Inst. 2005, 97, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-I.; Yeh, S.-H.; Chen, P.-J.; Iloeje, U.H.; Jen, C.-L.; Su, J.; Wang, L.-Y.; Lu, S.-N.; You, S.-L.; Chen, D.-S.; et al. Associations Between Hepatitis B Virus Genotype and Mutants and the Risk of Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2008, 100, 1134–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, J.; Akahane, T.; Nakayama, H.; Kimura, O.; Kobayashi, T.; Kisara, N.; Sato, T.; Morosawa, T.; Izuma, M.; Kakazu, E.; et al. Comparison of hepatitis B virus genotypes B and C among chronically hepatitis B virus-infected patients who received nucleos(t)ide analogs: A multicenter retrospective study. Hepatol. Res. 2019, 49, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.; Chen, P.; Lai, M.; Chen, D.-S. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003, 124, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.C.; Burak, K.W.; Coffin, C.S. Impact of Hepatitis B Virus Genetic Variation, Integration, and Lymphotropism in Antiviral Treatment and Oncogenesis. Microorganisms 2020, 8, 1470. [Google Scholar] [CrossRef]
- Livingston, S.E.; Simonetti, J.P.; McMahon, B.J.; Bulkow, L.R.; Hurlburt, K.J.; Homan, C.E.; Snowball, M.M.; Cagle, H.H.; Williams, J.L.; Chulanov, V.P. Hepatitis B Virus Genotypes in Alaska Native People with Hepatocellular Carcinoma: Preponderance of Genotype F. J. Infect. Dis. 2007, 195, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, C.-F.; Li, T.-C.; Huang, H.-Y.; Lin, J.-H.; Chen, W.-S.; Shyu, W.-C.; Wu, H.-C.; Peng, C.-Y.; Su, I.-J.; Jeng, L.-B. Next-Generation Sequencing-Based Quantitative Detection of Hepatitis B Virus Pre-S Mutants in Plasma Predicts Hepatocellular Carcinoma Recurrence. Viruses 2020, 12, 796. [Google Scholar] [CrossRef]
- Takahashi, K.; Akahane, Y.; Hino, K.; Ohta, Y.; Mishiro, S. Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: Comparative analysis of 40 full-length isolates. Arch. Virol. 1998, 143, 2313–2326. [Google Scholar] [CrossRef]
- Li, Y.; Xia, Y.; Cheng, X.; Kleiner, D.E.; Hewitt, S.M.; Sproch, J.; Li, T.; Zhuang, H.; Liang, T.J. Hepatitis B Surface Antigen Activates Unfolded Protein Response in Forming Ground Glass Hepatocytes of Chronic Hepatitis B. Viruses 2019, 11, 386. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-W.; Lin, Y.-J.; Lin, P.-W.; Wu, H.-C.; Hsu, K.-H.; Yen, C.-J.; Chan, S.-H.; Huang, W.; Su, I.-J. A clustered ground-glass hepatocyte pattern represents a new prognostic marker for the recurrence of hepatocellular carcinoma after surgery. Cancer 2011, 117, 2951–2960. [Google Scholar] [CrossRef]
- Wang, H.-C.; Huang, W.; Lai, M.-D.; Su, I.-J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Su, I.-J.; Wang, H.-C.; Wu, H.-C.; Huang, W.-Y. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.-F.; Wu, H.-C.; Su, I.-J.; Jeng, L.-B. Hepatitis B Virus Pre-S Mutants as Biomarkers and Targets for the Development and Recurrence of Hepatocellular Carcinoma. Viruses 2020, 12, 945. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Jeng, L.-B.; Chan, W.-L.; Su, I.-J.; Teng, C.-F. Hepatitis B Virus Pre-S Gene Deletions and Pre-S Deleted Proteins: Clinical and Molecular Implications in Hepatocellular Carcinoma. Viruses 2021, 13, 862. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Wu, H.-C.; Chen, C.-F.; Fausto, N.; Lei, H.-Y.; Su, I.-J. Different Types of Ground Glass Hepatocytes in Chronic Hepatitis B Virus Infection Contain Specific Pre-S Mutants that May Induce Endoplasmic Reticulum Stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Su, I.-J.; Wang, L.H.-C.; Hsieh, W.-C.; Wu, H.-C.; Teng, C.-F.; Tsai, H.-W.; Huang, W. The emerging role of hepatitis B virus Pre-S2 deletion mutant proteins in HBV tumorigenesis. J. Biomed. Sci. 2014, 21, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mathai, A.M.; Alexander, J.; Kuo, F.-Y.; Torbenson, M.; Swanson, P.; Yeh, M.M. Type II ground-glass hepatocytes as a marker of hepatocellular carcinoma in chronic hepatitis B. Hum. Pathol. 2013, 44, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 20 May 2021).
