
Activation of DNA Damage Response Pathways during Lytic Replication of KSHV

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Induction of Lytic Reactivation in KSHV-Infected Cell Lines
2.3. Inhibition of DDR Kinases during Lytic Replication
2.4. Infection of EA.hy926 Cells with TRE-BCBL-1-RTA-Derived KSHV Virus
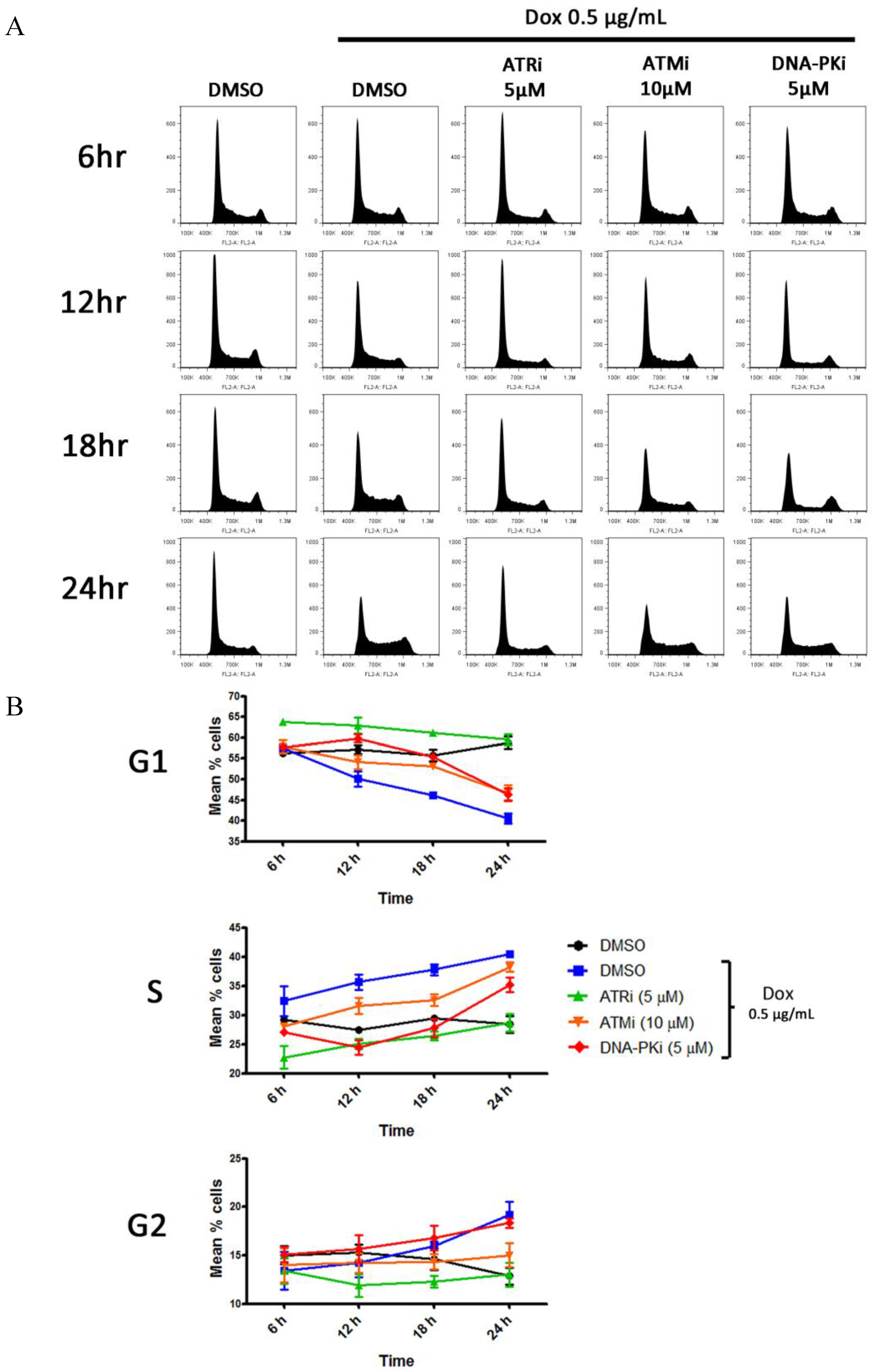
2.5. Cell Cycle Analysis
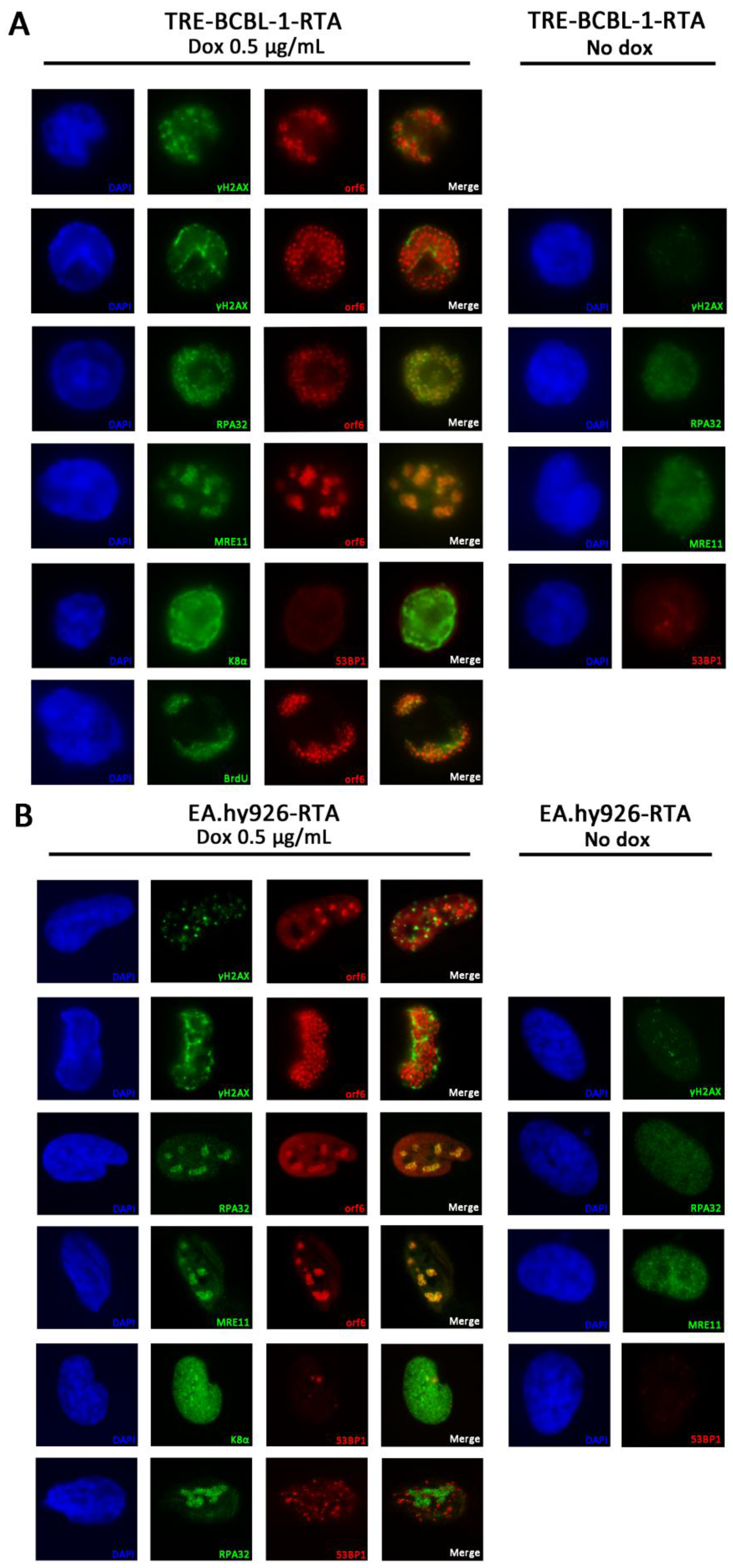
2.6. Immunofluorescence Microscopy (IF)
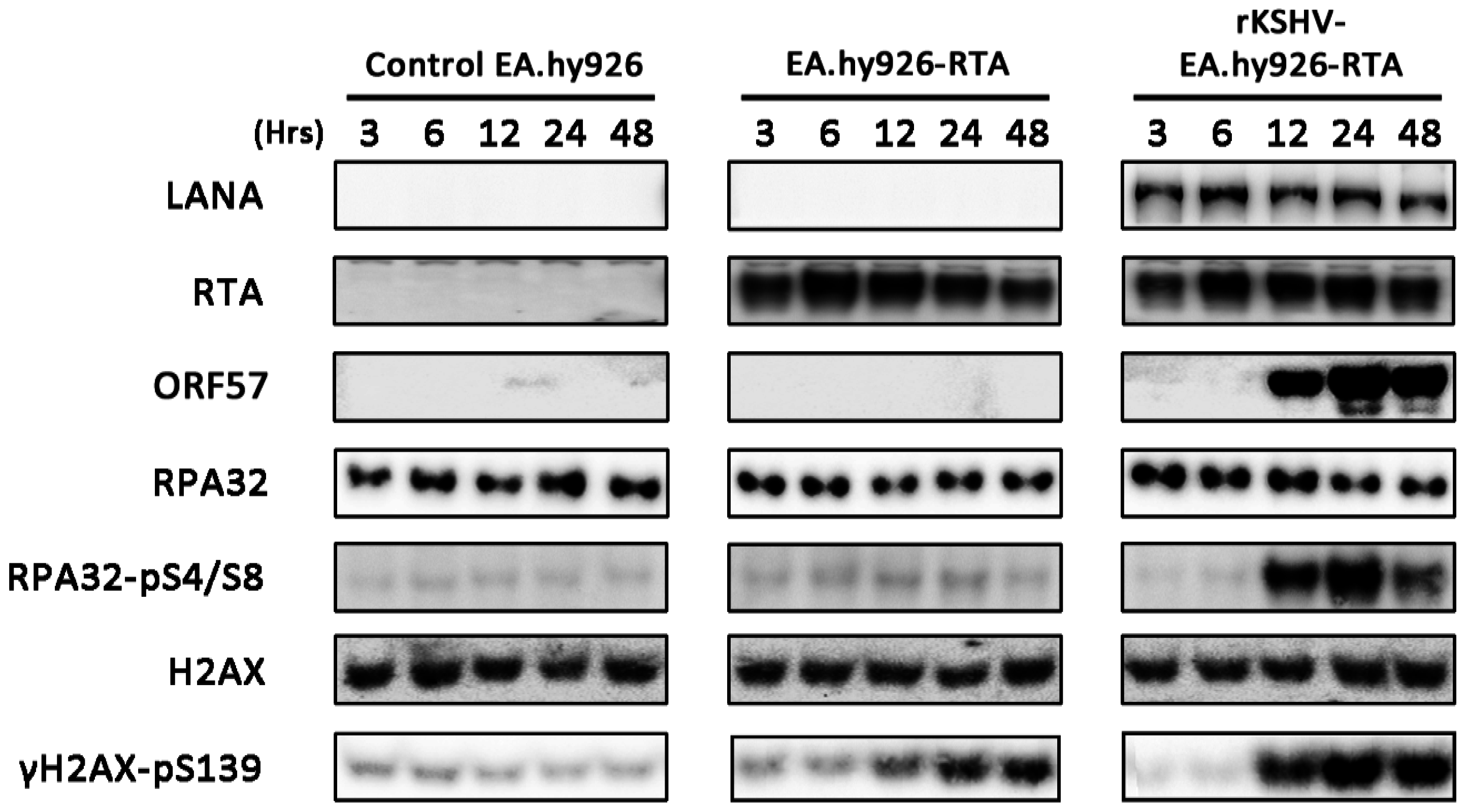
2.7. Western Blot Analysis
3. Results
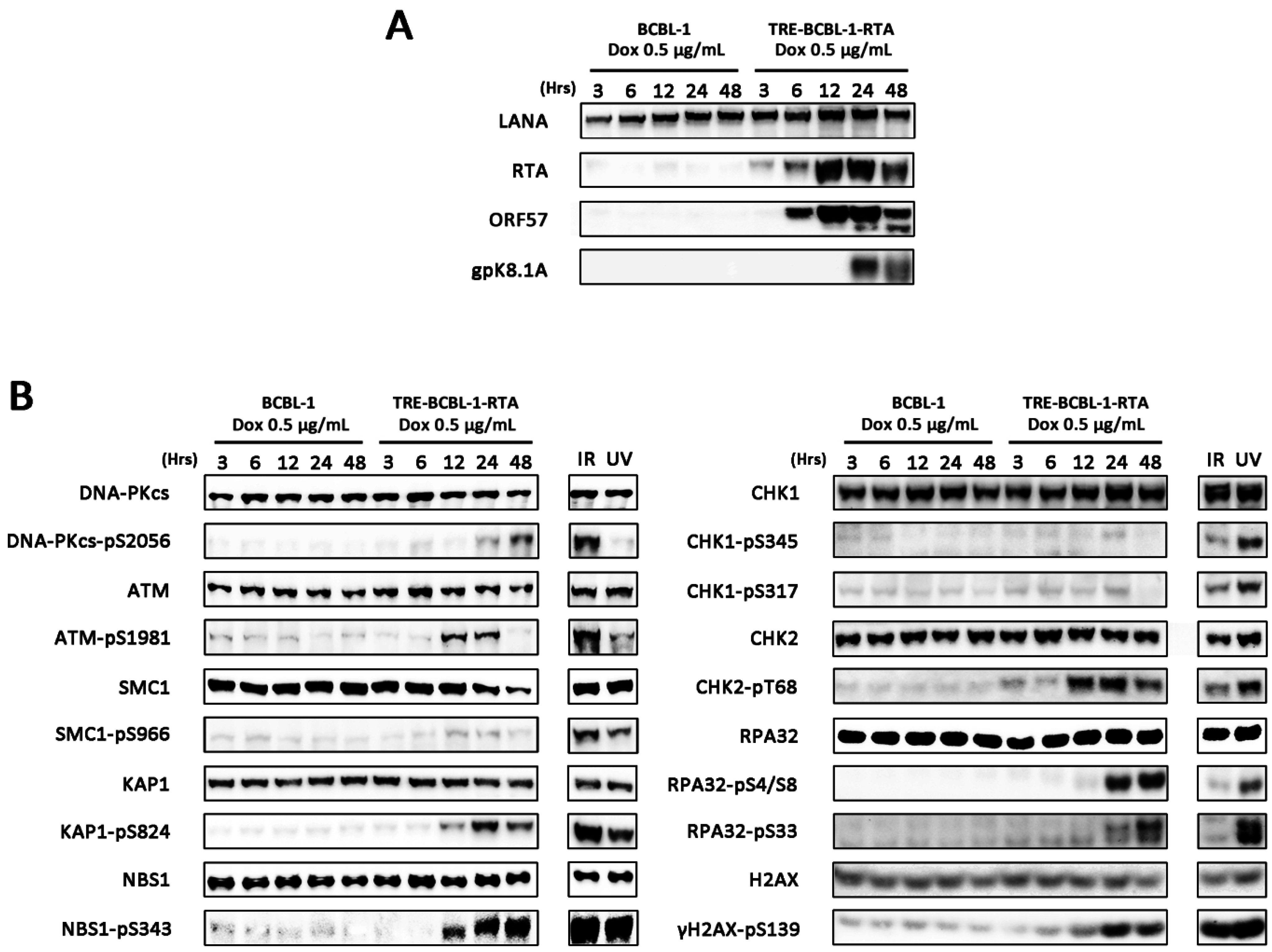
3.1. Lytic Reactivation of KSHV in B Cells Activates DDR Pathways

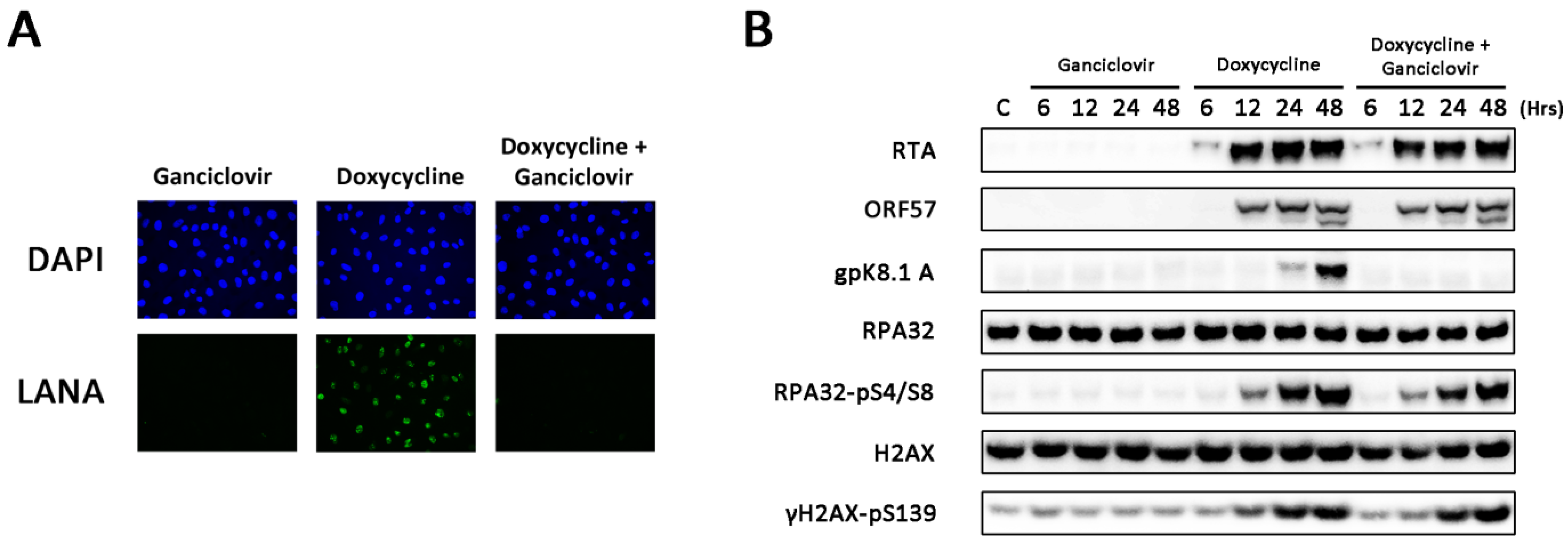
3.2. Amplification of Viral DNA and Late Viral Gene Expression Is Not Required for DDR Activation

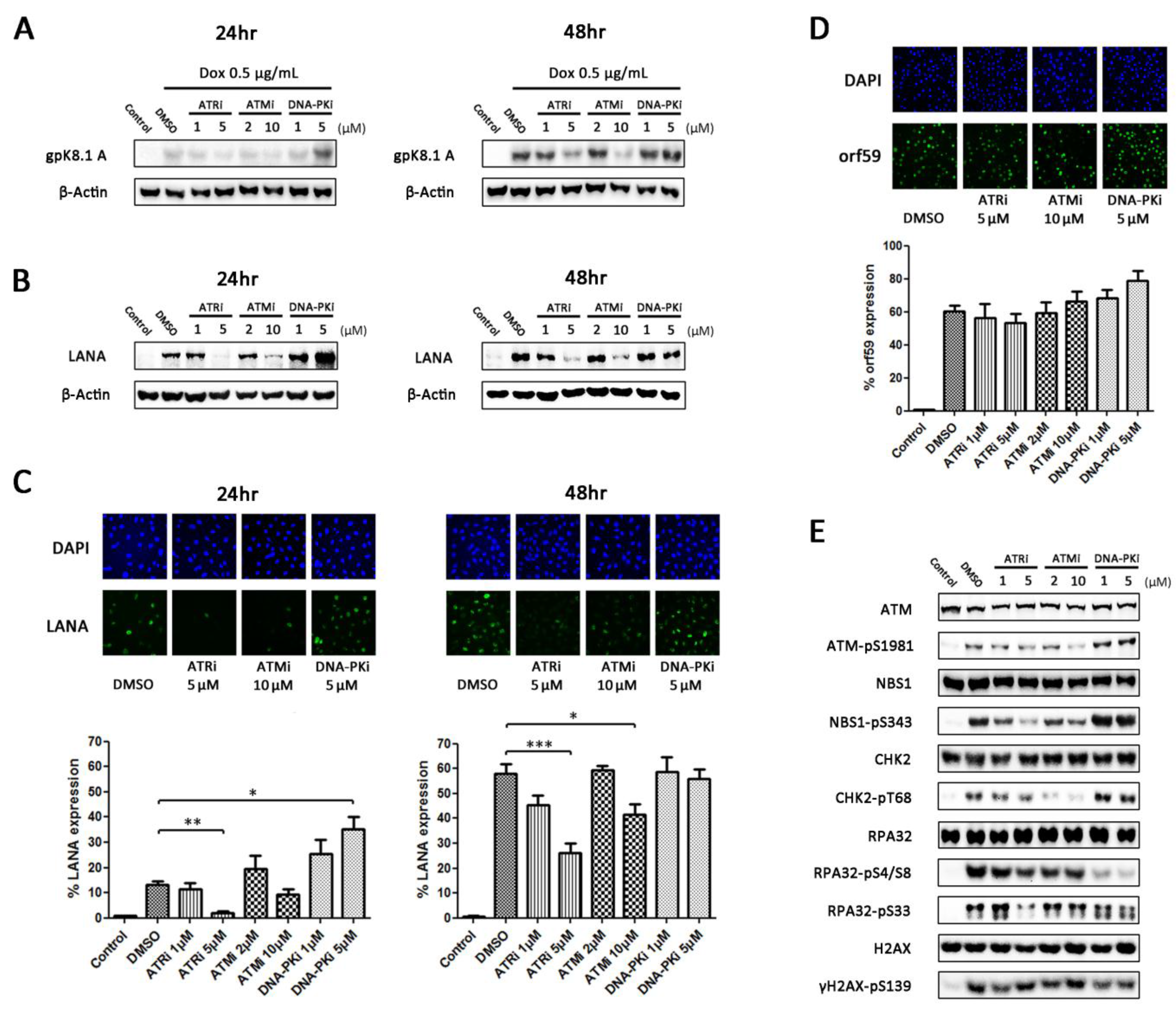
