
Integrating High throughput Sequencing into Survey Design Reveals Turnip Yellows Virus and Soybean Dwarf Virus in Pea (Pisum Sativum) in the United Kingdom

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
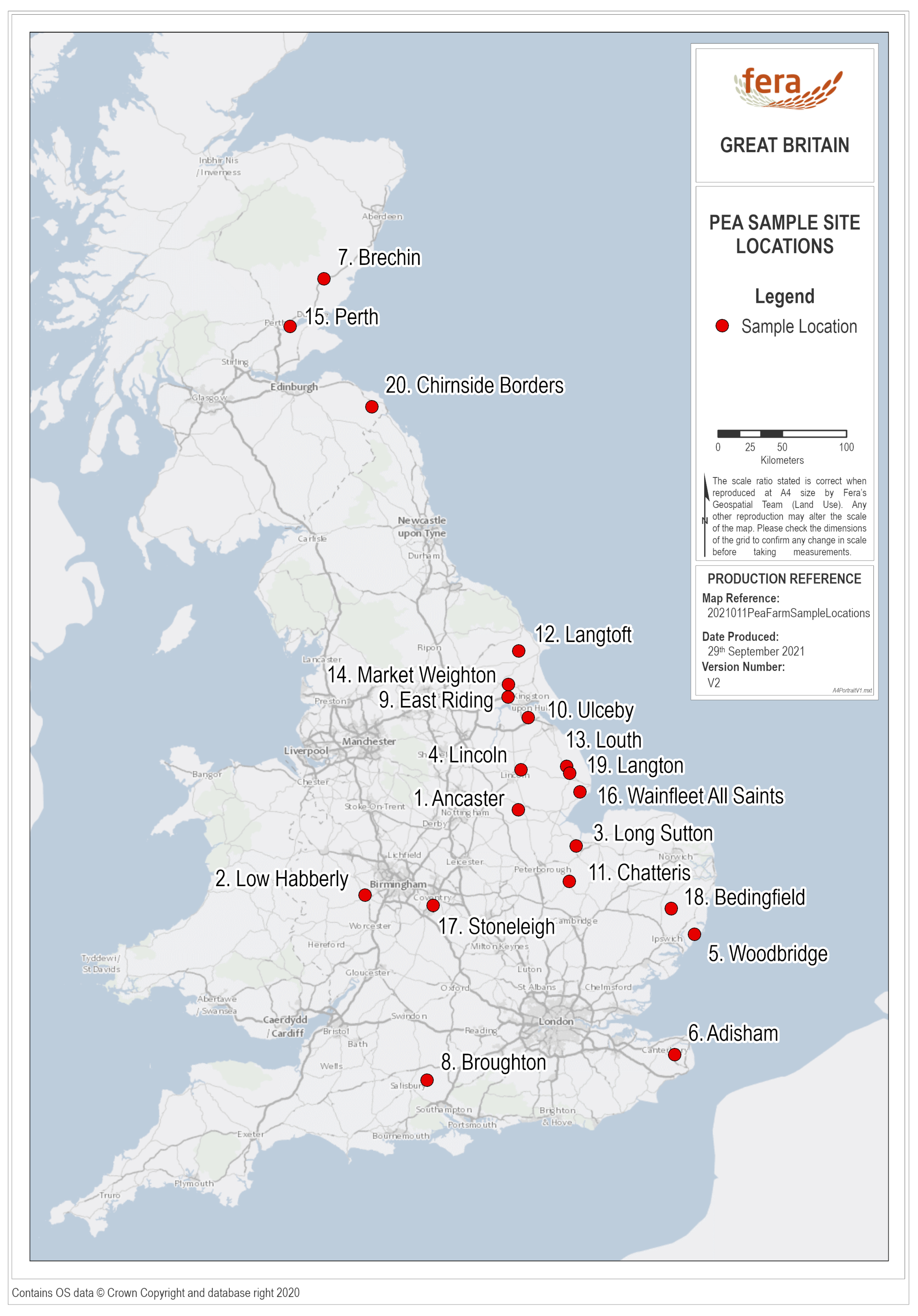
2.1. Field Sampling
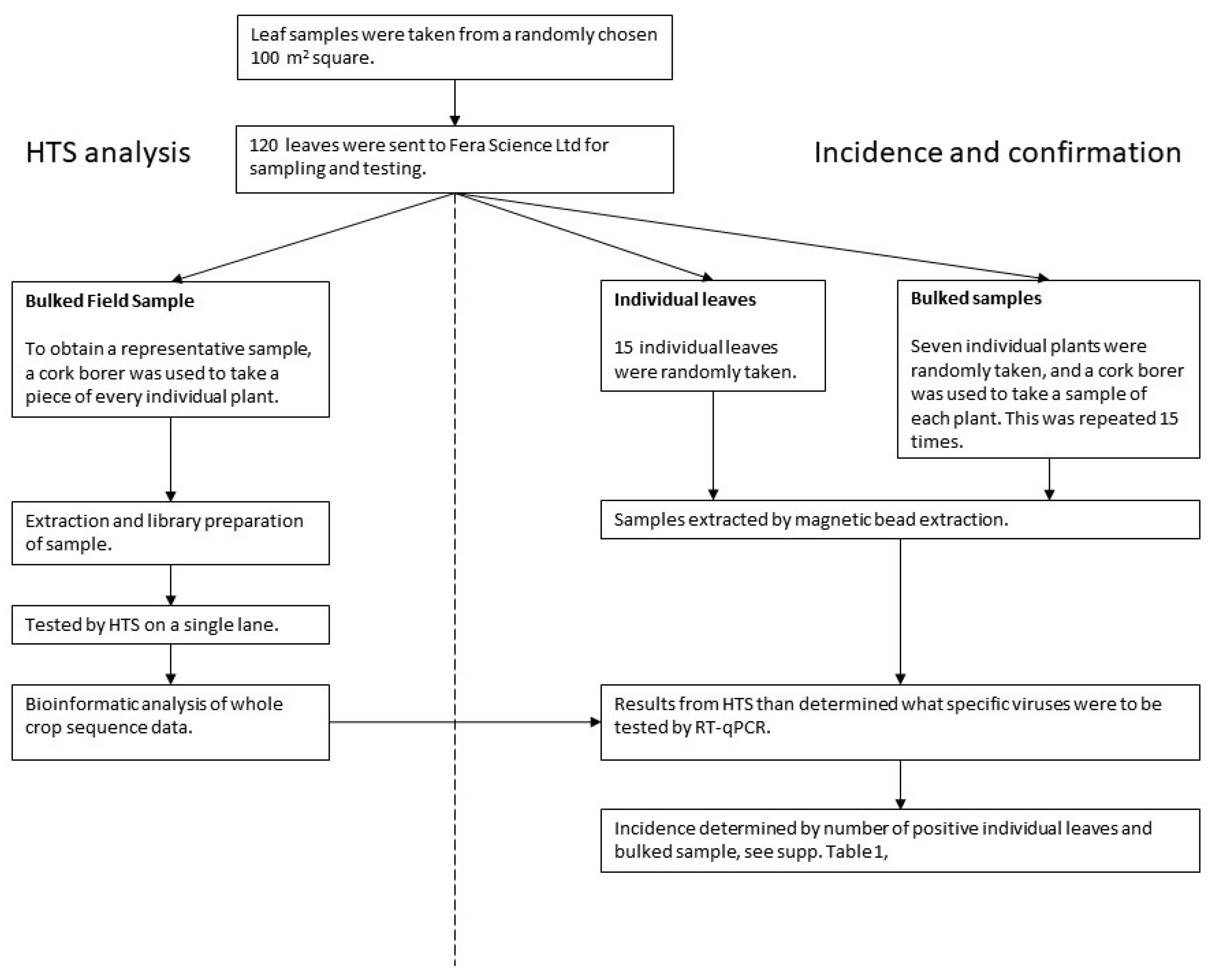
2.2. Sub-Sampling for Virus Analysis
- (a)
- HTS analysis–Bulk field sample (BFS): A representative sample of the whole field was obtained by using a cork borer to take a piece of every leaf. This bulk field sample was tested by HTS.
- (b)
- Confirmation and incidence–Individual leaves and bulked samples: Subsamples were also taken from the initial field sample for both confirmation of HTS detections and estimating the incidence of viruses. (i) Fifteen individual leaves were randomly chosen, and (ii) fifteen bulked samples of seven leaves were randomly chosen. A cork-borer was used to obtain a piece of each leaf within the bulked sample.