- WHO. Who Global Hepatitis Report; WHO: Geneva, Switzerland, 2017; ISBN 9789241565455. [Google Scholar]
- Komatsu, H.; Inui, A.; Sogo, T.; Tateno, A.; Shimokawa, R.; Fujisawa, T. Tears from Children with Chronic Hepatitis B Virus (HBV) Infection Are Infectious Vehicles of HBV Transmission: Experimental Transmission of HBV by Tears, Using Mice with Chimeric Human Livers. J. Infect. Dis. 2012, 206, 478–485. [Google Scholar] [CrossRef]
- Kidd-Ljunggren, K.; Holmberg, A.; Bläckberg, J.; Lindqvist, B. High levels of hepatitis B virus DNA in body fluids from chronic carriers. J. Hosp. Infect. 2006, 64, 352–357. [Google Scholar] [CrossRef]
- Heiberg, I.L.; Hoegh, M.; Ladelund, S.; Niesters, H.; Hogh, B. Hepatitis B Virus DNA in Saliva from Children with Chronic Hepatitis B Infection: Implications for Saliva as a Potential Mode of Horizontal Transmission. Pediatr. Infect. Dis. J. 2010, 29, 465–467. [Google Scholar] [CrossRef]
- Bereket-Yucel, S.; Konukman, F. Risk of hepatitis B infections in Olympic wrestling. Br. J. Sports Med. 2007, 41, 306–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Eijk, A.A.; Niesters, H.; Hansen, B.; Pas, S.D.; Richardus, J.H.; Mostert, M.; Janssen, H.L.; Schalm, S.W.; De Man, R.A. Paired, quantitative measurements of hepatitis B virus DNA in saliva, urine and serum of chronic hepatitis B patients. Eur. J. Gastroenterol. Hepatol. 2005, 17, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Heiberg, I.L.; Hogh, B. Horizontal transmission of hepatitis B virus--why discuss when we can vaccinate? J. Infect. Dis. 2012, 206, 464–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, G.S.; Thompson, A.J. Viral Hepatitis B: Clinical and Epidemiological Characteristics. Cold Spring Harb. Perspect. Med. 2014, 4, a024935. [Google Scholar] [CrossRef] [Green Version]
- Terrault, N.A.; Lok, A.S.; McMahon, B.J.; Chang, K.-M.; Hwang, J.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef] [PubMed]
- Beasley, R.P.; Hwang, L.-Y.; Lin, C.-C.; Leu, M.-L.; Stevens, C.E.; Szmuness, W.; Chen, K.-P. Incidence of Hepatitis B Virus Infections in Preschool Children in Taiwan. J. Infect. Dis. 1982, 146, 198–204. [Google Scholar] [CrossRef]
- Coursaget, P.; Yvonnet, B.; Chotard, J.; Vincelot, P.; Sarr, M.; Diouf, C.; Chiron, J.P.; Diop-Mar, I. Age- and sex-related study of hepatitis B virus chronic carrier state in infants from an endemic area (Senegal). J. Med. Virol. 1987, 22, 1–5. [Google Scholar] [CrossRef]
- Likhitsup, A.; Lok, A.S. Understanding the Natural History of Hepatitis B Virus Infection and the New Definitions of Cure and the Endpoints of Clinical Trials. Clin. Liver Dis. 2019, 23, 401–416. [Google Scholar] [CrossRef]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T–lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N.; Nagaoka, T.; Yamashiro, M.; Mochizuki, K.; Kaneko, A.; Yamamoto, K.; Omura, M.; Hikiji, K.; Kato, M. Long-term histologic and virologic outcomes of acute self-limited hepatitis B. Hepatology 2003, 37, 1172–1179. [Google Scholar] [CrossRef]
- Ahn, S.H.; Park, Y.N.; Park, J.Y.; Chang, H.-Y.; Lee, J.M.; Shin, J.E.; Han, K.-H.; Park, C.; Moon, Y.M.; Chon, C.Y. Long-term clinical and histological outcomes in patients with spontaneous hepatitis B surface antigen seroclearance. J. Hepatol. 2005, 42, 188–194. [Google Scholar] [CrossRef]
- Hsu, C.; Tsou, H.-H.; Lin, S.-J.; Wang, M.-C.; Yao, M.; Hwang, W.-L.; Kao, W.-Y.; Chiu, C.-F.; Lin, S.-F.; Lin, J.; et al. Chemotherapy-induced hepatitis B reactivation in lymphoma patients with resolved HBV infection: A prospective study. Hepatology 2013, 59, 2092–2100. [Google Scholar] [CrossRef]
- Seto, W.-K.; Chan, T.S.-Y.; Hwang, Y.-Y.; Wong, D.K.-H.; Fung, J.; Liu, K.S.-H.; Gill, H.; Lam, Y.-F.; Lau, E.H.; Cheung, K.-S.; et al. Hepatitis B reactivation in occult viral carriers undergoing hematopoietic stem cell transplantation: A prospective study. Hepatology 2017, 65, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Mysore, K.R.; Leung, D.H. Hepatitis B and C. Clin. Liver Dis. 2018, 22, 703–722. [Google Scholar] [CrossRef]
- Shouval, D.; Shibolet, O. Immunosuppression and HBV Reactivation. Semin. Liver Dis. 2013, 33, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H. Reactivation of hepatitis B. Hepatology 2009, 49, S156–S165. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-T.; Zhao, X.-Q.; Li, G.-P.; Chen, Y.-Z.; Wang, L.; Han, M.-F.; Li, W.-N.; Chen, T.; Chen, G.; Xu, N.; et al. Immune response pattern varies with the natural history of chronic hepatitis B. World J. Gastroenterol. 2019, 25, 1950–1963. [Google Scholar] [CrossRef]
- Busca, A.; Kumar, A. Innate immune responses in hepatitis B virus (HBV) infection. Virol. J. 2014, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Chisari, F.V. Hepatitis B virus transgenic mice: Models of viral immunobiology and pathogenesis. In Current Topics in Micro-biology and Imunology; Oldstone, M.B.A., Ed.; Springer: Berlin/Heidelberg, Germany, 1996; Volume 206, pp. 149–173. ISBN 978-3-642-85208-4. [Google Scholar]
- Tseng, T.-C.; Huang, L.-R. Immunopathogenesis of Hepatitis B Virus. J. Infect. Dis. 2017, 216, S765–S770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisicaro, P.; Valdatta, C.; Boni, C.; Massari, M.; Mori, C.; Zerbini, A.; Orlandini, A.; Sacchelli, L.; Missale, G.; Ferrari, C. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut 2009, 58, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal Analysis of Early Immune Responses in Patients with Acute Hepatitis B Virus Infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef]
