
Comparative Computational Modeling of the Bat and Human Immune Response to Viral Infection with the Comparative Biology Immune Agent Based Model

Abstract
:1. Introduction
- It can be difficult to intuit the overall effects of a complex set of interactions, and therefore a dynamic representation of this knowledge can uncover unanticipated or paradoxical dynamic effects, particularly when positive and negative feedback are present [9].
- While these models may be complex, they are necessarily selective abstractions of the real world (as are the diagrams they are based on). Since the model will only exhibit behaviors possible from the interactions they embody, they can be used to determine the sufficiency of a particular hypothesis/theory in terms of explaining observations from the real world. While they cannot falsify a particular hypothesis, they can suggest where additional features of the biology need to be added to achieve a particular desired behavior.
- Toward this end, computational models of this type can be used for examining new hypotheses, be they components or interactions within the system itself or putative interventions [9]. Examining the plausibility of such modifications can suggest new experiments or observations that might need to be performed.
- These types of computational models can be used as experimental objects, where simulation experiments can be performed that evaluate the new hypotheses noted in #3. Since simulation experiments are not subject to the cost and logistical constraints that apply to wet-lab experiments and can often be run at a considerably greater scale (in terms of number of individuals examined), they can be a useful adjunct to pre-testing and design of subsequent real-world experiments [9].
- As computational embodiments of knowledge/hypothesis structures, these models can serve as “bridging” knowledge structures that can represent what is conserved from one individual or zoological context to another. Rather than relying on a list of components and features, a computational model for dynamic knowledge representation can encapsulate what is functionally “similar” from one species/organism/individual and note what the effect might be for what is explicitly “different.” [11].
2. Materials and Methods
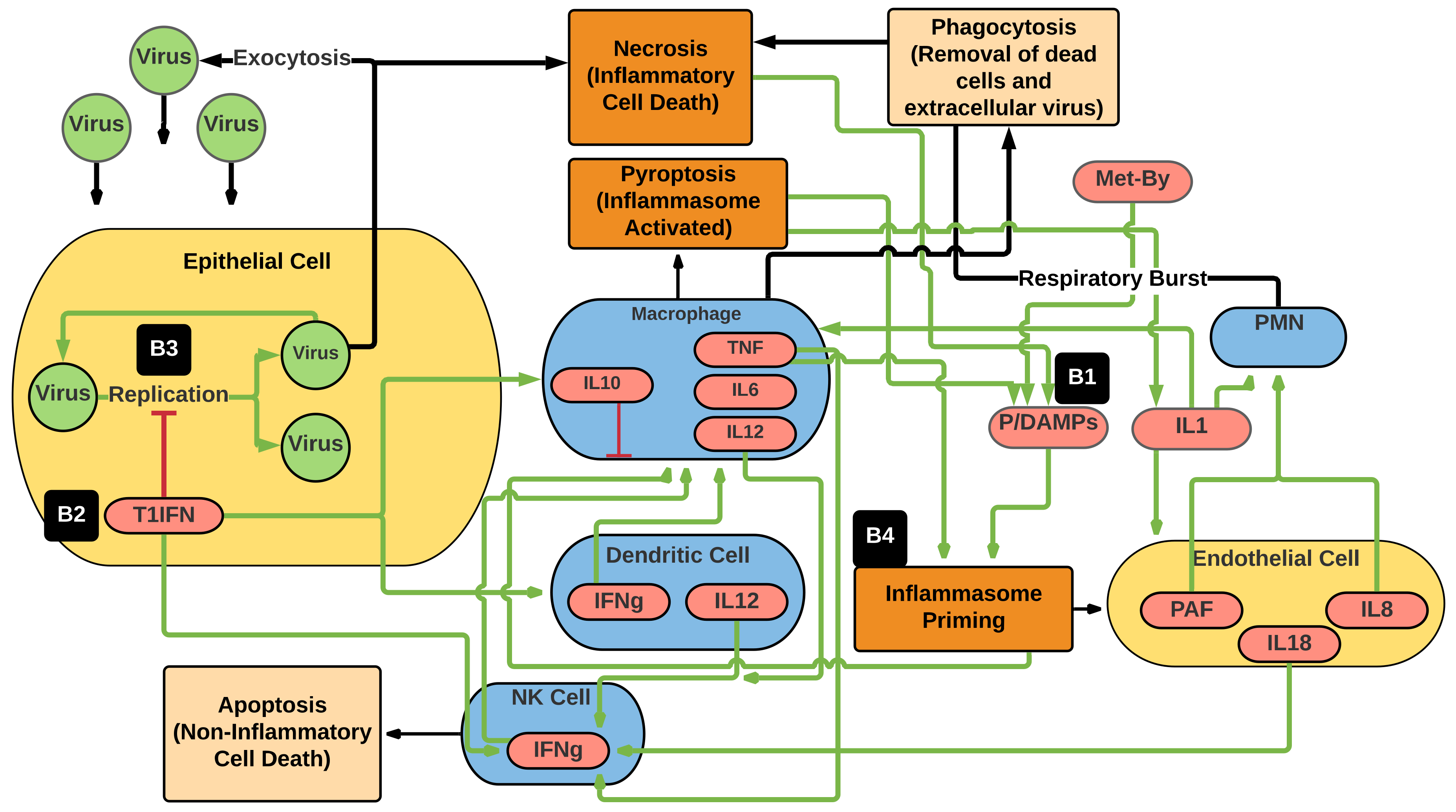
2.1. Overview and Abstraction Level
2.2. Model Implementation
- “Diffuse”: This primitive simulates diffusion of a patch variable to its surrounding 8 patches. The argument for the primitive is the percentage of the value of the center patch that is removed and then evenly distributed among the surrounding 8 patches.
- “Uphill”: This primitive is essentially a chemotactic function, where an agent surveys the surrounding 8 patches and moves toward the patch with the highest target variable. Note that within the CBIABM, this primitive is decomposed into several lines of code to allow chemotaxis to be driven by combinations of mediators. In the CBIABM, all mobile cells share the same movement rate.
