
Impact of Adenovirus E4-ORF3 Oligomerization and Protein Localization on Cellular Gene Expression

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Infection
2.2. Immunofluoresence and Western Blot Analysis
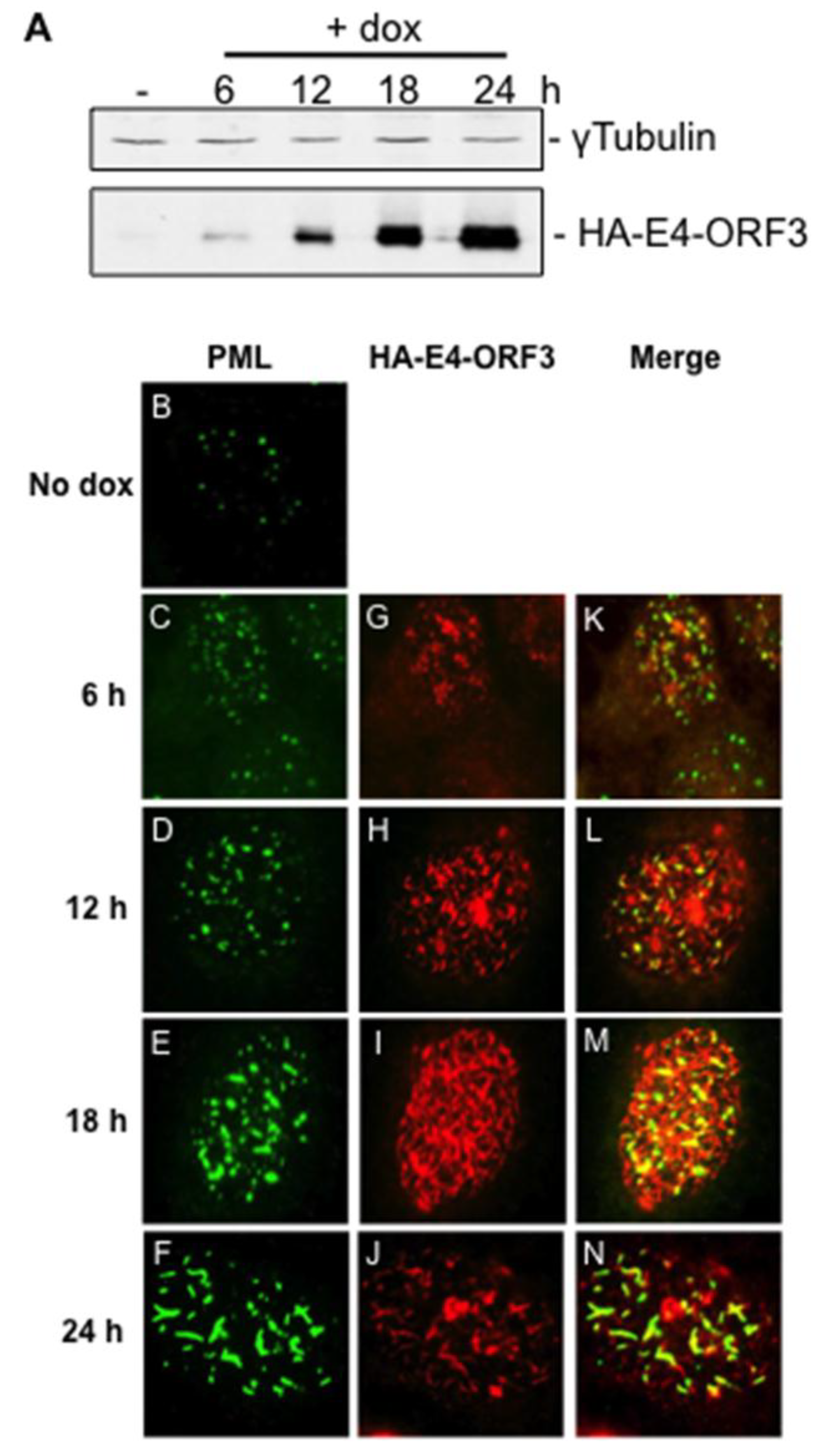
2.3. Creation of an E4-ORF3 Inducible Cell Line
2.4. Microarray Analysis
2.5. RT-qPCR
2.6. Generation of Knockdown Cell Lines
3. Results
3.1. Creation and Characterization of a Tet-Inducible E4-ORF3 Cell Line

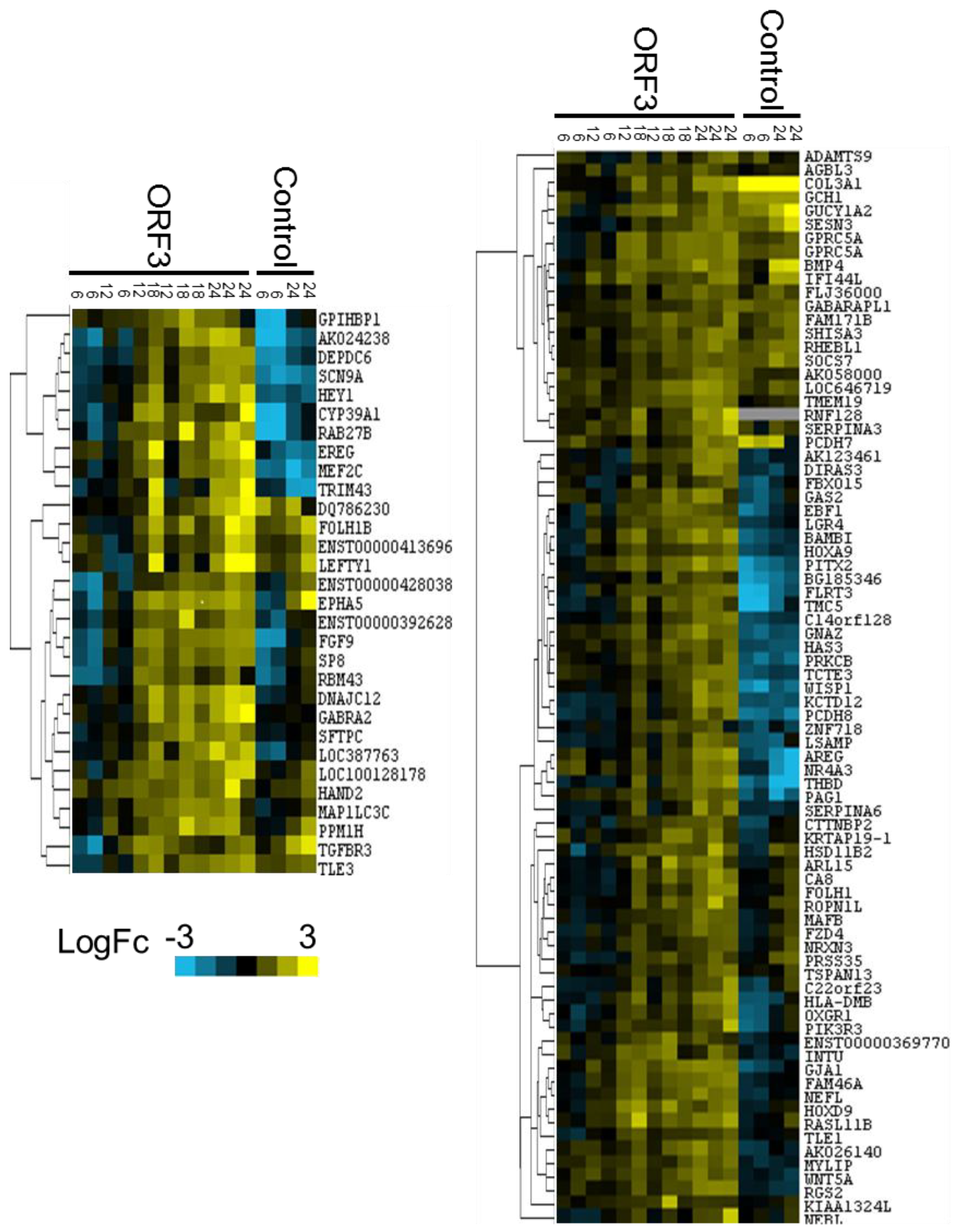
3.2. Microarray Experiments and Quality Control
3.3. Data Analysis and Target Gene Selection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Count | % | p value |
|---|---|---|---|---|
| Biological process | Cell surface receptor-linked signal transduction | 59 | 18.97 | 1.01E-09 |
| Response to wounding | 29 | 9.32 | 5.77E-08 | |
| Inflammatory response | 19 | 6.11 | 8.57E-06 | |
| Defense response | 25 | 8.04 | 4.95E-05 | |
| Cell-cell signaling | 25 | 8.04 | 6.65E-05 | |
| Immune response | 25 | 8.04 | 1.94E-04 | |
| G protein coupled receptor signaling pathway | 28 | 9.00 | 3.44E-04 | |
| Cell migration | 14 | 4.50 | 8.35E-04 | |
| Cellular component | Extracellular region | 74 | 23.79 | 2.86E-11 |
| Extracellular space | 36 | 11.58 | 1.42E-08 | |
| Integral to plasma membrane | 50 | 16.08 | 2.03E-08 | |
| Intrinsic to plasma membrane | 50 | 16.08 | 4.20E-08 | |
| Extracellular region part | 42 | 13.50 | 1.10E-07 | |
| Plasma membrane part | 62 | 19.93 | 1.49E-04 | |