- Webster, G.J.; Reignat, S.; Maini, M.; Whalley, S.A.; Ogg, G.S.; King, A.; Brown, D.; Amlot, P.L.; Williams, R.; Vergani, D.; et al. Incubation Phase of Acute Hepatitis B in Man: Dynamic of Cellular Immune Mechanisms. Hepatology 2000, 32, 1117–1124. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Ishikawa, T.; Hobbs, M.V.; Matzke, B.; Schreiber, R.; Chisari, F.V. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 1996, 4, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-γ and Tumor Necrosis Factor-α Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8 + T Cells Mediate Viral Clearance and Disease Pathogenesis during Acute Hepatitis B Virus Infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, C.; Penna, A.; Bertoletti, A.; Valli, A.; Antoni, A.D.; Giuberti, T.; Cavalli, A.; Petit, M.A.; Fiaccadori, F. Cellular immune response to hepatitis B virus-encoded antigens in acute and chronic hepatitis B virus infection. J. Immunol. 1990, 145, 3442–3449. [Google Scholar]
- Guidotti, L.G.; Rochford, R.; Chung, J.; Shapiro, M.; Purcell, R.; Chisari, F. Viral Clearance without Destruction of Infected Cells During Acute HBV Infection. Science 1999, 284, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-C.; Sung, P.S.; Park, S.-H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.L.; Gardiner, L.; Nishimura, S.; Shinkai, K.; Locksley, R.; Ganem, D. Activation of a Nonclassical NKT Cell Subset in a Transgenic Mouse Model of Hepatitis B Virus Infection. Immunity 2002, 16, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, P.; Haque, M.; Humphries-Bickley, T.; Ravi, S.; Song, J. Hepatitis B Virus Immunopathology, Model Systems, and Current Therapies. Front. Immunol. 2017, 8, 436. [Google Scholar] [CrossRef] [Green Version]
- Chisari, F.; Isogawa, M.; Wieland, S. Pathogenesis of hepatitis B virus infection. Pathol. Biol. 2010, 58, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Stross, L.; Günther, J.; Gasteiger, G.; Asen, T.; Graf, S.; Aichler, M.; Esposito, I.; Busch, D.H.; Knolle, P.A.; Sparwasser, T.; et al. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology 2012, 56, 873–883. [Google Scholar] [CrossRef]
- Suresh, M.; Czerwinski, S.; Murreddu, M.G.; Kallakury, B.V.; Ramesh, A.; Gudima, S.O.; Menne, S. Innate and adaptive immunity associated with resolution of acute woodchuck hepatitis virus infection in adult woodchucks. PLoS Pathog. 2019, 15, e1008248. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengsch, B.; Martin, B.; Thimme, R. Restoration of HBV-specific CD8+ T cell function by PD-1 blockade in inactive carrier patients is linked to T cell differentiation. J. Hepatol. 2014, 61, 1212–1219. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral Intrahepatic T-Cell Responses Can Be Restored by Blocking Programmed Death-1 Pathway in Chronic Hepatitis B. Gastroenterol. 2010, 138, 682–693. [Google Scholar] [CrossRef]
- Schurich, A.; Khanna, P.; Lopes, A.R.; Han, K.J.; Peppa, D.; Micco, L.; Nebbia, G.; Kennedy, P.T.; Geretti, A.-M.; Dusheiko, G.; et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011, 53, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the Tim-3/Galectin-9 Pathway of T Cell Exhaustion in Chronic Hepatitis B Virus Infection. PLoS ONE 2012, 7, e47648. [Google Scholar] [CrossRef]
- Raziorrouh, B.; Schraut, W.; Gerlach, T.; Nowack, D.; Grüner, N.H.; Ulsenheimer, A.; Zachoval, R.; Wächtler, M.; Spannagl, M.; Haas, J.; et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T-cell function. Hepatology 2010, 52, 1934–1947. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
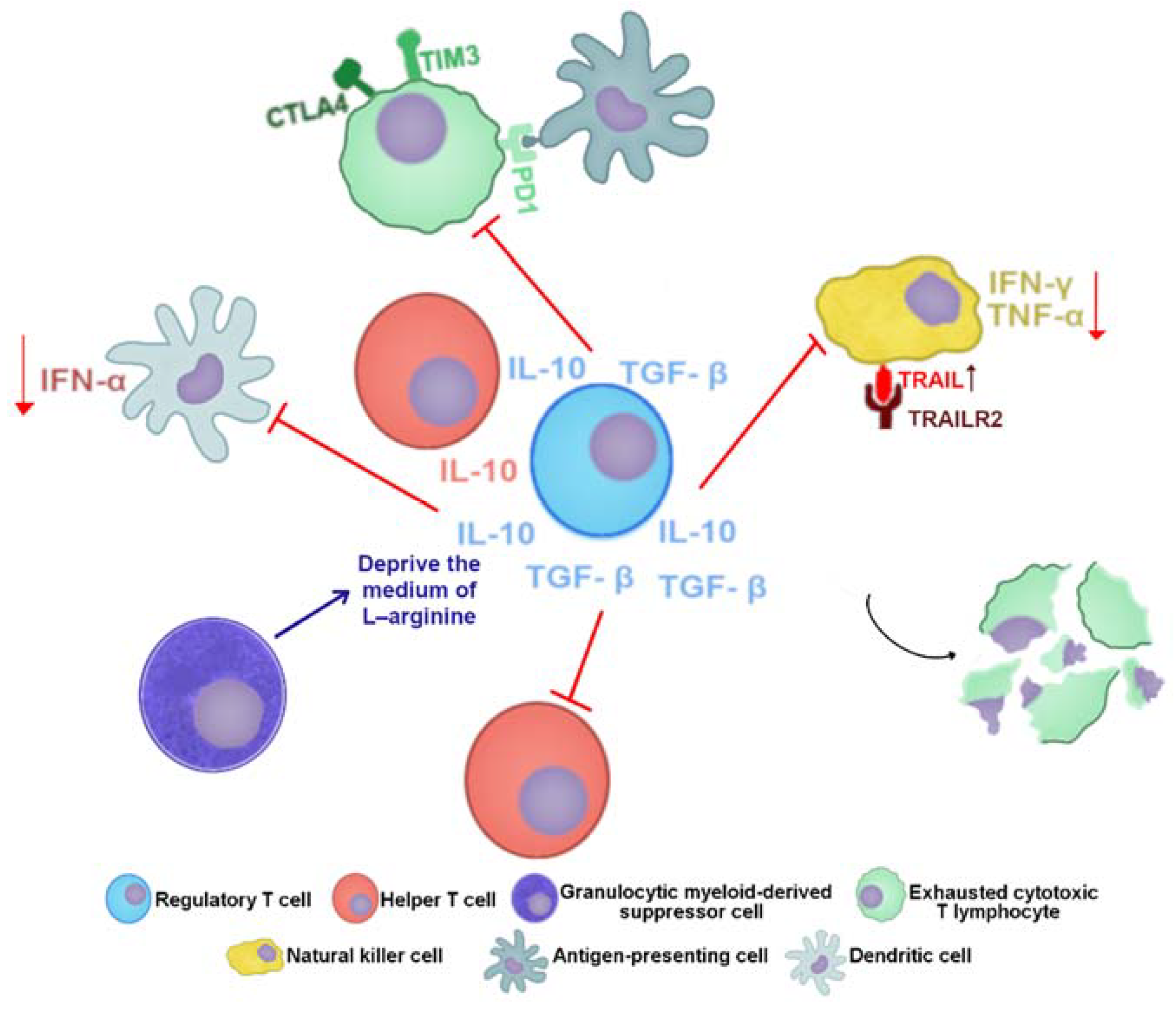
- Stoop, J.N.; Van Der Molen, R.G.; Baan, C.C.; van der Laan, L.; Kuipers, E.J.; Kusters, J.G.; Janssen, H.L.A. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology 2005, 41, 771–778. [Google Scholar] [CrossRef]
- Park, J.-J.; Wong, D.K.; Wahed, A.; Lee, W.M.; Feld, J.J.; Terrault, N.; Khalili, M.; Sterling, R.K.; Kowdley, K.V.; Bzowej, N.; et al. Hepatitis B Virus–Specific and Global T-Cell Dysfunction in Chronic Hepatitis B. Gastroenterology 2016, 150, 684–695.e5. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.R.; Kellam, P.; Das, A.; Dunn, C.; Kwan, A.; Turner, J.; Peppa, D.; Gilson, R.J.; Gehring, A.; Bertoletti, A.; et al. Bim-mediated deletion of antigen-specific CD8+ T cells in patients unable to control HBV infection. J. Clin. Investig. 2008, 118, 1835–1845. [Google Scholar] [CrossRef]
- Peppa, D.; Gill, U.S.; Reynolds, G.; Easom, N.J.; Pallett, L.J.; Schurich, A.; Micco, L.; Nebbia, G.; Singh, H.D.; Adams, D.; et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell–mediated deletion. J. Exp. Med. 2013, 210, 99–114. [Google Scholar] [CrossRef]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M. Natural Killer Cell Functional Dichotomy in Chronic Hepatitis B and Chronic Hepatitis C Virus Infections. Gastroenterology 2009, 137, 1151–1160.e7. [Google Scholar] [CrossRef]