2.3. Historical Isolate
2.4. HTS–RNA Extraction, Library Preparation and Analysis
2.5. Sub-Sample RNA Extraction, RT-qPCR, and RT-PCR Confirmation
2.6. Estimating Prevalence Based on Tests Using Different Pool Sizes
2.7. HTS–Increased Depth Testing
3. Results
3.1. Bulked Field Sample Inferred Viruses and Sub-Sample Real-Time RT-PCR Confirmation/Incidence
3.2. Symptomatic Samples from Sites HTS Inferred Viruses and Real-Time RT-PCR Confirmation
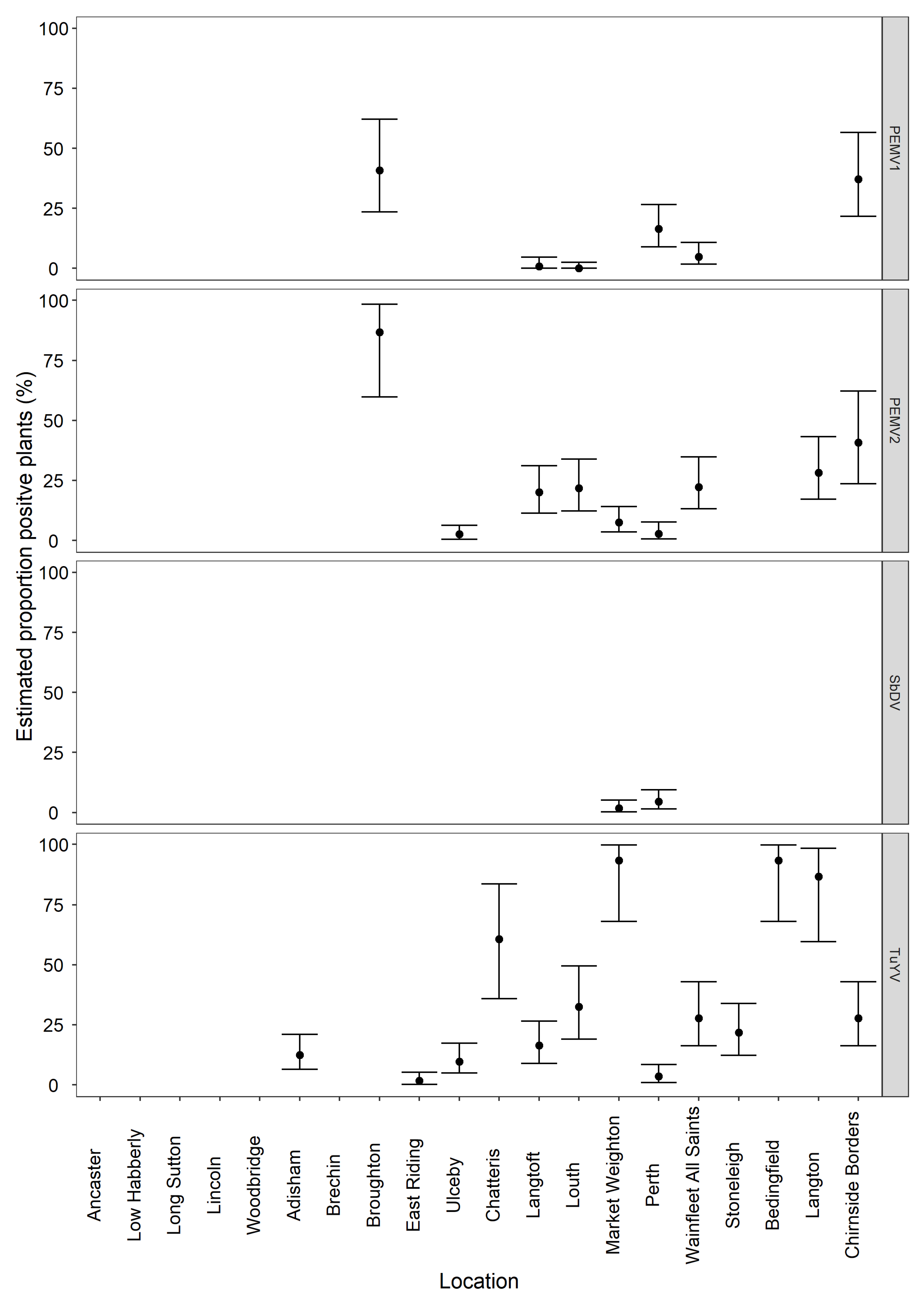
3.3. General Surveillance Samples HTS Inferred Viruses and Real-Time RT-PCR Confirmation
3.4. HTS-Increased Depth Testing
3.5. Historic Isolate
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fox, A.; Fowkes, A.R. Pea Viruses: Investigating the Current Knowledge on Distribution and Control of Pea Viruses; AHDB: Warwickshire, UK, 2017. [Google Scholar]
- Defra. Farming Statistics—Final Crop Areas, Yields, Livestock Populations and Agricultural Workforce at 1 June 2020 United Kingdom. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/946161/structure-jun2020final-uk-22dec20.pdf (accessed on 3 November 2021).