| Molecular function | Growth factor activity | 14 | 4.50 | 3.75E-06 |
| Polysaccharide binding | 13 | 4.18 | 1.36E-05 | |
| Pattern binding | 13 | 4.18 | 1.36E-05 | |
| Cytokine activity | 14 | 4.50 | 2.47E-05 | |
| Glycosoaminoglycan binding | 12 | 3.86 | 2.86E-05 | |
| Ligand-gated ion channel activity | 11 | 3.54 | 6.42E-05 | |
| Ligand-gated channel activity | 11 | 3.54 | 6.42E-05 | |
| Heparin binding | 10 | 3.22 | 6.47E-05 | |
| Carbohydrate binding | 18 | 5.79 | 1.00E-04 | |
| Gated channel activity14 | 16 | 5.14 | 2.71E-04 |

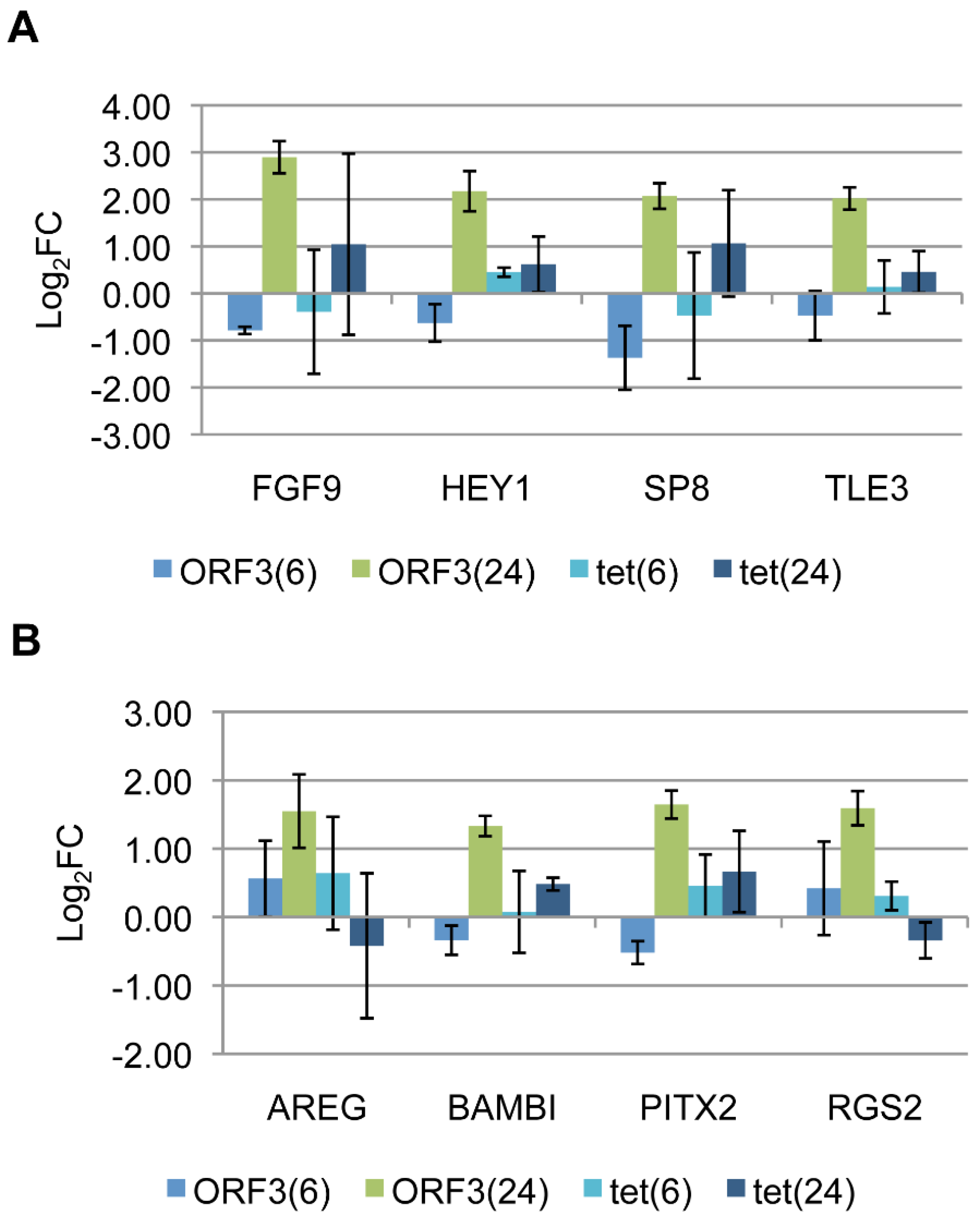
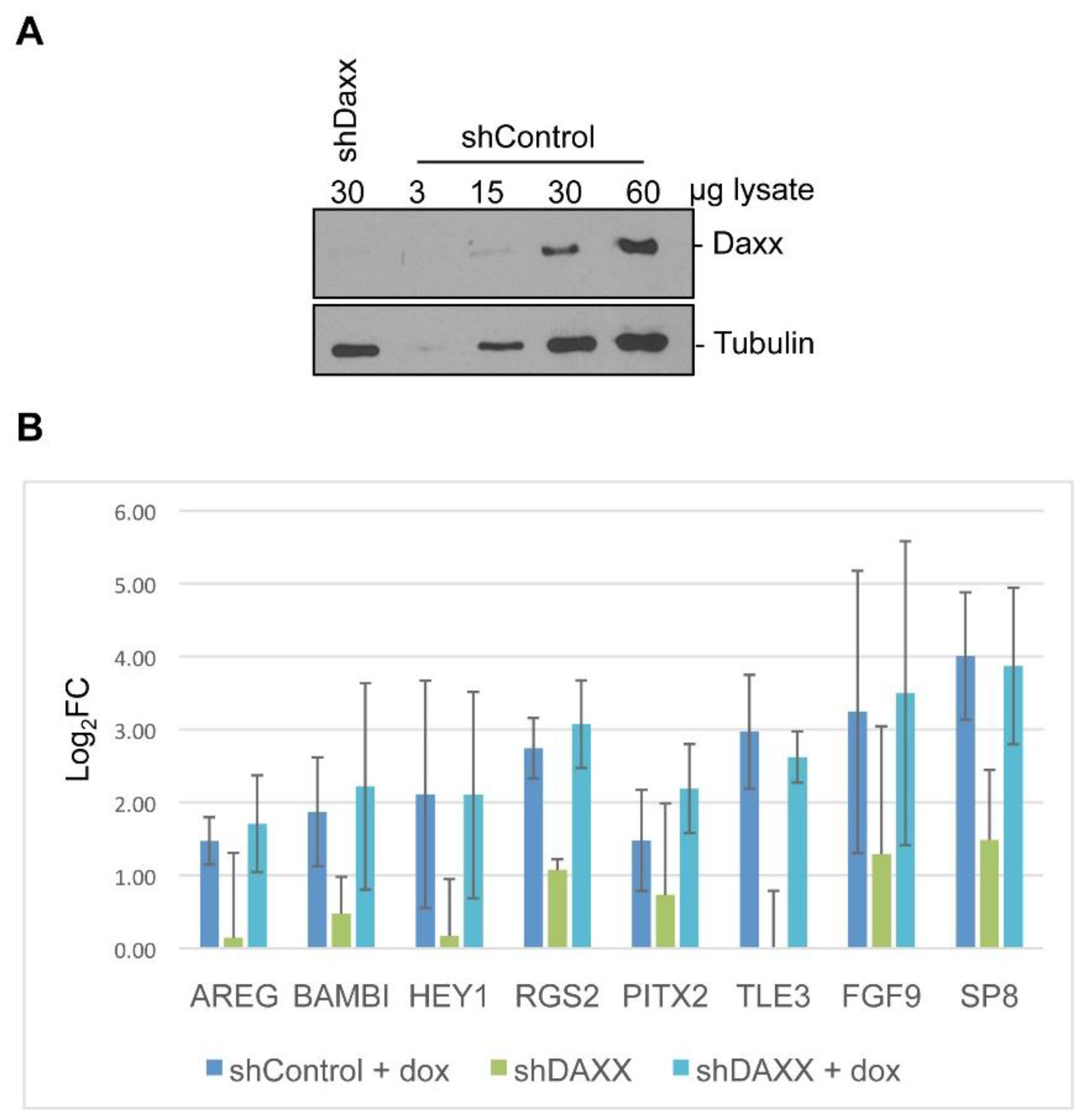
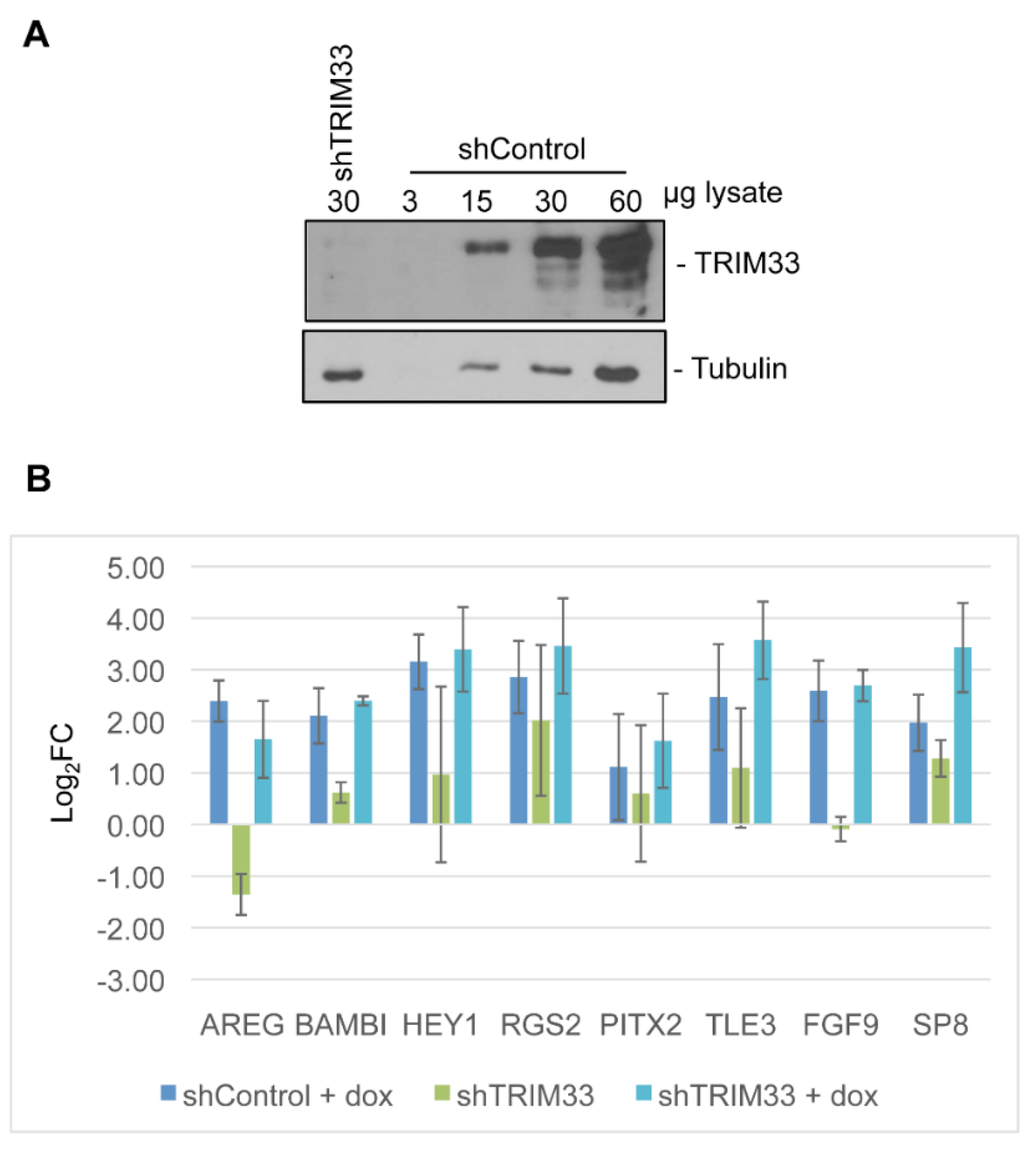
3.4. Target Gene Validation

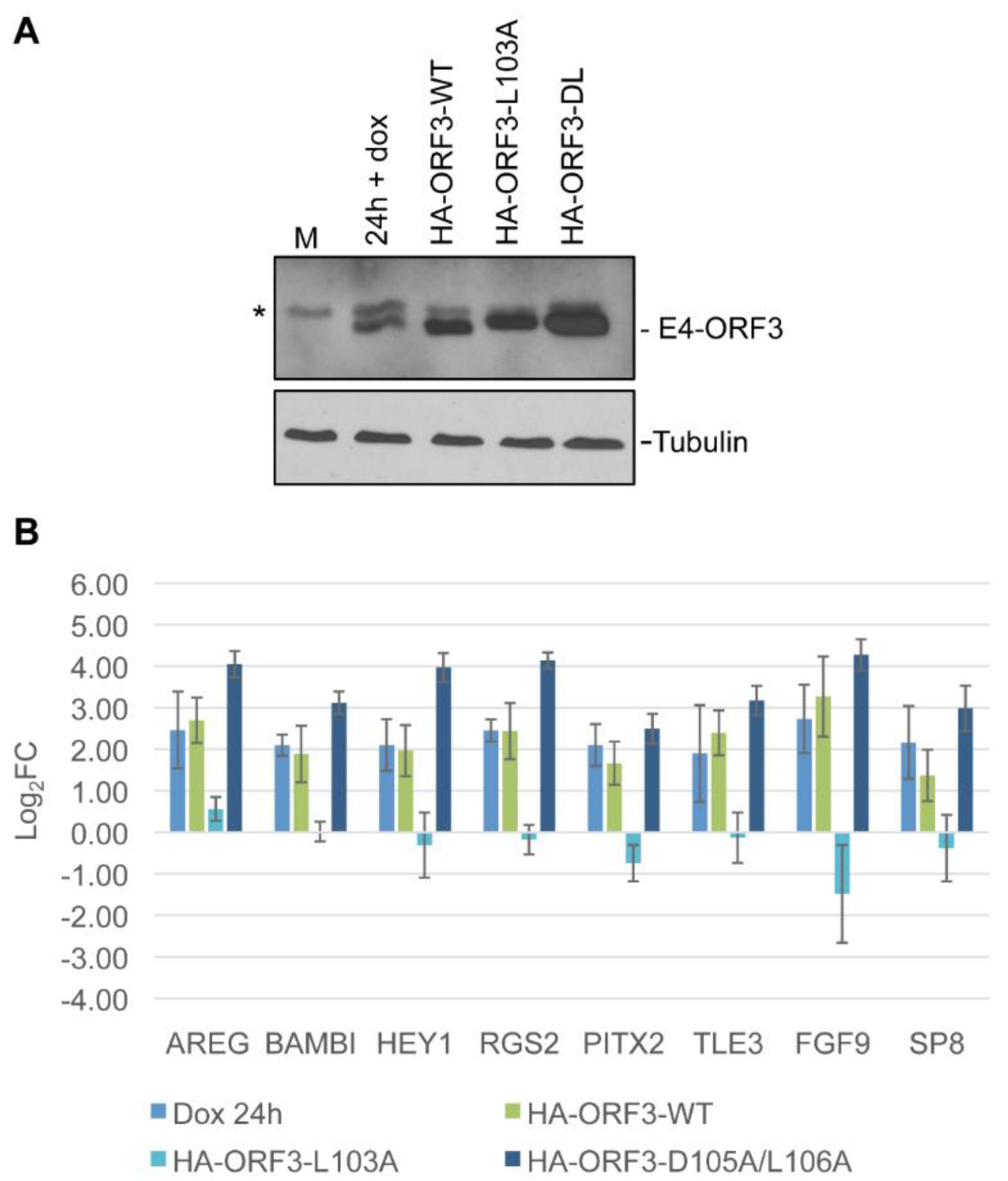
3.5. Linking Differential Gene Expression to E4-ORF3 Functions



4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bridge, E.; Ketner, G. Redundant control of adenovirus late gene expression by early region 4. J. Virol. 1989, 63, 631–638. [Google Scholar]
- Huang, M.M.; Hearing, P. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J. Virol. 1989, 63, 2605–2615. [Google Scholar] [PubMed]
- Sandler, A.B.; Ketner, G. Adenovirus early region 4 is essential for normal stability of late nuclear RNAs. J. Virol. 1989, 63, 624–630. [Google Scholar] [PubMed]
- Cheng, C.Y.; Blanchette, P.; Branton, P.E. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology 2007, 364, 36–44. [Google Scholar]