- The simulation is initialized;
- A specified amount of extracellular virus is placed in a random pattern onto the epithelial cell grid;
- The rules for the cells are executed. The implementation of NetLogo shuffles the order of execution within each class of agent but runs each class of agents in a specified sequence. Once the specified set of instructions for each step are completed, the process starts again until the stopping conditions are met;
- The total time course simulated in the experiments below is 14 days (though this can be modified).
- While the initial recruitment of immune response cells may enhance the initial containment of the viral inoculum, these cells accomplish that effect by the generation of proinflammatory mediators, which are the exact processes invoked in disease manifestation that bats are thought to avoid. Therefore, while the addition of this feature might alter the specific potential transition zone in the difference between bat and human disease manifestation is should not qualitatively alter the existence of such a zone, which is what these simulation experiments are intended to show.
- Similarly, while depletion and exhaustion of inflammatory cells are known to occur in severe human disease, these processes only occur after the inflammatory response to the viral inoculum has substantially progressed. Since the hypothesis underlying bat resistance to viral infection emphasizes early attenuation of the inflammatory response, this phenomenon does not affect those early dynamics and the sought-after qualitative difference between bat and human disease manifestation
- Alternatively, polymorphonuclear neutrophil (PMN) populations do dynamically change in the CBIABM. This is because there are no baseline PMNs in non-inflamed tissue, and therefore their recruitment must be explicitly represented (in contrast, macrophages, NK cells, and dendritic cells, though augmented by circulating precursors, are already present in tissue at baseline).
- 1.
- Epithelial cells. These are generic epithelial cells that represent the cells initially exposed and susceptible to viral infection. The epithelial cells also abstractly represent generic “tissue”, as the current CBIABM does not explicitly represent muscle or specific organs. One epithelial cell agent occupies a single grid space for a total of 2601 possible healthy epithelial cells in the system, and they do not move. The total health of the system is reflected by the percentage of healthy epithelial cells out of 2601, the variable called “%System-Health”. The epithelial cell agents have the following variables and functions:
- a.
- Susceptibility to infection: this variable represents how readily the epithelial cell can be infected by extracellular virus and abstracts its expression of potential receptors that can be targeted by various viruses. This is a constant in the current version of the CBIABM and is used to probabilistically (in relation to the number of extracellular viral particles on that specific gride space) determine whether an individual epithelial cell agent becomes infected.
- b.
- Susceptibility to reactive oxygen species/cytotoxic compounds: this variable represents how much damage the epithelial cell can sustain before it undergoes necrosis, a proinflammatory form of death that results in the production of additional danger signals (P/DAMPs).
- c.
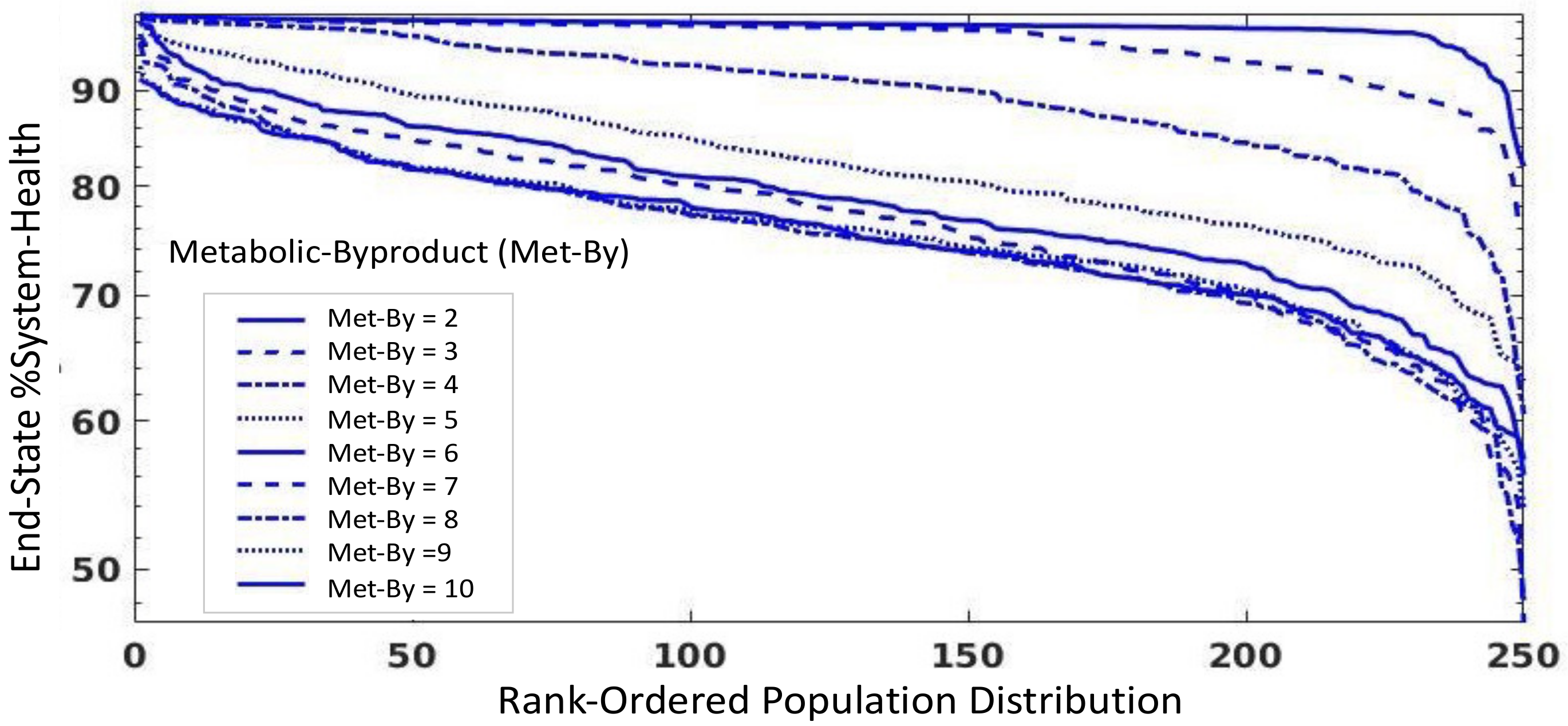
- Metabolic Byproduct (Met-By): this variable represents the amount of oxidative byproducts produced from baseline metabolism and are sensed as P/DAMPs. This value is 10-fold higher in bats versus humans, representing the increased metabolism in bats needed for flight [3]. This value is added to the P/DAMPSs present on a particular patch.