- Adams, I.; Skelton, A.; Macarthur, R.; Hodges, T.; Hinds, H.; Flint, L.; Nath, P.; Boonham, N.; Fox, A. Carrot yellow leaf virus is associated with carrot internal necrosis. PLoS ONE 2014, 9, e109125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, A.; Fowkes, A.; Skelton, A.; Harju, V.; Buxton–Kirk, A.; Kelly, M.; Forde, S.; Pufal, H.; Conyers, C.; Ward, R.; et al. Using High Throughput Sequencing in support of a plant health outbreak reveals novel viruses in Ullucus tuberosus (Basellaceae). Plant Pathol. 2019, 68, 576–587. [Google Scholar]
- Harju, V.; Skelton, A.; Forde, S.M.D.; Bennet, S.; Glover, R.; Monger, W.; Adams, I.P.; Boonham, N.; Fox, A. New virus detected on Nasturtium officinale (watercress). New Dis. Rep. 2012, 25, 1. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, Y.; Lüddecke, P.; Heidler, C.; Hartrick, J.; Sieg-Müller, A.; Hübert, C.; Wichura, A.; Ziebell, H. First report of Southern tomato virus in German tomatoes. New Dis. Rep. 2019, 40, 1. [Google Scholar] [CrossRef] [Green Version]
- Monger, W.A.; Goodfellow, H.A.; Back, E.J. First report of Arabis mosaic virus in potato (Solanum tuberosum), identified by nanopore sequencing. New Dis. Rep. 2020, 41, 29. [Google Scholar] [CrossRef]
- Adams, I.; Fox, A.; Boonham, N.; Massart, S.; De Jonghe, K. The impact of high throughput sequencing on plant health diagnostics. Eur. J. Plant Pathol. 2018, 152, 909–919. [Google Scholar] [CrossRef]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for routine plant virus diagnostics: State of the art and challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [Green Version]
- Ibaba, J.D.; Gubba, A. High-Throughput Sequencing Application in the Diagnosis and Discovery of Plant-Infecting Viruses in Africa, A Decade Later. Plants 2020, 9, 1376. [Google Scholar] [CrossRef]
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.-S.; Rumbou, A.; Saldarelli, P.; Škorić, D. A framework for the evaluation of biosecurity, commercial, regulatory, and scientific impacts of plant viruses and viroids identified by NGS technologies. Front. Microbiol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmos, A.; Boonham, N.; Candresse, T.; Gentit, P.; Giovani, B.; Kutnjak, D.; Liefting, L.; Maree, H.; Minafra, A.; Moreira, A. High–throughput sequencing technologies for plant pest diagnosis: Challenges and opportunities. EPPO Bull. 2018, 48, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Maina, S.; Zheng, L.; Rodoni, B.C. Targeted Genome Sequencing (TG-Seq) Approaches to Detect Plant Viruses. Viruses 2021, 13, 583. [Google Scholar] [CrossRef]
- Alcalá-Briseño, R.I.; Casarrubias-Castillo, K.; López-Ley, D.; Garrett, K.A.; Silva-Rosales, L. Network Analysis of the Papaya Orchard Virome from Two Agroecological Regions of Chiapas, Mexico. mSystems 2020, 5, e00423-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coetzee, B.; Freeborough, M.-J.; Maree, H.J.; Celton, J.-M.; Rees, D.J.G.; Burger, J.T. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Czotter, N.; Molnar, J.; Szabó, E.; Demian, E.; Kontra, L.; Baksa, I.; Szittya, G.; Kocsis, L.; Deak, T.; Bisztray, G. NGS of virus-derived small RNAs as a diagnostic method used to determine viromes of hungarian vineyards. Front. Microbiol. 2018, 9, 122. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, Y.Z.A.; Herz, K.; Hartrick, J.; Fletcher, J.; Blouin, A.; MacDiarmid, R.M.; Ziebell, H. Investigating the pea virome in Germany–old friends and new players in the field (s). Front. Microbiol. 2020, 11, 2605. [Google Scholar] [CrossRef]