- Cheng, C.Y.; Gilson, T.; Dallaire, F.; Ketner, G.; Branton, P.E.; Blanchette, P. The E4orf6/E1B55K E3 ubiquitin ligase complexes of human adenoviruses exhibit heterogeneity in composition and substrate specificity. J. Virol. 2011, 85, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Harada, J.N.; Shevchenko, A.; Pallas, D.C.; Berk, A.J. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 2002, 76, 9194–9206. [Google Scholar] [CrossRef] [PubMed]
- Querido, E.; Blanchette, P.; Yan, Q.; Kamura, T.; Morrison, M.; Boivin, D.; Kaelin, W.G.; Conaway, R.C.; Conaway, J.W.; Branton, P.E. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001, 15, 3104–3117. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Rohleder, K.J.; Hanakahi, L.A.; Ketner, G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 2007, 81, 7034–7040. [Google Scholar] [CrossRef] [PubMed]
- Dallaire, F.; Blanchette, P.; Groitl, P.; Dobner, T.; Branton, P.E. Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J. Virol. 2009, 83, 5329–5338. [Google Scholar] [CrossRef] [PubMed]
- Orazio, N.I.; Naeger, C.M.; Karlseder, J.; Weitzman, M.D. The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J. Virol. 2011, 85, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Doucas, V.; Ishov, A.M.; Romo, A.; Juguilon, H.; Weitzman, M.D.; Evans, R.M.; Maul, G.G. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 1996, 10, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.D.; Kwiatkowski, W.; Deerinck, T.J.; Noske, A.; Blain, K.Y.; Land, H.S.; Soria, C.; Powers, C.J.; May, A.P.; Shu, X.; et al. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 2012, 151, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Patsalo, V.; Yondola, M.A.; Luan, B.; Shoshani, I.; Kisker, C.; Green, D.F.; Raleigh, D.P.; Hearing, P. Biophysical and functional analyses suggest that adenovirus E4-ORF3 protein requires higher-order multimerization to function against promyelocytic leukemia protein nuclear bodies. J. Biol. Chem. 2012, 287, 22573–22583. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Ornelles, D.A. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 2005, 24, 7686–7696. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.Y.; Hearing, P. Adenovirus regulates sumoylation of Mre11-Rad50-Nbs1 components through a paralog-specific mechanism. J. Virol. 2012, 86, 9656–9665. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Hearing, P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 2005, 79, 6207–6215. [Google Scholar] [CrossRef] [PubMed]
- Soria, C.; Estermann, F.E.; Espantman, K.C.; O'Shea, C.C. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 2010, 466, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Vink, E.I.; Yondola, M.A.; Wu, K.; Hearing, P. Adenovirus E4-ORF3-dependent relocalization of TIF1alpha and TIF1gamma relies on access to the Coiled-Coil motif. Virology 2012, 422, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Yondola, M.A.; Hearing, P. The adenovirus E4 ORF3 protein binds and reorganizes the TRIM family member transcriptional intermediary factor 1 alpha. J. Virol. 2007, 81, 4264–4271. [Google Scholar] [CrossRef] [PubMed]