- d.
- Total cell membrane: this variable represents how much cell membrane the cell has that can be consumed by viral exocytosis before the cell dies by necrosis (proinflammatory death) and release of danger signals (P/DAMPS).
- e.
- Apoptosis: This function is initiated by sensing of viral infection and represents programmed cell death to shorten the time (and therefore total amount) of viral production by an infected epithelial cell. This function is also accelerated by interactions with NK cells as a representation of NK cells’ antiviral effect. As a result of apoptosis, almost no epithelial cells progress to membrane consumption death (1c above), though may be altered in future versions where for simulations of viruses known to interrupt apoptosis. Notably, cells that die by apoptosis do not propagate inflammation; this is distinct from cells that die via necrosis (1d above), which release P/DAMPs until they are cleared by phagocytosis by macrophages.
- f.
- Production of type 1 interferons (T1IFN): primary inflammatory and antiviral mediator produced by infected epithelial cells in humans and at baseline in bats.
- g.
- Regeneration: This is an abstracted healing function that allows the regrowth of new epithelial cells into empty patches from where dead epithelial cells (either apoptotic or necrosed) have been cleared by phagocytosis. This process is simulated to take 3 days.
- 2.
- Natural killer cells (NK cells): These are mobile immune cells that are a major component of innate antiviral activity. They migrate toward infected epithelial cells and accelerate their apoptosis. Their functions and properties include:
- a.
- Chemotaxis to T1IFN. This means they move up a gradient of T1IFN.
- b.
- Production of interferon-gamma (IFNg) in the presence of T1IFNs, interleukin-12 (IL-12) and interleukin-18 (IL-18).
- c.
- Accelerate apoptosis in infected epithelial cells via abstraction of perforin and granzyme function.
- d.
- NK cell populations are held constant for the duration of the simulation (see discussion/rationale in the preceding text).
- 3.
- Macrophages: These are mobile immune cells that respond to signals produced by infected epithelial cells and immune cells (NK cells, dendritic cells, polymorphonuclear neutrophils, and other macrophages). These cells are central regulators of the innate immune response; they also phagocytose extracellular virus and cellular debris from dead epithelial cells, clearing space for new epithelial cells to regenerate into. Their functions and variables include:
- a.
- Chemotaxis to T1IFNs and P/DAMPS;
- b.
- Have a differential activation level representing the ability to perform proinflammatory functions (M1 phenotype) or anti-inflammatory functions (M2 phenotype). The activation level is determined by a balance between proinflammatory signals: T1IFN, P/DAMPs, IFNg, and IL-1 versus anti-inflammatory signals: Interleukin-10 (IL-10);
- i.
- M1 Macrophages produce interleukin-8 (IL-8) and Il-12, and if their inflammasome is activated, tumor necrosis factor (TNF), interleukin-6 (IL-6), IL-10 and IL-1 and IL-18;
- ii.
- M2 macrophages produce IL-10.
- c.
- Have an abstracted inflammasome that becomes activated through two steps:
- i.
- Priming: Occurs when exposed to P/DAMPs or TNF;
- ii.
- Activation: If already primed, if sufficient extracellular virus is phagocytosed, this triggers the production of precursors for IL-1 and IL-18, allows M1 production of TNF, IL-6, and IL-10, and initiates the pyroptosis pathway.Both priming and activation have defined thresholds; these are set at different levels between humans (lower) and bats (higher).
- d.
- Can undergo pyroptosis: as discussed above, pyroptosis is an inflammogenic form of cell death. The precursors to IL-1 and IL-18 are released as active cytokines upon pyroptosis, as well as the production of P/DAMPs representing the release of extracellular DNA at cell death. Pyroptosis occurs ~2 h after inflammasome activation [8,23,24,25]. Note that we have elected to keep macrophage populations steady for the duration of the simulation; therefore, when a macrophage undergoes pyroptosis, it is immediately replaced by the creation of a naïve macrophage placed randomly in the world grid. This is a modeling decision that abstracts but qualitatively reproduces steady-state depletion/recruitment dynamics.
- e.
- Perform phagocytosis: this is the endocytosis of extracellular viruses and cellular debris, clearing away damaged cells to allow for epithelial regrowth. However, it is recognized that there is a limit to the amount of material a macrophage can phagocytose; therefore, there is a variable that determines the upper limit of this amount: when this limit is reached, the macrophage is considered “exhausted” and is unable to clear any more material [26].
- 4.
- Dendritic Cells: These are antigen-presenting cells that have similar functions to macrophages but are a key component in the transition from innate to adaptive immunity: they are primarily responsible for presenting antigen to naïve T-cell subtypes and inducing antigen-specific differentiation of various T-cell subtypes. However, in the current version of the CBIABM, their role in inducing adaptive immunity is not represented, and rather they serve primarily to chemotax in response to T1IFN and produce IL-6, IL-12, and IFNg. As with macrophages and NK cells, dendritic cell populations are held stable for the duration of the simulation.
- 5.
- Endothelium: The majority of the initial viral response is considered to take place in the epithelial tissue at risk, though the vascular supply to that tissue means that there is close proximity with the endothelial cells lining those vessels. The activation of the endothelium is a key step in the transition from what would otherwise be an inflammatory process restricted in local tissue to an expansion toward systemic effects. As such, endothelial activation is a critical tipping point that affects the manifestation of the disease [27]. The inflammatory role of the endothelium is represented in the CBIABM by projecting a layer of endothelial cells “on top of” each epithelial cell and having those cells being able to be activated by signals generated on the patch on which it sits. Endothelial cells are activated by a combination of inflammatory signals: IL-1 and TNF. This activation involves the function of the endothelial inflammasome, which in humans activates a series of signaling and adhesion functions that eventually lead to the recruitment of polymorphonuclear neutrophils (PMNs) from the bloodstream to the area of inflammation. We hypothesize that, similar to their immune cell inflammasomes, bats have a reduced degree of responsiveness in their endothelial inflammasomes; this decreased function is reflected in the different values assigned to the endothelial activation threshold between human and bat simulations. In addition to the simulated induction of adhesion molecules, activated endothelium produces platelet-activating factor (PAF), which is a chemotaxis signal for polymorphonuclear neutrophils (PMNs) [28].