- Han, J.; Domier, L.L.; Cassone, B.J.; Dorrance, A.; Qu, F. Assessment of Common Soybean-Infecting Viruses in Ohio, USA, Through Multi-site Sampling and High-Throughput Sequencing. Plant Health Prog. 2016, 17, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.; Saha, P.; Wiley, G.; Quan, J.; White, J.; Lai, H.; Chavarría, F.; Shen, G.; Roe, B. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19 (Suppl. 1), 81–88. [Google Scholar] [CrossRef]
- Shates, T.M.; Sun, P.; Malmstrom, C.M.; Dominguez, C.; Mauck, K.E. Addressing Research Needs in the Field of Plant Virus Ecology by Defining Knowledge Gaps and Developing Wild Dicot Study Systems. Front. Microbiol. 2019, 9, 3305. [Google Scholar] [CrossRef]
- Sõmera, M.; Massart, S.; Tamisier, L.; Sooväli, P.; Sathees, K.; Kvarnheden, A. A Survey Using High-Throughput Sequencing Suggests That the Diversity of Cereal and Barley Yellow Dwarf Viruses Is Underestimated. Front. Microbiol. 2021, 12, 673218. [Google Scholar] [CrossRef] [PubMed]
- Piombo, E.; Abdelfattah, A.; Droby, S.; Wisniewski, M.; Spadaro, D.; Schena, L. Metagenomics Approaches for the Detection and Surveillance of Emerging and Recurrent Plant Pathogens. Microorganisms 2021, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Rozado, Z.; Adams, I.P.; Skelton, A.; Dickinson, M.; Boonham, N. Investigating the Viral Causes of Internal Necrosis in Carrot; International Society for Horticultural Science (ISHS): Leuven, Belgium, 2017; pp. 245–250. [Google Scholar]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33) [Software]. 2011. Available online: https://github.com/najoshi/sickle (accessed on 2 July 2018).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Weller, S.; Elphinstone, J.; Smith, N.; Boonham, N.; Stead, D. Detection of Ralstonia solanacearum Strains with a Quantitative, Multiplex, Real-Time, Fluorogenic PCR (TaqMan) Assay. Appl. Environ. Microbiol. 2000, 66, 2853–2858. [Google Scholar] [CrossRef] [Green Version]
- Thekkeveetil, T.; Hobbs, H.A.; Wang, Y.; Kridelbaugh, D.; Donnelly, J.; Hartman, G.L.; Domier, L.L. First Report of Soybean dwarf virus in Soybean in Northern Illinois. Plant Dis. 2007, 91, 1686. [Google Scholar] [CrossRef] [PubMed]
- Buxton-Kirk, A.; Adams, I.; Frew, L.; Ward, R.; Kelly, M.; Forde, S.; Skelton, A.; Harju, V.; Baucas, N.S.; Bas-Ilan, M.A.G.; et al. First report of Turnip yellows virus in cabbage in the Philippines. New Dis. Rep. 2021, 44, e12020. [Google Scholar] [CrossRef]
- Abraham, A.D.; Menzel, W.; Vetten, H.J.; Saucke, H. First Report of Soybean dwarf virus (Genus Luteovirus) Infecting Faba Bean and Clover in Germany. Plant Dis. 2007, 91, 1059. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, G. Exact Confidence Intervals for Proportions Estimated by Group Testing. Biometrics 1996, 52, 1134–1146. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 14 November 2021).