- Ullman, A.J.; Hearing, P. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J. Virol. 2008, 82, 7325–7335. [Google Scholar] [CrossRef] [PubMed]
- Ullman, A.J.; Reich, N.C.; Hearing, P. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J. Virol. 2007, 81, 4744–4752. [Google Scholar] [CrossRef] [PubMed]
- Karen, K.A.; Hoey, P.J.; Young, C.S.; Hearing, P. Temporal regulation of the Mre11-Rad50-Nbs1 complex during adenovirus infection. J. Virol. 2009, 83, 4565–4573. [Google Scholar] [CrossRef] [PubMed]
- Weiden, M.D.; Ginsberg, H.S. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 1994, 91, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.S.; Schwartz, R.A.; Ferenchak, K.; Carson, C.T.; McSharry, B.P.; Wilkinson, G.W.; Weitzman, M.D. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J. Virol. 2008, 82, 8362–8372. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Hearing, P. Distinct roles of the Adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 2003, 77, 5295–5304. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.S.; Bridge, E. Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology 2008, 374, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.C.; Chelbi-Alix, M.K. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 2011, 31, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Laukens, K.; Dang, T.H.; van Ostade, X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int. J. Biol. Sci. 2010, 6, 51–67. [Google Scholar]
- Cammas, F.; Khetchoumian, K.; Chambon, P.; Losson, R. TRIM involvement in transcriptional regulation. Adv. Exp. Med. Biol. 2012, 770, 59–76. [Google Scholar] [PubMed]
- Ikeda, K.; Inoue, S. TRIM proteins as RING finger E3 ubiquitin ligases. Adv. Exp. Med. Biol. 2012, 770, 27–37. [Google Scholar] [PubMed]
- Napolitano, L.M.; Meroni, G. TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life 2012, 64, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Zacchigna, L.; Cordenonsi, M.; Soligo, S.; Adorno, M.; Rugge, M.; Piccolo, S. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 2005, 121, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, Y.; Dadakhujaev, S.; Chandhoke, A.S.; Huynh, M.A.; Oldenborg, A.; Ikeuchi, M.; Deng, L.; Bennett, E.J.; Harper, J.W.; Bonni, A.; Bonni, S. TIF1gamma protein regulates epithelial-mesenchymal transition by operating as a small ubiquitin-like modifier (SUMO) E3 ligase for the transcriptional regulator SnoN1. J. Biol. Chem. 2014, 289, 25067–25078. [Google Scholar] [CrossRef] [PubMed]
- Khetchoumian, K.; Teletin, M.; Tisserand, J.; Mark, M.; Herquel, B.; Ignat, M.; Zucman-Rossi, J.; Cammas, F.; Lerouge, T.; Thibault, C.; Metzger, D.; Chambon, P.; Losson, R. Loss of Trim24 (Tif1alpha) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat. Genet. 2007, 39, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Delva, L.; Rachez, C.; Cenciarelli, C.; Gandini, D.; Zhang, H.; Kalantry, S.; Freedman, L.P.; Pandolfi, P.P. A RA-dependent, tumour-growth suppressive transcription complex is the target of the PML-RARalpha and T18 oncoproteins. Nat. Genet. 