- Peppa, D.; Micco, L.; Javaid, A.; Kennedy, P.T.F.; Schurich, A.; Dunn, C.; Pallant, C.; Ellis, G.; Khanna, P.; Dusheiko, G.; et al. Blockade of Immunosuppressive Cytokines Restores NK Cell Antiviral Function in Chronic Hepatitis B Virus Infection. PLoS Pathog. 2010, 6, e1001227. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Fu, B.; Gao, Y.; Liao, X.; Sun, R.; Tian, Z.; Wei, H. TGF-β1 Down-Regulation of NKG2D/DAP10 and 2B4/SAP Expression on Human NK Cells Contributes to HBV Persistence. PLoS Pathog. 2012, 8, e1002594. [Google Scholar] [CrossRef]
- Van Der Molen, R.G.; Sprengers, D.; Binda, R.S.; De Jong, E.C.; Niesters, H.; Kusters, J.G.; Kwekkeboom, J.; Janssen, H.L.A. Functional impairment of myeloid and plasmacytoid dendritic cells of patients with chronic hepatitis B. Hepatology 2004, 40, 738–746. [Google Scholar] [CrossRef]
- Pallett, L.J.; Gill, U.S.; Quaglia, A.; Sinclair, L.V.; Jover-Cobos, M.; Schurich, A.; Singh, K.P.; Thomas, N.; Das, A.; Chen, A.; et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 2015, 21, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Kuo, C.-F.; Akbari, O.; Ou, J.-H.J. Maternal-Derived Hepatitis B Virus e Antigen Alters Macrophage Function in Offspring to Drive Viral Persistence after Vertical Transmission. Immunity 2016, 44, 1204–1214. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Li, L.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Interferon-γ facilitates hepatic antiviral T cell retention for the maintenance of liver-induced systemic tolerance. J. Exp. Med. 2016, 213, 1079–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhang, Z.; Luan, Y.; Zou, Z.; Sun, Y.; Li, Y.; Jin, L.; Zhou, C.; Fu, J.; Gao, B.; et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology 2014, 59, 1331–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.Q.; Zhang, J.Y.; Zhang, H.; Zou, Z.S.; Wang, F.S.; Jia, J.H. Increased Th17 cells contribute to disease progression in patients with HBV-associated liver cirrhosis. J. Viral Hepat. 2012, 19, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Oketani, M.; Uto, H.; Ido, A.; Tsubouchi, H. Management of hepatitis B virus-related acute liver failure. Clin. J. Gastroenterol. 2014, 7, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichai, P.; Samuel, D. Management of Fulminant Hepatitis B. Curr. Infect. Dis. Rep. 2019, 21, 25. [Google Scholar] [CrossRef]
- Hughes, S.A.; Wedemeyer, H.; Harrison, P.M. Hepatitis delta virus. Lancet 2011, 378, 73–85. [Google Scholar] [CrossRef]
- Kusakabe, A.; Tanaka, Y.; Mochida, S.; Nakayama, N.; Inoue, K.; Sata, M.; Isoda, N.; Kang, J.H.; Sumino, Y.; Yatsuhashi, H.; et al. Case-control study for the identification of virological factors associated with fulminant hepatitis B. Hepatol. Res. 2009, 39, 648–656. [Google Scholar] [CrossRef]
- Chen, Z.; Diaz, G.; Pollicino, T.; Zhao, H.; Engle, R.E.; Schuck, P.; Shen, C.-H.; Zamboni, F.; Long, Z.; Kabat, J.; et al. Role of humoral immunity against hepatitis B virus core antigen in the pathogenesis of acute liver failure. PNAS 2018, 115, E11369–E11378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Yan, W.-Z.; Zhao, C.-Y.; Che, H.-H.; Liu, X.-Y.; Liu, Z.-Z.; Wang, Y.-D.; Wang, W.; Li, M.; Gao, J. Increased CD4+CD25+ regulatory T cells correlate with poor short-term outcomes in hepatitis B virus-related acute-on-chronic liver failure patients. J. Microbiol. Immunol. Infect. 2015, 48, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wu, Z.-B.; Ye, Y.-N.; Liu, J.; Zhang, G.-L.; Su, Y.-J.; He, H.-L.; Zheng, Y.-B.; Gao, Z.-L. Plasma Interleukin-10: A Likely Predictive Marker for Hepatitis B Virus-Related Acute-on-Chronic Liver Failure. Hepat. Mon. 2014, 14, e19370. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Tang, K.; Li, Q.; Yuan, G.; Cao, W.; Lu, W. Changes of IL-1, TNF-Alpha, IL-12 and IL-10 Levels with Chronic Liver Failure. Surg. Sci. 2011, 2, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Mourtzikou, A.; Alepaki, M.; Stamouli, M.; Pouliakis, A.; Skliris, A.; Karakitsos, P. Evaluation of serum levels of IL-6, TNF-α, IL-10, IL-2 and IL-4 in patients with chronic hepatitis. Inmunología 2014, 33, 41–50. [Google Scholar] [CrossRef]
- Song, L.H.; Binh, V.Q.; Duy, D.N.; Kun, J.F.; Bock, T.C.; Kremsner, P.G.; Luty, A. Serum cytokine profiles associated with clinical presentation in Vietnamese infected with hepatitis B virus. J. Clin. Virol. 2003, 28, 93–103. [Google Scholar] [CrossRef]
- Falasca, K.; Ucciferri, C.; Dalessandro, M.; Zingariello, P.; Mancino, P.; Petrarca, C.; Pizzigallo, E.; Conti, P.; Vecchiet, J. Cytokine patterns correlate with liver damage in patients with chronic hepatitis B and C. Ann. Clin. Lab. Sci. 2006, 36, 144–150. [Google Scholar]
- Poovorawan, K.; Tangkijvanich, P.; Chirathaworn, C.; Wisedopas, N.; Treeprasertsuk, S.; Komolmit, P.; Poovorawan, Y. Circulating Cytokines and Histological Liver Damage in Chronic Hepatitis B Infection. Hepat. Res. Treat. 2013, 2013, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ming, D.; Yu, X.; Guo, R.; Deng, Y.; Li, J.; Lin, C.; Su, M.; Lin, Z.; Su, Z. Elevated TGF-β1/IL-31 Pathway Is Associated with the Disease Severity of Hepatitis B Virus–Related Liver Cirrhosis. Viral Immunol. 2015, 28, 209–216. [Google Scholar] [CrossRef]