- binGroup: Evaluation and Experimental Design for Binomial Group Testing, R Package Version 2.2-1. 2018. Available online: https://cran.r-project.org/package=binGroup (accessed on 14 November 2021).
- Filardo, F.; Nancarrow, N.; Kehoe, M.; McTaggart, A.R.; Congdon, B.; Kumari, S.; Aftab, M.; Trębicki, P.; Rodoni, B.; Thomas, J.; et al. Genetic diversity and recombination between turnip yellows virus strains in Australia. Arch. Virol. 2021, 166, 813–829. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BBMap: A Fast, Accurate, Splice-Aware Aligner. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 2 July 2018).
- MacDiarmid, R.; Rodoni, B.; Melcher, U.; Ochoa-Corona, F.; Roossinck, M. Biosecurity implications of new technology and discovery in plant virus research. PLoS Pathog. 2013, 9, e1003337. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.; McGrann, G.; Clark, B. Turnip Yellows Virus (Syn Beet Western Yellows Virus): An Emerging Threat to European Oilseed Rape Production; HGCA: Cape Town, South Africa, 2008. [Google Scholar]
- Aftab, M.; Nancarrow, N.; Trębicki, P. Temperate Pulse Viruses: Turnip Yellows Virus (TuYV). Available online: http://agriculture.vic.gov.au/agriculture/pests-diseases-and-weeds/plant-diseases/grains-pulses-and-cereals/turnip-yellows-virus-tuyv (accessed on 18 May 2020).
- Fletcher, J.D. Surveys of virus diseases in pea, lentil, dwarf and broad bean crops in South Island, New Zealand. N. Z. J. Crop Hortic. Sci. 1993, 21, 45–52. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.A.; Ziebell, H. Two Divergent Isolates of Turnip Yellows Virus from Pea and Rapeseed and First Report of Turnip Yellows Virus-Associated RNA in Germany. Microbiol. Resour. Announc. 2019, 8, e00214-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asare-Bediako, E.; Jones, J.E.; Hambidge, A.J.; Stevens, M.; Mead, A.; Jenner, C.E.; Walsh, J.A. The incidence of turnip yellows virus in oilseed rape crops (Brassica napus L.) in three different regions of England over three consecutive growing seasons and the relationship with the abundance of flying Myzus persicae. Ann. Appl. Biol. 2020, 176, 130–137. [Google Scholar] [CrossRef]
- Ainsworth, G.C. The identification of certain viruses found infecting Leguminous plants in Great Britain. Ann. Appl. Biol. 1940, 27, 218–226. [Google Scholar] [CrossRef]
- McEwen, F.L.; Schroeder, W.T.; Davis, A.C. Host Range and Transmission of the Pea Enation Mosaic Virus. J. Econ. Entomol. 1957, 50, 770–775. [Google Scholar] [CrossRef]
- Timmerman-Vaughan, G.; Larsen, R.; Murray, S.; McPhee, K.; Coyne, C. Analysis of the accumulation of Pea enation mosaic virus genomes in seed tissues and lack of evidence for seed transmission in pea (Pisum sativum). Phytopathology 2009, 99, 1281–1288. [Google Scholar] [CrossRef]
- Demler, S.A.; de Zoeten, G.A. The Nucleotide Sequence and Luteovirus-like Nature of RNA 1 of an Aphid Non-transmissible Strain of Pea Enation Mosaic Virus. J. Gen. Virol. 1991, 72, 1819–1834. [Google Scholar] [CrossRef]
- Demler, S.A.; Rucker, D.G.; de Zoeten, G.A. The chimeric nature of the genome of pea enation mosaic virus: The independent replication of RNA 2. J. Gen. Virol. 1993, 74, 1–14. [Google Scholar] [CrossRef]
- Demler, S.A.; Rucker, D.G.; Nooruddin, L.; de Zoeten, G.A. Replication of the satellite RNA of pea enation mosaic virus is controlled by RNA 2-encoded functions. J. Gen. Virol. 1994, 75, 1399–1406. [Google Scholar] [CrossRef]