3.3. Inhibition of DDR Kinases Results in Alterations in Production of Infectious Virus

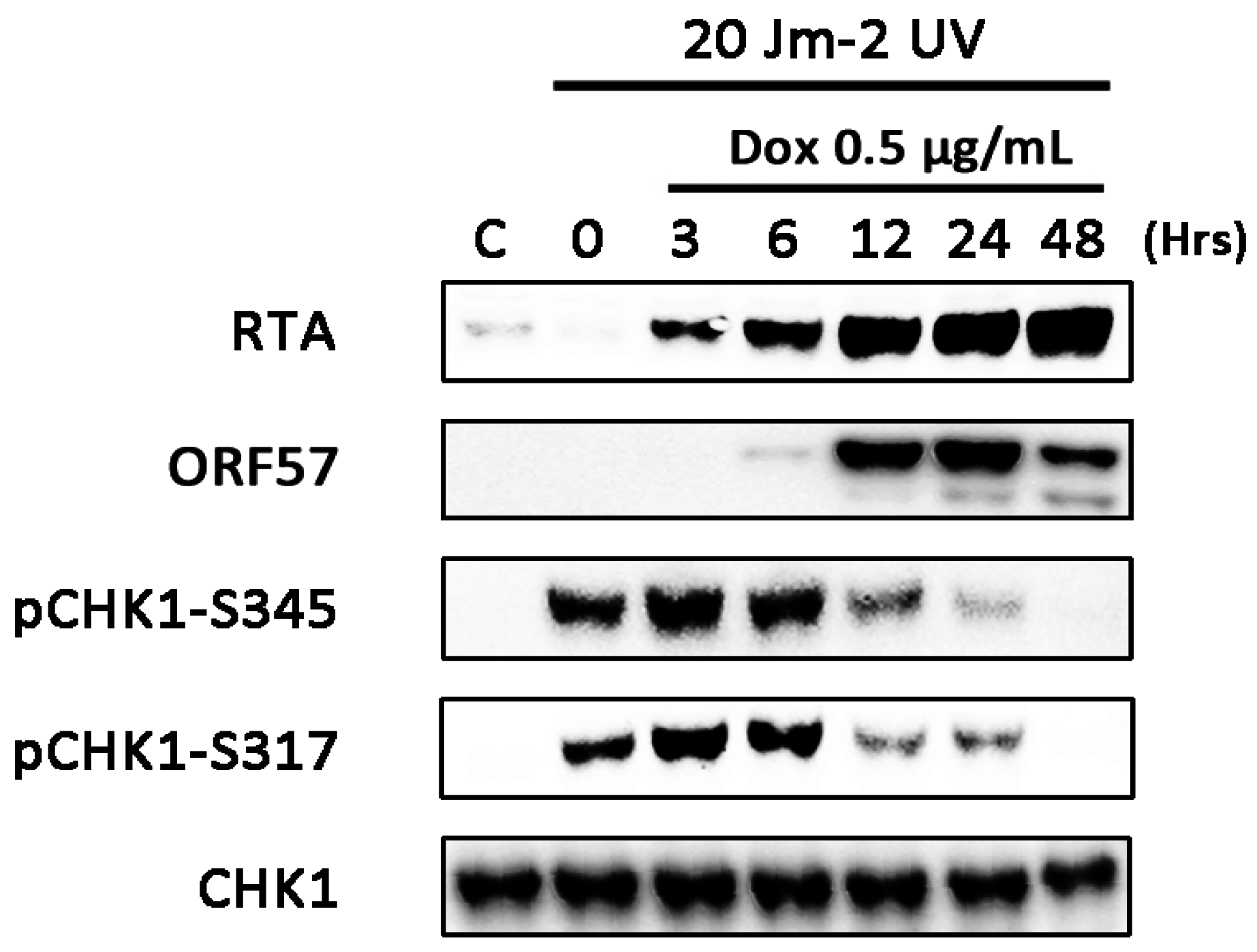
3.4. CHK1 Activation is Inhibited at Later Times during Lytic Replication

3.5. DDR Activation Does Not Result in a G1 Block Following Lytic Reactivation

3.6. RPA32 and MRE11 Localise to Sites of Viral Replication

3.7. Lytic Reactivation of KSHV in an Endothelial Cell Line Activates the DDR

4. Discussion
Acknowledgments
Author Contributions
Conflict of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in aids-associated kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in aids-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; D’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. Kshv reactivation and novel implications of protein isomerization on lytic switch control. Viruses 2015, 7, 72–109. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Kuppermann, B.D.; Wolitz, R.A.; Palestine, A.G.; Li, H.; Robinson, C.A. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche ganciclovir study group. N. Engl. J. Med. 1999, 340, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Ganem, D. Inefficient establishment of kshv latency suggests an additional role for continued lytic replication in kaposi sarcoma pathogenesis. J. Clin. Investig. 2004, 113, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-pk phosphorylation of rpa32 ser4/ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair Amst. 2014, 21, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of rpa-ssdna complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.; Chen, J. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003, 278, 30057–30062. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. Topbp1 activates the atr-atrip complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Dutta, D.; Ansari, M.A.; Dutta, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J. Virol. 2014, 88, 2821–2834. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, J.; Liao, Q.; Wu, Y.; Peng, C.; Chen, X. Lytic infection of kaposi’s sarcoma-associated herpesvirus induces DNA double-strand breaks and impairs non-homologous end joining. J. Gen. Virol. 2013, 94, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.R.; Noerenberg, M.; Whitehouse, A. A novel mechanism inducing genome instability in kaposi’s sarcoma-associated herpesvirus infected cells. PLoS Pathog. 2014, 10, e1004098. [Google Scholar] [CrossRef] [PubMed]
- Meerbrey, K.L.; Hu, G.; Kessler, J.D.; Roarty, K.; Li, M.Z.; Herschkowitz, J.I.; Burrows, A.E.; Fang, J.E.; Ciccia, A.; Sun, T.; et al. The pinducer lentiviral toolkit for inducible rna interference in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 3665–3670. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; O’Hearn, P.M. Use of the red fluorescent protein as a marker of kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology 2004, 325, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Lu, M.; Gwack, Y.; Souvlis, J.; Zeichner, S.L.; Jung, J.U. Global changes in kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible rta transactivator. J. Virol. 2003, 77, 4205–4220. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.; Boehme, R. Antiviral activity and mechanism of action of ganciclovir. Rev. Infect. Dis. 1988, 10, S490–S494. [Google Scholar] [CrossRef] [PubMed]