- 6.
- Polymorphonuclear neutrophils (PMNs): these are the primary circulating immune/white blood cells. They serve a central role in response to bacterial infections, but in general, their role in containing viral infections is less pronounced. However, they can be recruited to sites of viral infection if the endothelium in the region of the infection becomes activated and initiates the sequence of PMN adhesion and migration through the blood vessel walls into the tissue. The process of PMN adhesion and migration is abstracted in the CBIABM and is modeled to take about 6–12 h from initial endothelial activation through the production of endothelial adhesion molecules, the adhesion of PMNs to those molecules through to the migration of PMNs into the tissue. Once in the tissue, they will chemotax toward IL-8 and PAF and subsequently undergo a respiratory burst that produces proteolytic and cytotoxic compounds that induce epithelial cell necrosis (a proinflammatory cell death) [29]. Respiratory burst is associated with the death of the PMN. As noted above, PMN populations are dynamically shifting via recruitment via activated endothelium and therefore depleted by the respiratory burst process.
- They are consumed by binding to receptors on the cellular agents.
- There is a percentage degradation of the value of each mediator per step.
- There is an arbitrary lower threshold of the value of each mediator, at which point the value is set to 0.
2.3. Simulation Experiments
- Bats have higher baseline production of P/DAMPS to simulate the enhanced metabolic rate needed for flight (reflected by the difference in “metabolic byproduct” value)
- Bats have baseline production of type 1 interferons
- Bats have enhanced intracellular antiviral effect of type 1 interferons
- Bats have reduced inflammasome priming and activation (reflected by higher values for the variables “inflammasome-priming-threshold”, “inflammasome-activation-threshold”, and “bat endothelial activation” versus “human-endothelial activation”).
- The world size was chosen to consist of a 51 × 51 grid. This was arbitrary and made for computational efficiency based on our experience constructing this type of model.
- The time frame was selected such that one step of the CBIABM represents 10 min; this provided a frame of reference for how the various cellular rules manifested their dynamics.