- Smith, K.M. A Textbook of Plant Virus Diseases; Elsevier Science: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Syller, J. Molecular and biological features of umbraviruses, the unusual plant viruses lacking genetic information for a capsid protein. Physiol. Mol. Plant Pathol. 2003, 63, 35–46. [Google Scholar] [CrossRef]
- Luoto, A.; Lehtonen, M.; Valkonen, J.; Santala, J. First report of Soybean dwarf virus infecting white clover (Trifolium repens) in Finland. Plant Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Congdon, B.S.; Coutts, B.A.; Renton, M.; Jones, R.A.C. Pea seed-borne mosaic virus: Stability and Wind-Mediated Contact Transmission in Field Pea. Plant Dis. 2016, 100, 953–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briddon, R.W.; Ghabrial, S.; Lin, N.S.; Palukaitis, P.; Scholthof, K.B.G.; Vetten, H.J. Satellites and other virus-dependent nucleic acids. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Elsevier Academic Press: Oxford, UK, 2011. [Google Scholar]
- Barker, H.; Smith, H.G. The Luteoviridae; CABI: Egham, UK, 1999. [Google Scholar]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next Generation Sequencing for Detection and Discovery of Plant Viruses and Viroids: Comparison of Two Approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef] [Green Version]
- Hammond, J.; Adams, I.P.; Fowkes, A.R.; McGreig, S.; Botermans, M.; van Oorspronk, J.J.; Westenberg, M.; Verbeek, M.; Dullemans, A.M.; Stijger, C.C. Sequence analysis of 43-year old samples of Plantago lanceolata show that Plantain virus X is synonymous with Actinidia virus X and is widely distributed. Plant Pathol. 2021, 70, 249–258. [Google Scholar] [CrossRef]
- IPPC. ISPM No. 31 Methodologies for Sampling of Consignments; ISPM: Sao Paulo, Brazil, 2008; p. 79. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Set | Sequence (5′–3′) | Source | |
|---|---|---|---|
| Real-time RT-PCR | |||
| Pea enation mosaic virus-1 | PEMV-1-4838 F | YCT AAT ACC TAG TAG CAA AGG GAA GTT T | This study |
| PEMV-1-4815 | CCC CAA CAT CCA TCA GCT TTT C | ||
| PEMV-1-4891 Pe1 | FAM-TCC AAG CAG TGA AGA GCA TTG GYG-TAMRA | ||
| PEMV-1-4891 Pe2 | FAM-TCC AAG CAG TGA AGA GTA TTG GAG-TAMRA | ||
| Pea enation mosaic virus-2 | PEMV-2-1347 F | CTG AAA AGA TAA AYT TCA CAG CCA AA | This study |
| PEMV-2-1395 R | AAT CGT GGA TCC CTA GGC TGT A | ||
| PEMV-2-1375 Pe | FAM-TGA CCC CGC CCC TCG TGT G-TAMRA | ||
| Pea seed-borne mosaic virus | PSbMV-9129 F1 | TGA CAT HTC AAA CAC TCG AGC AA | This study |
| PSbMV-9129 F2 | TGA CAT HTC AAA CAC TCG CGC AA | ||
| PSbMV-9190 R1 | TGT CWC CAA YCC CRT ACT CTT G | ||
| PSbMV-9155 Pe1 | FAM-AGA GCC AGT TYG ATA AYT GGT GGA GGG-TAMRA | ||
| PSbMV-9155 Pe2 | FAM-AGA GCC AAT TYG ATA AYT GGT GGA GAG-TAMRA | ||
| Soybean dwarf virus | SbDV-F | TGG CTA TTA TAG AAT GGT GCG TAA AC | [30] |
| SbDV-R | GCC ATG GAA ATG AGG GAA TG | ||
| SbDV-Pe | FAM-AGC ATA TCC AAA GAC GC-MGB | ||
| Turnip yellows virus | TuYV-F2 | GCC GCT TGT TTC TCA GTT CTG | [31] |
| TuYV-R2 | GAC TAA CCA CGA GTA AAG AAG CTC AA | ||
| TuYV-P2 | FAM-ACG AGT TGC GGC ACG ATC CAG C-TAMRA | ||
| RT-PCR | |||
| Soybean dwarf virus | SbDV-F | GTC TAC CTA AAA ATT TCA AAG AAT CTG | [32] |
| SbDV-R | CGG ACC CGG TTC TCC GTC TA | ||