- Yu, X.; Guo, R.; Ming, D.; Deng, Y.; Su, M.; Lin, C.; Li, J.; Lin, Z.; Su, Z. The Transforming Growth Factor β1/Interleukin-31 Pathway Is Upregulated in Patients with Hepatitis B Virus-Related Acute-on-Chronic Liver Failure and Is Associated with Disease Severity and Survival. Clin. Vaccine Immunol. 2015, 22, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, S.; Li, S.; Yang, N.; Tang, X.; Zhang, S.; Hu, D.; Lu, M. Deregulation of Regulatory T Cells in Acute-on-Chronic Liver Failure: A Rat Model. Mediat. Inflamm. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Zhang, G.-L.; Xie, D.-Y.; Lin, B.-L.; Xie, C.; Ye, Y.-N.; Peng, L.; Zhang, S.-Q.; Zhang, Y.-F.; Lai, Q.; Zhu, J.-Y.; et al. Imbalance of interleukin-17-producing CD4 T cells/regulatory T cells axis occurs in remission stage of patients with hepatitis B virus-related acute-on-chronic liver failure. J. Gastroenterol. Hepatol. 2013, 28, 513–521. [Google Scholar] [CrossRef]
- Liang, X.-S.; Li, C.-Z.; Zhou, Y.; Yin, W.; Liu, Y.-Y.; Fan, W.-H. Changes in circulating Foxp3+regulatory T cells and interleukin-17-producing T helper cells during HBV-related acute-on-chronic liver failure. World J. Gastroenterol. 2014, 20, 8558–8571. [Google Scholar] [CrossRef]
- Niu, Y.-H.; Yin, D.-L.; Liu, H.-L.; Yi, R.-T.; Yang, Y.-C.; Xue, H.-A.; Chen, T.-Y.; Zhang, S.-L.; Lin, S.-M.; Zhao, Y.-R. Restoring the Treg cell to Th17 cell ratio may alleviate HBV-related acute-on-chronic liver failure. World J. Gastroenterol. 2013, 19, 4146–4154. [Google Scholar] [CrossRef]
- Hu, F.; Bi, S.; Yan, H.; Shi, Y.; Sheng, J. Associations between hepatitis B virus basal core promoter/pre-core region mutations and the risk of acute-on-chronic liver failure: A meta-analysis. Virol. J. 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Xu, Z.; Liu, Y.; Li, X.; Bai, S.; Ding, N.; Zhong, Y.; Wang, L.; Mao, P.; Zoulim, F.; et al. Hepatitis B virus genotype and basal core promoter/precore mutations are associated with hepatitis B-related acute-on-chronic liver failure without pre-existing liver cirrhosis. J. Viral Hepat. 2010, 17, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Zhou, B.; Gao, H.; Ma, S.; Yang, G.; Xu, M.; Abbott, W.G.; Chen, J.; Sun, J.; Wang, Z.; et al. Hepatitis B virus genotype B with G1896A and A1762T/G1764A mutations is associated with hepatitis B related acute-on-chronic liver failure. J. Med. Virol. 2011, 83, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Han, M.; Chen, F.; Xu, Y.; Chen, E.; Wang, X.; Liu, Y.; Sun, J.; Hou, J.; Ning, Q.; et al. Hepatitis B virus genotype B and mutations in basal core promoter and pre-core/core genes associated with acute-on-chronic liver failure: A multicenter cross-sectional study in China. Hepatol. Int. 2014, 8, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-T.; Huang, S.-Y.; Chen, L.; Liu, F.; Cai, X.-H.; Guo-Qing, Z.; Wang, M.-J.; Han, Y.; Su-Yuan, H.; Jiang, J.-H.; et al. Characterization of Full-Length Genomes of Hepatitis B Virus Quasispecies in Sera of Patients at Different Phases of Infection. J. Clin. Microbiol. 2015, 53, 2203–2214. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Cao, Q.; Xiong, Y.; Zhang, E.; Lu, M. Interaction between Hepatitis B Virus and Toll-Like Receptors: Current Status and Potential Therapeutic Use for Chronic Hepatitis B. Vaccines 2018, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Luangsay, S.; Ait-Goughoulte, M.; Michelet, M.; Floriot, O.; Bonnin, M.; Gruffaz, M.; Rivoire, M.; Fletcher, S.; Javanbakht, H.; Lucifora, J.; et al. Expression and functionality of Toll- and RIG-like receptors in HepaRG cells. J. Hepatol. 2015, 63, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-L.; Liao, F. Melanoma Differentiation–Associated Gene 5 Senses Hepatitis B Virus and Activates Innate Immune Signaling To Suppress Virus Replication. J. Immunol. 2013, 191, 3264–3276. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA Sensor RIG-I Dually Functions as an Innate Sensor and Direct Antiviral Factor for Hepatitis B Virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. PNAS 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhao, X.; Wang, Z.; Shu, W.; Li, L.; Li, Y.; Guo, Z.; Gao, B.; Xiong, S. Nuclear Sensor Interferon-Inducible Protein 16 Inhibits the Function of Hepatitis B Virus Covalently Closed Circular DNA by Integrating Innate Immune Activation and Epigenetic Suppression. Hepatology 2020, 71, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach-Rivière, L.; Bergez, M.; Mönch, S.; Qu, B.; Riess, M.; Vondran, F.W.; Liese, J.; Hornung, V.; Urban, S.; König, R. Hepatitis B Virus DNA is a Substrate for the cGAS/STING Pathway but is not Sensed in Infected Hepatocytes. Viruses 2020, 12, 592. [Google Scholar] [CrossRef] [PubMed]
- Breiner, K.M.; Schaller, H.; Knolle, P.A. Endothelial cell–mediated uptake of a hepatitis B virus: A new concept of liver targeting of hepatotropic microorganisms. Hepatology 2001, 34, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Li, J.-M.; Liu, M.-K.; Zhang, T.-T.; Wang, D.-P.; Zhou, W.-H.; Hu, L.-Z.; Lv, W.-L. Pathological process of liver sinusoidal endothelial cells in liver diseases. World J. Gastroenterol. 2017, 23, 7666–7677. [Google Scholar] [CrossRef]