- The initial populations of macrophages, NK cells, and dendritic cells were set at 50, 25, and 50, respectively. Again, arbitrary and chosen based on our experience constructing this type of model.
3. Results
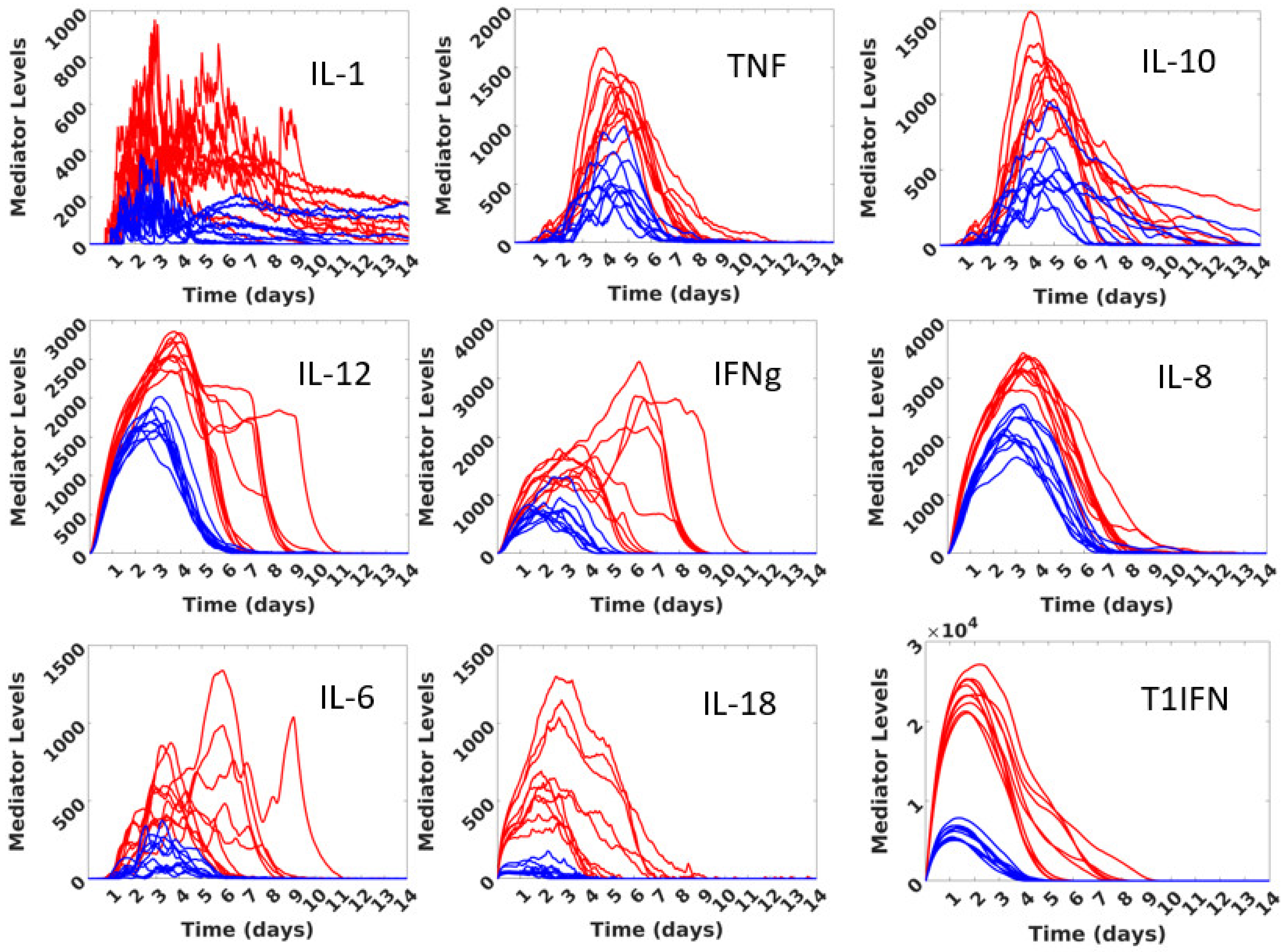
3.1. Initial Calibration
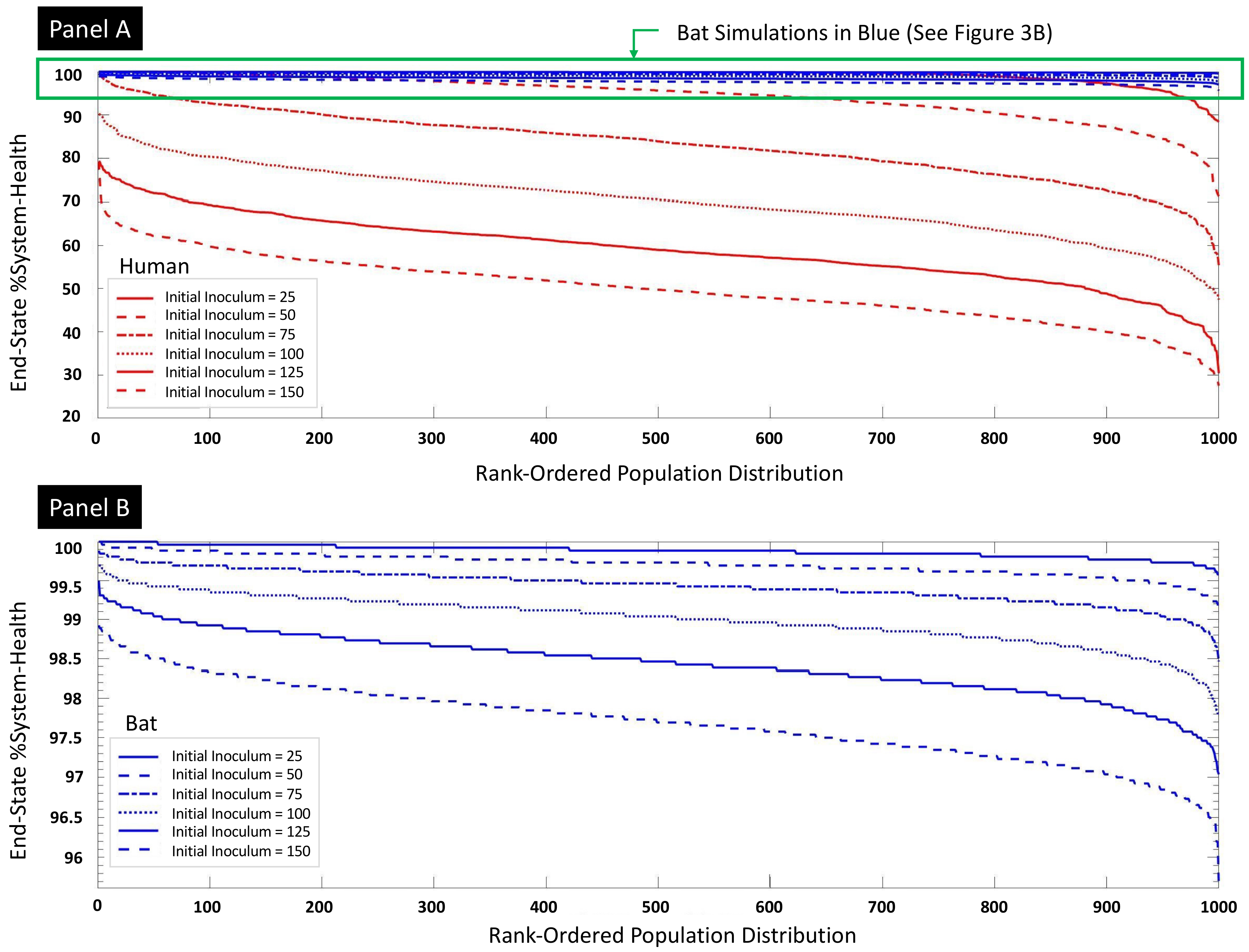
3.2. Parameter Sweep of Initial Viral Inoculum
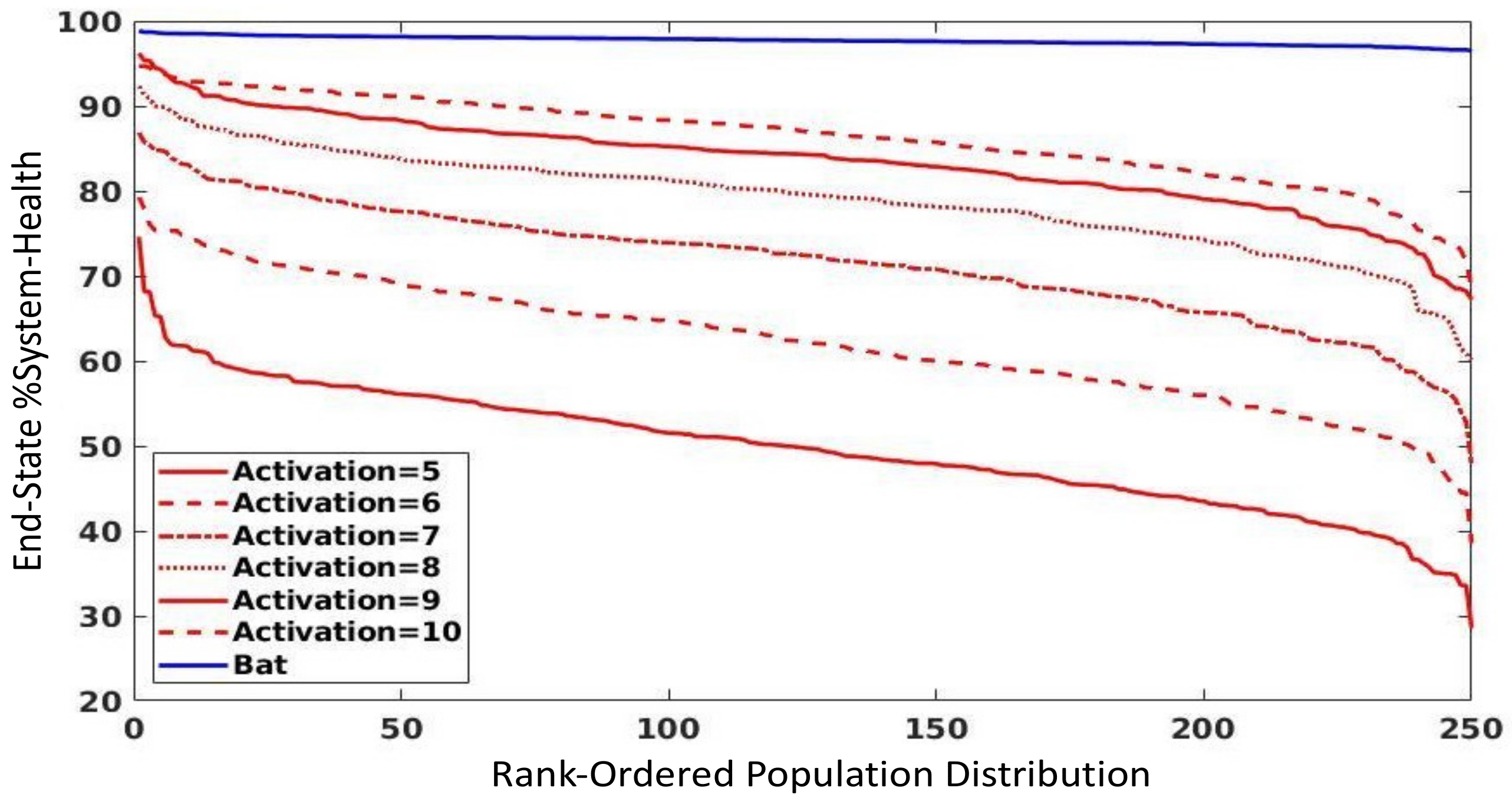
3.3. Parameter Sweep of Endothelial Activation Threshold
3.4. Investigation into Effect of Metabolic Stress on Potential Viral Spillover
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mediator | Modeled Functions and References |
|---|---|
| Pathogen/Damage-Associated Molecular Patterns (P/DAMPS) | Produced when cells die by some other means than apoptosis (pyroptosis or necrosis). Persists in the presence of non-apoptosis dead cells until phagocytosed by macrophages. Functions a chemoattractant for macrophages, shifts macrophages to proinflammatory phenotype, primes inflammasomes [24,25]. Added to by “metabolic byproduct” (“Met-By”) as simulated extracellular DNA from cellular damage from metabolic stress. Diffuse at 0.1, Evaporate at 0.90. |
| Reactive Oxygen Species (ROS) | Produced during PMN respiratory burst, damages epithelial cells [29]. Diffuse at 0.1, Evaporate at 0.90. |
| Platelet-Activating Factor (PAF) | Made by activated endothelial cells, functions as chemotaxis and adhesion activation/recruitment signal for PMNs [28,29]. Diffuse at 0.1, Evaporate at 0.90. |
| Tumor Necrosis Factor (TNF) | Produced by proinflammatory macrophages, functions to activate the inflammasome in macrophages, activates endothelium, consumed by epithelial cells [6,7,8,29]. Diffuse at 0.2, Evaporate at 0.99. |
| Interleukin-1 (IL-1) | Produced by inflammasome priming and activation in macrophages, primarily released at pyroptosis, produced by PMNs, functions to activate endothelium and shift macrophages to proinflammatory phenotype, along with TNF stimulates infected epithelial cells to produce IL-6 [3,4,6,7,44]. Consumed by macrophages. Diffuses at 0.2, Evaporates at 0.99. |
| Interleukin-18 (IL-18) | Produced by infected epithelial cells. Produced by inflammasome priming and activation in macrophages and primarily released at pyroptosis, facilitate NK cells to produce IFNg (in conjunction with T1IFN and IL-12), consumed by NK cells [3,4,6,7,8,50]. Diffuses at 0.2, Evaporates at 0.99. |