| Site | Sub- Sample | HTS Result | Results of Real-Time RT-PCR Confirmation | |||
|---|---|---|---|---|---|---|
| TuYV | PEMV-1 | PEMV-2 | PSbMV | |||
| Market Rasen | Fragment of turnip yellows virus associated RNA (TuYVaRNA) | Positive | Positive | Positive | Nt | |
| Ramsey-1 | TuYV, PEMV-1, PEMV-2 | Positive | Positive | Positive | Nt | |
| Ramsey-2 | TuYV, PEMV-2 | Positive | Positive | Positive | Nt | |
| March | TuYV, PEMV-1, PEMV-2, Pea seed-borne mosaic virus (PSbMV), Fragments of TuYVaRNA | Positive | Positive | Positive | Positive | |
| Ramsey-3 | TuYV, PEMV-1, PEMV-2, PSbMV | Positive | Positive | Positive | Positive | |
| Ramsey-4 | TuYV, PEMV-2 | Positive | Positive | Positive | Nt | |
| The Deepings | TuYV, PEMV-1, PEMV-2, PEMV satRNA, bean yellow mosaic virus (BYMV) | Positive | Positive | Positive | Nt | |
| Cambridge-1 | TuYV, PEMV-1, PEMV-2, PSbMV | Positive | Positive | Positive | Positive | |
| Cambridge-2 | PEMV-1, PEMV-2, PEMV satRNA | Negative | Positive | Positive | Nt | |
| Market Weighton | 1 | TuYV, PEMV-1, PEMV-2 | Positive | Positive | Positive | Nt |
| Market Weighton | 2 | Positive | Negative | Negative | Nt | |
| Market Weighton | 3 | Positive | Positive | Positive | Nt | |
| Market Weighton | 4 | Positive | Negative | Positive | Nt | |
| Market Weighton | 5 | Positive | Negative | Positive | Nt | |
| Market Weighton | 6 | Positive | Negative | Negative | Nt | |
| Market Weighton | 7 | Positive | Positive | Negative | Nt | |
| Market Weighton | 8 | Positive | Negative | Negative | Nt | |
| Wainfleet | 1 | TuYV, PEMV-1, PEMV-2, PEMV satRNA | Positive | Negative | Positive | Nt |
| Wainfleet | 2 | Positive | Positive | Positive | Nt | |
| Wainfleet | 3 | Positive | Positive | Positive | Nt | |
| Wainfleet | 4 | Positive | Positive | Positive | Nt | |
| Wainfleet | 5 | Positive | Positive | Positive | Nt | |
| Wainfleet | 6 | Positive | Positive | Positive | Nt | |
| Eye | 1 | TuYV, PEMV-2 | Positive | Negative | Negative | Nt |
| Eye | 2 | Positive | Negative | Negative | Nt | |
| Eye | 3 | Positive | Negative | Positive | Nt | |
| Langton | 1 | TuYV | Positive | Negative | Negative | Nt |
| Langton | 2 | Positive | Negative | Positive | Nt | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fowkes, A.R.; McGreig, S.; Pufal, H.; Duffy, S.; Howard, B.; Adams, I.P.; Macarthur, R.; Weekes, R.; Fox, A. Integrating High throughput Sequencing into Survey Design Reveals Turnip Yellows Virus and Soybean Dwarf Virus in Pea (Pisum Sativum) in the United Kingdom. Viruses 2021, 13, 2530. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122530
Fowkes AR, McGreig S, Pufal H, Duffy S, Howard B, Adams IP, Macarthur R, Weekes R, Fox A. Integrating High throughput Sequencing into Survey Design Reveals Turnip Yellows Virus and Soybean Dwarf Virus in Pea (Pisum Sativum) in the United Kingdom. Viruses. 2021; 13(12):2530. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122530
Chicago/Turabian StyleFowkes, Aimee R., Sam McGreig, Hollie Pufal, Shona Duffy, Becky Howard, Ian P. Adams, Roy Macarthur, Rebecca Weekes, and Adrian Fox. 2021. "Integrating High throughput Sequencing into Survey Design Reveals Turnip Yellows Virus and Soybean Dwarf Virus in Pea (Pisum Sativum) in the United Kingdom" Viruses 13, no. 12: 2530. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122530