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Van Montfoort, N.; Van Der Aa, E.; Bosch, A.V.D.; Brouwers, H.; Vanwolleghem, T.; Janssen, H.L.A.; Javanbakht, H.; Buschow, S.; Woltman, A.M. Hepatitis B Virus Surface Antigen Activates Myeloid Dendritic Cells via a Soluble CD14-Dependent Mechanism. J. Virol. 2016, 90, 6187–6199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Galindo, D.A.; Sánchez-Ávila, F.; Bobadilla-Morales, L.; Gómez-Quiróz, P.; Bueno-Topete, M.; Armendáriz-Borunda, J.; Sánchez-Orozco, L.V. New amino acid changes in drug resistance sites and HBsAg in hepatitis B virus genotype H. J. Med. Virol. 2015, 87, 985–992. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, L.; Dai, Y.; Hu, Y.; Xu, B.; Zhou, Y.-H. Conservative Evolution of Hepatitis B Virus Precore and Core Gene During Immune Tolerant Phase in Intrafamilial Transmission. Virol. Sin. 2020, 35, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Desmond, C.P.; Bartholomeusz, A.; Gaudieri, S.; Revill, P.A.; Lewin, S.R. A systematic review of T-cell epitopes in hepatitis B virus: Identification, genotypic variation and relevance to antiviral therapeutics. Antivir. Ther. 2008, 13, 161–175. [Google Scholar]
- Stirk, H.; Thornton, J.; Howard, C. Atopological Model for Hepatitis B Surface Antigen. Intervirology 1992, 33, 148–158. [Google Scholar] [CrossRef]
- Wu, C.; Deng, W.; Deng, L.; Cao, L.; Qin, B.; Li, S.; Wang, Y.; Pei, R.; Yang, D.; Lu, M.; et al. Amino Acid Substitutions at Positions 122 and 145 of Hepatitis B Virus Surface Antigen (HBsAg) Determine the Antigenicity and Immunogenicity of HBsAg and InfluenceIn VivoHBsAg Clearance. J. Virol. 2012, 86, 4658–4669. [Google Scholar] [CrossRef] [Green Version]
- Theamboonlers, A.; Chongsrisawat, V.; Jantaradsamee, P.; Poovorawan, Y. Variants within the “a” Determinant of HBs Gene in Children and Adolescents with and without Hepatitis B Vaccination as Part of Thailand’s Expanded Program on Immunization (EPI). Tohoku J. Exp. Med. 2001, 193, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Luongo, M.; Critelli, R.; Grottola, A.; Gitto, S.; Bernabucci, V.; Bevini, M.; Vecchi, C.; Montagnani, G.; Villa, E. Acute hepatitis B caused by a vaccine-escape HBV strain in vaccinated subject: Sequence analysis and therapeutic strategy. J. Clin. Virol. 2015, 62, 89–91. [Google Scholar] [CrossRef]
- Carman, W.; Karayiannis, P.; Waters, J.; Thomas, H.; Zanetti, A.; Manzillo, G.; Zuckerman, A. Vaccine-induced escape mutant of hepatitis B virus. Lancet 1990, 336, 325–329. [Google Scholar] [CrossRef]
- Howard, C.R.; Allison, L.M. Hepatitis B Surface Antigen Variation and Protective Immunity. Intervirology 1995, 38, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hossain, G.; Ueda, K. Investigation of a Novel Hepatitis B Virus Surface Antigen (HBsAg) Escape Mutant Affecting Immunogenicity. PLoS ONE 2017, 12, e0167871. [Google Scholar] [CrossRef]
- Lazarevic, I. Clinical implications of hepatitis B virus mutations: Recent advances. World J. Gastroenterol. 2014, 20, 7653–7664. [Google Scholar] [CrossRef]
- Horvat, R.T. Diagnostic and Clinical Relevance of HBV Mutations. Lab. Med. 2011, 42, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Ou, S.-H.; Chen, C.-R.; Ge, S.-X.; Pei, B.; Lin, Y.-C.; Ni, H.-Y.; Huang, C.-H.; Yeo, A.E.T.; Shih, J.W.K.; et al. Molecular Characteristics of Occult Hepatitis B Virus from Blood Donors in Southeast China. J. Clin. Microbiol. 2009, 48, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Lv, J.; Feng, Y.; Liu, J.; Jingjing, L.; Xu, A.; Zhang, L. Temporal trend of hepatitis B surface mutations in the post-immunization period: 9 years of surveillance (2005–2013) in eastern China. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Nomura, F.; Itoga, S.; Isobe, K.; Nakai, T. Prevalence of vaccine-induced escape mutants of hepatitis B virus in the adult population in China: A prospective study in 176 restaurant employees. J. Gastroenterol. Hepatol. 2001, 16, 1373–1377. [Google Scholar] [CrossRef]
- Di Lello, F.A.; Ridruejo, E.; Martínez, A.P.; Pérez, P.S.; Campos, R.H.; Flichman, D.M. Molecular epidemiology of hepatitis B virus mutants associated with vaccine escape, drug resistance and diagnosis failure. J. Viral Hepat. 2018, 26, 552–560. [Google Scholar] [CrossRef]
- Mello, F.C.A.; Martel, N.; Gomes, S.A.; Araujo, N.M. Expression of Hepatitis B Virus Surface Antigen Containing Y100C Variant Frequently Detected in Occult HBV Infection. Hepat. Res. Treat. 2011, 2011, 1–4. [Google Scholar] [CrossRef]
- Inuzuka, T.; Ueda, Y.; Arasawa, S.; Takeda, H.; Matsumoto, T.; Osaki, Y.; Uemoto, S.; Seno, H.; Marusawa, H. Expansion of viral variants associated with immune escape and impaired virion secretion in patients with HBV reactivation after resolved infection. Sci. Rep. 2018, 8, 18070. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.; Cheng, N.; Zlotnick, A.; Stahl, S.; Wingfield, P.; Belnap, D.; Kanngiesser, U.; Noah, M.; Steven, A. Hepatitis B virus capsid: Localization of the putative immunodominant loop (residues 78 to 83) on the capsid surface, and implications for the distinction between c and e-antigens. J. Mol. Biol. 1998, 279, 1111–1121. [Google Scholar] [CrossRef]