| Interleukin-6 (IL-6) | Produced by proinflammatory macrophages, dendritic cells, and infected epithelial cells [51,52], diffuses at 0.2, evaporates at 0.99. |
| Interleukin-8 (IL-8) | Produced by proinflammatory macrophages, functions as chemotactic compound for PMNs [29]. Diffuses at 0.3, evaporates at 0.99. |
| Interleukin-10 (IL-10) | Produced by both pro- and anti-inflammatory macrophages, function is to shift balance of macrophages from proinflammatory to anti-inflammatory phenotypes [53,54]. Consumed by macrophages. Diffuses at 0.2, evaporates at 0.99. |
| Interleukin-12 (IL-12) | Produced by dendritic cells and proinflammatory macrophages, function to facilitate NK cell production of IFNg (in conjunction with IL-18 and T1IFN) [55,56]. Consumed by NK cells. Diffuses at 0.2, Evaporates at 0.99. |
| Type 1 Interferons (T1IFN) | Produced by infected epithelial cells and NK cells, functions as chemotactic compound for NK cells, dendritic cells, and macrophages, facilitates production of IFNg by NK cells (in conjunction with IL-12 and IL-18), induces production of IL-12, IFNg, and (in conjunction with IL-1) IL6, has antiviral effect by reducing viral replication in infected epithelial cells, constitutively produced in bats, only induced in humans, enhances apoptosis in response to viral infection [3,4,57,58,59]. Consumed by macrophages and dendritic cells. Diffuses at 0.1, Evaporates at 0.99. |
| Interferon-gamma (IFNg) | Produced by NK cells, proinflammatory macrophages, dendritic cells, functions to shift macrophages toward proinflammatory phenotype [58,60,61]. Consumed by macrophages. Diffuses at 0.2, evaporates at 0.99. |
- They are produced by cellular agents based on those agents’ rules.
- They all diffuse via the NetLogo primitive “diffuse.” This command takes a percentage of the value of the variable on each patch and divides it among the surrounding 8 patches. For instance, “Diffuse mediator 0.1” takes 10% of the value of that mediator and divides it among the surrounding 8 patches.
- Mediators are qualitatively divided into those with more local effects (diffuse 0.1) and those with slightly more diffuse effects (diffuse 0.2). The exception is IL-8, which has a diffuse of 0.3; this was done because of how PMNs arrive on the screen in order to make sure they were able to have a gradient to follow.