1999, 23, 287–295. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massague, J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Agricola, E.; Randall, R.A.; Gaarenstroom, T.; Dupont, S.; Hill, C.S. Recruitment of TIF1gamma to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 2011, 43, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Wang, Z.; Zaromytidou, A.I.; Zhang, X.H.; Chow-Tsang, L.F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A poised chromatin platform for TGF-beta access to master regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Nevels, M.; Tauber, B.; Kremmer, E.; Spruss, T.; Wolf, H.; Dobner, T. Transforming potential of the adenovirus type 5 E4orf3 protein. J. Virol. 1999, 73, 1591–1600. [Google Scholar] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar]
- Smyth, G.K.; Speed, T. Normalization of cDNA microarray data. Methods 2003, 31, 265–273. [Google Scholar] [CrossRef] [PubMed]
- de Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res. 2009, 37, 1–13. [Google Scholar]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Cui, W.; Taub, D.D.; Gardner, K. qPrimerDepot: A primer database for quantitative real time PCR. Nucl. Acids Res. 2007, 35, D805–D809. [Google Scholar]
- Lefever, S.; Vandesompele, J.; Speleman, F.; Pattyn, F. RTPrimerDB: The portal for real-time PCR primers and probes. Nucl. Acids Res. 2009, 37 (Suppl. 1), D942–D945. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucl. Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Leis, M.; Stamminger, T. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 2006, 80, 8006–8018. [Google Scholar] [CrossRef] [PubMed]
- Salomoni, P. The PML-Interacting Protein DAXX: Histone Loading Gets into the Picture. Front. Oncol. 2013, 3, e152. [Google Scholar] [CrossRef]
- Lowe, S.W.; Ruley, H.E. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993, 7, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Debbas, M.; White, E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993, 7, 546–554. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vink, E.I.; Zheng, Y.; Yeasmin, R.; Stamminger, T.; Krug, L.T.; Hearing, P. Impact of Adenovirus E4-ORF3 Oligomerization and Protein Localization on Cellular Gene Expression. Viruses 2015, 7, 2428-2449. https://0-doi-org.brum.beds.ac.uk/10.3390/v7052428
Vink EI, Zheng Y, Yeasmin R, Stamminger T, Krug LT, Hearing P. Impact of Adenovirus E4-ORF3 Oligomerization and Protein Localization on Cellular Gene Expression. Viruses. 2015; 7(5):2428-2449. https://0-doi-org.brum.beds.ac.uk/10.3390/v7052428
Chicago/Turabian StyleVink, Elizabeth I., Yueting Zheng, Rukhsana Yeasmin, Thomas Stamminger, Laurie T. Krug, and Patrick Hearing. 2015. "Impact of Adenovirus E4-ORF3 Oligomerization and Protein Localization on Cellular Gene Expression" Viruses 7, no. 5: 2428-2449. https://0-doi-org.brum.beds.ac.uk/10.3390/v7052428