- Sendi, H.; Mehrab-Mohseni, M.; Shahraz, S.; Norder, H.; Alavian, S.M.; Noorinayer, B.; Zali, M.R.; Pumpens, P.; Bonkovsky, H.L.; Magnius, L.O. CTL escape mutations of core protein are more frequent in strains of HBeAg negative patients with low levels of HBV DNA. J. Clin. Virol. 2009, 46, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zhang, E.; Yang, D.; Lu, M. Contribution of Toll-like receptors to the control of hepatitis B virus infection by initiating antiviral innate responses and promoting specific adaptive immune responses. Cell. Mol. Immunol. 2014, 12, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, R.-J.; Chen, X.-W.; Lu, M.-J. Control of hepatitis B virus replication by interferons and Toll-like receptor signaling pathways. World J. Gastroenterol. 2014, 20, 11618–11629. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.-I.; Hirata, Y.; Kameyama, T.; Tokunaga, Y.; Nishito, Y.; Hirabayashi, K.; Yano, J.; Ochiya, T.; Tateno, C.; Tanaka, Y.; et al. Targeted Induction of Interferon-λ in Humanized Chimeric Mouse Liver Abrogates Hepatotropic Virus Infection. PLoS ONE 2013, 8, e59611. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, W.P.; Chan, H.L.Y.; Lampertico, P.; Hou, J.; Tangkijvanich, P.; Reesink, H.W.; Zhang, W.; Mangia, A.; Tanwandee, T.; Montalto, G.; et al. Genome-wide Association Study Identifies Genetic Variants Associated with Early and Sustained Response to (Pegylated) Interferon in Chronic Hepatitis B Patients: The GIANT-B Study. Clin. Infect. Dis. 2019, 69, 1969–1979. [Google Scholar] [CrossRef]
- Konerman, M.A.; Lok, A.S. Interferon Treatment for Hepatitis B. Clin. Liver Dis. 2016, 20, 645–665. [Google Scholar] [CrossRef]
- Shen, F.; Jieliang, C.; Wang, Y.; Sozzi, V.; Revill, P.A.; Liu, J.; Gao, L.; Yang, G.; Lu, M.; Sutter, K.; et al. Hepatitis B virus sensitivity to interferon-α in hepatocytes is more associated with cellular interferon response than with viral genotype. Hepatology 2018, 67, 1237–1252. [Google Scholar] [CrossRef] [Green Version]
- Alcantara, F.F.; Tang, H.; McLachlan, A. Functional characterization of the interferon regulatory element in the enhancer 1 region of the hepatitis B virus genome. Nucleic Acids Res. 2002, 30, 2068–2075. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.-J.; Chen, E.-Q.; Yang, J.-H.; Zhou, T.-Y.; Liu, L.; Tang, H. A mutation in the interferon regulatory element of HBV may influence the response of interferon treatment in chronic hepatitis B patients. Virol. J. 2012, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Lu, H.; Xu, L.; Idris, N.F.B.; Li, Y.; Hu, J.; Huang, A.; Tu, Z. The response of hepatitis B virus genotype to interferon is associated with a mutation in the interferon-stimulated response element. Medicine 2019, 98, e18442. [Google Scholar] [CrossRef]
- Chen, J.; Wu, M.; Wang, F.; Zhang, W.; Wang, W.; Zhang, X.; Zhang, J.; Liu, Y.; Liu, Y.; Feng, Y.; et al. Hepatitis B virus spliced variants are associated with an impaired response to interferon therapy. Sci. Rep. 2015, 5, 16459. [Google Scholar] [CrossRef] [Green Version]
- Brook, M.G.; McDonald, J.A.; Karayiannis, P.; Caruso, L.; Forster, G.; Harris, J.R.; Thomas, H.C. Randomised controlled trial of interferon alfa 2A (rbe) (Roferon-A) for the treatment of chronic hepatitis B virus (HBV) infection: Factors that influence response. Gut 1989, 30, 1116–1122. [Google Scholar] [CrossRef]
- Perrillo, R. Benefits and risks of interferon therapy for hepatitis B. Hepatology 2009, 49, S103–S111. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Shen, H.-C.; Jia, N.-N.; Wang, H.; Lin, L.-Y.; An, B.-Y.; Gui, H.-L.; Guo, S.-M.; Cai, W.; Yu, H.; et al. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect. 2009, 11, 515–523. [Google Scholar] [CrossRef]
- Xu, N.; Yao, H.-P.; Lv, G.-C.; Chen, Z. Downregulation of TLR7/9 leads to deficient production of IFN-α from plasmacytoid dendritic cells in chronic hepatitis B. Inflamm. Res. 2012, 61, 997–1004. [Google Scholar] [CrossRef]
- Vincent, I.E.; Zannetti, C.; Lucifora, J.; Norder, H.; Protzer, U.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trepo, C.; Hasan, U.; et al. Hepatitis B Virus Impairs TLR9 Expression and Function in Plasmacytoid Dendritic Cells. PLoS ONE 2011, 6, e26315. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Broering, R.; Trippler, M.; Poggenpohl, L.; Fiedler, M.; Gerken, G.; Lu, M.; Schlaak, J.F. Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J. Viral Hepat. 2014, 21, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-W.; Lin, S.-C.; Wei, S.-C.; Hu, J.-T.; Chang, H.-Y.; Huang, S.-H.; Chen, D.-S.; Chen, P.-J.; Hsu, P.-N.; Yang, S.-S.; et al. Reduced Toll-like receptor-3 expression in chronic hepatitis B patients and its restoration by interferon therapy. Antivir. Ther. 2013, 18, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.W.; Huang, M.P.; Zhong, B. Intrahepatic Toll-Like Receptor 3 in Chronic HBV Infection Subjects: Asymptomatic Carriers, Active Chronic Hepatitis, Cirrhosis, and Hepatocellular Carcinoma. Zahedan J. Res. Med. Sci. 2016, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Meng, Z.; Jiang, M.; Pei, R.; Trippler, M.; Broering, R.; Bucchi, A.; Sowa, J.-P.; Dittmer, U.; Yang, D.; et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2008, 49, 1132–1140. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, Y.; Shi, B.; Zhang, X.; Wang, J.; Zhang, Z.; Shen, F.; Zhang, Q.; Sun, S.; Yuan, Z. HBsAg inhibits TLR9-mediated activation and IFN-α production in plasmacytoid dendritic cells. Mol. Immunol. 2009, 46, 2640–2646. [Google Scholar] [CrossRef]
- Chen, Z.; Cheng, Y.; Xu, Y.; Liao, J.; Zhang, X.; Hu, Y.; Zhang, Q.; Wang, J.; Zhang, Z.; Shen, F.; et al. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin. Immunol. 2008, 128, 400–408. [Google Scholar] [CrossRef]