- All mediators also undergo an “evaporation” function, where the mediator is decreased by a percentage amount; for example, set mediator × 0.9 decreases the mediator 10% each step. This is to simulate non-represented degradation of the mediator.
- Mediators are qualitatively divided into those with slightly longer lifespans (× 0.99) and those with shorter (× 0.90).
- Some mediators are actively consumed by binding to cellular receptors.
- All the mediators have a lower limit of detection below which the value of that mediator on a patch is set to 0. This is done because the Evaporation function is a percent decrement, and therefore, these values will never reach 0. This is done for two reasons: 1. Because the edges of the model wrap, there are artifacts in mediator levels when non-zero mediators persistently diffuse across the edges, and 2. For computational efficiency, so that all the patches do not need to compute minimal values of mediators. This value is the same for all the mediators and is set at <0.1.
Appendix B
| Mediator vs. Cell Types | T1IFN | TNF | IL-1 | IL-6 | IL-8 | IL-10 | IL-12 | IL-18 | IFNg | ROS | PAF | P/DAMPS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Epithelial Cells | + | + | + | + | ||||||||
| Macrophages | + | + | + | + | + | + | ||||||
| NK Cells | + | |||||||||||
| Dendritic Cells | + | + | + | |||||||||
| PMNs | + | + | ||||||||||
| Endothelial Cells | + |
Appendix C

References
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.-P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nat. Cell Biol. 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Han, H.-J.; Wen, H.-L.; Zhou, C.-M.; Chen, F.-F.; Luo, L.-M.; Liu, J.-W.; Yu, X.-J. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 2015, 205, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.T.; Ahn, M.; Goh, G.; Anderson, D.E.; Wang, L.-F. Lessons from the host defences of bats, a unique viral reservoir. Nat. Cell Biol. 2021, 589, 363–370. [Google Scholar] [CrossRef]
- Banerjee, A.; Baker, M.L.; Kulcsar, K.; Misra, V.; Plowright, R.; Mossman, K. Novel insights into immune systems of bats. Front. Immunol. 2020, 11, 26. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Misra, V. Immune system modulation and viral persistence in bats: Understanding viral spillover. Viruses 2019, 11, 192. [Google Scholar] [CrossRef] [Green Version]
- Vaccari, J.C.D.R.; Dietrich, W.D.; Keane, R.W. The Inflammasome in Times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 1–22. [Google Scholar] [CrossRef] [PubMed]
- An, G. Introduction of an agent-based multi-scale modular architecture for dynamic knowledge representation of acute inflammation. Theor. Biol. Med. Model. 2008, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- An, G. Dynamic Knowledge representation using agent-based modeling: Ontology instantiation and verification of conceptual models. In Systems Biology; Humana Press: Totowa, NJ, USA, 2009; Volume 500, pp. 445–468. [Google Scholar] [CrossRef]
- An, G. The crisis of reproducibility, the denominator problem and the scientific role of multi-scale modeling. Bull. Math. Biol. 2018, 80, 3071–3080. [Google Scholar] [CrossRef] [Green Version]
- Box, G. Science and statistics. J. Am. Stat. Assoc. 1976, 71, 791–799. [Google Scholar] [CrossRef]
- Bonabeau, E. Agent-based modeling: Methods and techniques for simulating human systems. Proc. Natl. Acad. Sci. USA 2002, 99, 7280–7287. [Google Scholar] [CrossRef] [Green Version]
- An, G. In silico experiments of existing and hypothetical cytokine-directed clinical trials using agent-based modeling. Crit. Care Med. 2004, 32, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- An, G. Agent-based computer simulation and sirs: Building a bridge between basic science and clinical trials. Shock 2001, 16, 266–273. [Google Scholar] [CrossRef]
- Cockrell, C.; An, G. Sepsis reconsidered: Identifying novel metrics for behavioral landscape characterization with a high-performance computing implementation of an agent-based model. J. Theor. Biol. 2017, 430, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia-Juarez, J.; Ganguli, S.; Kirschner, D. Identifying control mechanisms of granuloma formation during M. tuberculosis infection using an agent-based model. J. Theor. Biol. 2004, 231, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, R.; Holcombe, M. The epitheliome project: Multiscale agent-based modeling of epithelial cells. In Proceedings of the 3rd IEEE International Symposium on Biomedical Imaging: Nano to Macro, Arlington, VA, USA, 6–9 April 2006; IEEE: Manhattan, NY, USA, 2006; pp. 816–819. [Google Scholar] [CrossRef]
- Lee, J.-J.; Talman, L.; Peirce, S.; Holmes, J.W. Spatial scaling in multiscale models: Methods for coupling agent-based and finite-element models of wound healing. Biomech. Model. Mechanobiol. 2019, 18, 1297–1309. [Google Scholar] [CrossRef]
- Walpole, J.; Mac Gabhann, F.; Peirce, S.M.; Chappell, J.C. Agent-based computational model of retinal angiogenesis simulates microvascular network morphology as a function of pericyte coverage. Microcirculation 2017, 24, e12393. [Google Scholar] [CrossRef]
- Metzcar, J.; Wang, Y.; Heiland, R.; Macklin, P. A review of cell-based computational modeling in cancer biology. JCO Clin. Cancer Inform. 2019, 3, 1–13. [Google Scholar] [CrossRef]
- Tisue, S.; Wilensky, U. Netlogo: A simple environment for modeling complexity. In Proceedings of the International Conference on Complex Systems, Boston, MA, USA, 16–23 May 2004; Volume 21, pp. 16–21. [Google Scholar]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.S.; Kanneganti, T.-D. Caspases in cell death, inflammation, and pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [Green Version]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Zent, C.S.; Elliott, M.R. Maxed out macs: Physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 2016, 284, 1021–1039. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, N.M.; Steinberg, B.E.; Slutsky, A.S.; Lee, W.L. Broken barriers: A new take on sepsis pathogenesis. Sci. Transl. Med. 2011, 3, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, S.M.; Zimmerman, G.A.; Stafforini, D.M.; McIntyre, T.M. Platelet-activating factor and related lipid mediators. Annu. Rev. Biochem. 2000, 69, 419–445. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on neutrophil function in severe inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef] [Green Version]
- Cockrell, C.; An, G. Utilizing the heterogeneity of clinical data for model refinement and rule discovery through the application of genetic algorithms to calibrate a high-dimensional agent-based model of systemic inflammation. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Varga, S.M. The CD8 T cell response to respiratory virus infections. Front. Immunol. 2018, 9, 678. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2015, 13, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef] [PubMed]
- Falasca, L.; Agrati, C.; Petrosillo, N.; Di Caro, A.; Capobianchi, M.R.; Ippolito, G.; Piacentini, M. Molecular mechanisms of Ebola virus pathogenesis: Focus on cell death. Cell Death Differ. 2015, 22, 1250–1259. [Google Scholar] [CrossRef] [Green Version]
- Srikiatkhachorn, A.; Mathew, A.; Rothman, A.L. Immune-mediated cytokine storm and its role in severe dengue. Semin. Immunopathol. 2017, 39, 563–574. [Google Scholar] [CrossRef]
- Abegglen, L.M.; Caulin, A.F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M.S.; Kiso, W.K.; Schmitt, D.L.; Waddell, P.J.; Bhaskara, S.; et al. Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA 2015, 314, 1850–1860. [Google Scholar] [CrossRef]
- Sulak, M.; Fong, L.; Mika, K.; Chigurupati, S.; Yon, L.; Mongan, N.; Emes, R.; Lynch, V.J. Correction: TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. eLife 2016, 5, e11994. [Google Scholar] [CrossRef]
- Gentles, A.D.; Guth, S.; Rozins, C.; Brook, C.E. A review of mechanistic models of viral dynamics in bat reservoirs for zoonotic disease. Pathog. Glob. Health 2020, 114, 407–425. [Google Scholar] [CrossRef]
- Dimitrov, D.T.; Hallam, T.G.; Rupprecht, C.E.; Turmelle, A.S.; McCracken, G.F. Integrative models of bat rabies immunology, epizootiology and disease demography. J. Theor. Biol. 2007, 245, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.T.; Hallam, T.G.; Rupprecht, C.E.; McCracken, G.F. Adaptive modeling of viral diseases in bats with a focus on rabies. J. Theor. Biol. 2008, 255, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Brook, C.E.; Boots, M.; Chandran, K.; Dobson, A.P.; Drosten, C.; Graham, A.L.; Grenfell, B.T.; Müller, M.A.; Ng, M.; Wang, L.F.; et al. Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. eLife 2020, 9, e48401. [Google Scholar] [CrossRef]
- Dimitrov, D.T.; King, A.A. Modeling evolution and persistence of neurological viral diseases in wild populations. Math. Biosci. Eng. 2008, 5, 729–741. [Google Scholar] [CrossRef]
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iba, T.; Levy, J.H.; Levi, M.; Connors, J.M.; Thachil, J. Coagulopathy of coronavirus disease 2019. Crit. Care Med. 2020, 48, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Cockrell, R.C.; An, G. Examining the controllability of sepsis using genetic algorithms on an agent-based model of systemic inflammation. PLoS Comput. Biol. 2018, 14, e1005876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, B.K.; Yang, J.; Grathwohl, W.S.; Cockrell, C.; Santiago, C.; An, G.; Faissol, D.M. Deep reinforcement learning and simulation as a path toward precision medicine. J. Comput. Biol. 2019, 26, 597–604. [Google Scholar] [CrossRef]
- An, G.; Cockrell, C.; Day, J. Therapeutics as Control: Model-Based Control Discovery for Sepsis, in Complex Systems and Computational Biology Approaches to Acute Inflammation; Springer: New Yok, NY, USA, 2021; pp. 71–96. [Google Scholar]
- Goh, G.; Ahn, M.; Zhu, F.; Lee, L.B.; Luo, D.; Irving, A.T.; Wang, L.-F. Complementary regulation of caspase-1 and IL-1β reveals additional mechanisms of dampened inflammation in bats. Proc. Natl. Acad. Sci. USA 2020, 117, 28939–28949. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kishimoto, T. Targeting interleukin-6: All the way to treat autoimmune and inflammatory diseases. Int. J. Biol. Sci. 2012, 8, 1227–1236. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The role of interleukin 6 during viral infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef] [Green Version]
- Couper, K.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Brandstadter, J.; Yang, Y. Natural killer cell responses to viral infection. J. Innate Immun. 2011, 3, 274–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.B.; Salazar-Mather, T.P.; Dalod, M.; van Deusen, J.B.; Wei, X.-Q.; Liew, F.Y.; Caligiuri, M.A.; Durbin, J.E.; Biron, C.A. Coordinated and distinct roles for IFN-αβ, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 2002, 169, 4279–4287. [Google Scholar] [CrossRef] [Green Version]
- Murira, A.; Lamarre, A. Type-I interferon responses: From friend to foe in the battle against chronic viral infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, G.E.; Gaszewska-Mastarlarz, A.; Moskophidis, D. The role of alpha/beta and gamma interferons in development of immunity to influenza a virus in mice. J. Virol. 2000, 74, 3996–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef]
- Wu, C.; Xue, Y.; Wang, P.; Lin, L.; Liu, Q.; Li, N.; Xu, J.; Cao, X. IFN-γ Primes macrophage activation by increasing phosphatase and tensin homolog via downregulation of miR-3473b. J. Immunol. 2014, 193, 3036–3044. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cockrell, C.; An, G. Comparative Computational Modeling of the Bat and Human Immune Response to Viral Infection with the Comparative Biology Immune Agent Based Model. Viruses 2021, 13, 1620. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081620
Cockrell C, An G. Comparative Computational Modeling of the Bat and Human Immune Response to Viral Infection with the Comparative Biology Immune Agent Based Model. Viruses. 2021; 13(8):1620. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081620
Chicago/Turabian StyleCockrell, Chase, and Gary An. 2021. "Comparative Computational Modeling of the Bat and Human Immune Response to Viral Infection with the Comparative Biology Immune Agent Based Model" Viruses 13, no. 8: 1620. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081620