- Wang, H.; Ryu, W.-S. Hepatitis B Virus Polymerase Blocks Pattern Recognition Receptor Signaling via Interaction with DDX3: Implications for Immune Evasion. PLoS Pathog. 2010, 6, e1000986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B Virus Polymerase Disrupts K63-Linked Ubiquitination of STING to Block Innate Cytosolic DNA-Sensing Pathways. J. Virol. 2014, 89, 2287–2300. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wu, M.; Zhang, X.; Zhang, W.; Zhang, Z.; Chen, L.; He, J.; Zheng, Y.; Chen, C.; Wang, F.; et al. Hepatitis B virus polymerase impairs interferon-α-induced STA T activation through inhibition of importin-α5 and protein kinase C-δ. Hepatology 2013, 57, 470–482. [Google Scholar] [CrossRef]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Genome Size (bp) | Subgenotypes | Distinguishing Features |
|---|---|---|---|
| A | 3 221 | A1, A2, A4, and quasi-subgenotype A3 | 6 bp insertion at the 3′-end of core gene (Insertion of aa 153 and 154 in HBcAg) Unusual G1896A mutation Common BCP mutations |
| B | 3 215 | B1, B2, B4–B6, and quasi-subgenotype B3 | B1 and B5 are pure HBV/B strains B2, B3, and B4 are recombinants with genotype C in the core region |
| C | 3 215 | C1-C16 | Common BCP mutations |
| D | 3 182 | D1-D6 | 33 bp deletion at the 5′-end of pre-S1 region (Deletion of aa 1-11 in pre-S1) |
| E | 3 212 | - | 3 bp deletion at 5′-end of the pre-S1 region (Deletion of aa 11 in pre-S1) |
| F | 3 215 | F1-F4 | Unusual G1896A mutation |
| G | 3 248 | - | 36 bp insertion at position 190 in the core ORF (Insertion of 12 aa in HBcAg) 3 bp deletion at the 5′-end in the pre-S1 region (Deletion of aa 11 in pre-S1) Stop codons at positions 2 and 28 (G1896A) of the pre-core ORF render it unable to express HBeAg Usually found in coinfection with other genotypes that express HBeAg |
| H | 3 215 | - | Unusual G1896A mutation |
| I | 3 210 | I1 and I2 | Evolved as a recombinant of genotypes A, C, and G |
| J | 3 182 | Single HBV isolate identified in an elderly Japanese patient with HCC Highly divergent from others human HBV strains Likely a genotype C–gibbon Orthohepadnavirus recombinant strains |
| Phase | I | II | III | IV | V |
|---|---|---|---|---|---|
| HBeAg (+) Chronic HBV Infection | HBeAg (+) Chronic Hepatitis B | HBV Chronic Infection HBeAg (−) | Chronic Hepatitis B HBeAg (−) | HBsAg (−) | |
| HBeAg (+) Non-Inflammatory | HBeAg (+) Immune Active Phase | Inactive Carrier | HBeAg (−) Immune Active Phase | Occult Infection | |
| Previous term | Immune tolerant | Immune reactive HBeAg (+) | Inactive carrier | HBeAg (−) chronic hepatitis | Occult infection |
| HBeAg | +++ | ++ | − | − | − |
| Anti-HBe | - | + | + | + | + or − |
| HBsAg | ++++ | +++ | ++ or + Few patients develop spontaneous clearance (~1% annual) | ++ | − |
| Anti-HBs | - | +++ | ++ | + or − | + or − |
| HBV-DNA (IU/mL) | 107 | 104–107 | <2 × 103 ** or undetectable | >2 × 10 3 | Very low levels in liver and/or serum |
| ALT (IU/mL) | Normal | Elevated | Normal (~40) | Persistently or intermittently elevated | Normal |
| Hepaticdisease | None/minimal necroinflammation or fibrosis | Moderate/severe liver necroinflammation and accelerated progression of fibrosis | None/minimal necroinflammation and low fibrosis | Moderate/severe necroinflammation and fibrosis | Immunosuppression could lead to viral reactivation. Low risk of cirrhosis or HCC |
| NK cells innate response | Impaired production of IFN-γ and TNF-α | Impaired production of IFN-γ and TNF-α Phenotypically activated (↑ NKp44 in CD56bright and CD69 in CD56dim NK cells) | Restored production of IFN-γ | Phenotypically activated (↑ NKp44 in CD56bright and CD69 in CD56dim NK cells) | |
| Normal NK cells cytolytic activity without differences in CD107a, perforin, or granzyme B expression. | |||||
| Global and HBV specific T cell responses | ↓ IFN-γ # and TNF-α | ↓ IFN-γ # and TNF-α | ↑ HBsAg and HBcAg response producing IFN-γ | ↑↑ IFN-γ # and TNF-α | |
| Progress risk | 0.37% to 3.3% annual progression risk | ||||
| Others | Very low rate of spontaneous HBeAg loss Highly contagious. More frequent and prolonged in carriers infected by perinatal transmission Low efficacy of currently available treatments | Variable outcome, but most of them progress to stage III | Low risk of developing HCC or cirrhosis. Loss of HBsAg and/or spontaneous seroconversion occurs in 1–3% of cases | A low proportion of spontaneous remission of the disease | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Valdez, M.; Monroy-Ramírez, H.C.; Armendáriz-Borunda, J.; Sánchez-Orozco, L.V. Molecular Mechanisms during Hepatitis B Infection and the Effects of the Virus Variability. Viruses 2021, 13, 1167. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061167
Campos-Valdez M, Monroy-Ramírez HC, Armendáriz-Borunda J, Sánchez-Orozco LV. Molecular Mechanisms during Hepatitis B Infection and the Effects of the Virus Variability. Viruses. 2021; 13(6):1167. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061167
Chicago/Turabian StyleCampos-Valdez, Marina, Hugo C. Monroy-Ramírez, Juan Armendáriz-Borunda, and Laura V. Sánchez-Orozco. 2021. "Molecular Mechanisms during Hepatitis B Infection and the Effects of the Virus Variability" Viruses 13, no. 6: 1167. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061167