- Bresnahan, W.A.; Boldogh, I.; Thompson, E.A.; Albrecht, T. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late g1. Virology 1996, 224, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, A.; Iwahori, S.; Sato, Y.; Nakayama, S.; Isomura, H.; Murata, T.; Tsurumi, T. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in epstein-barr virus replication compartments. J. Virol. 2009, 83, 6641–6651. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Rosenke, K.; Czornak, K.; Fortunato, E.A. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007, 81, 1934–1950. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Dee, A.R.; Smith, S.; Schumacher, A.J.; Weller, S.K. Efficient herpes simplex virus 1 replication requires cellular atr pathway proteins. J. Virol. 2013, 87, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Tang, Q.; Maul, G.G.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-lyt-dependent DNA replication: Involvement of host cellular factors. J. Virol. 2008, 82, 2867–2882. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Henderson, C.; Adachi, Y. Phosphorylation and rapid relocalization of 53bp1 to nuclear foci upon DNA damage. Mol. Cell. Biol. 2001, 21, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.R.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and tip60 histone acetyltransferase to promote virus replication. Cell Host Microbe 2011, 10, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Hagemeier, S.R.; Barlow, E.A.; Meng, Q.; Kenney, S.C. The cellular ataxia telangiectasia-mutated kinase promotes epstein-barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J. Virol. 2012, 86, 13360–13370. [Google Scholar] [CrossRef] [PubMed]
- Hau, P.M.; Deng, W.; Jia, L.; Yang, J.; Tsurumi, T.; Chiang, A.K.; Huen, M.S.; Tsao, S.W. Role of atm in the formation of the replication compartment during lytic replication of epstein-barr virus in nasopharyngeal epithelial cells. J. Virol. 2015, 89, 652–668. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bekker-Jensen, S.; Mailand, N.; Lukas, C.; Bartek, J.; Lukas, J. Claspin operates downstream of TOPBP1 to direct atr signaling towards chk1 activation. Mol. Cell. Biol. 2006, 26, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-pk, atm and atr in rpa phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of atr in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Barton, O.; Noon, A.T.; Dahm, K.; Deckbar, D.; Goodarzi, A.A.; Lobrich, M.; Jeggo, P.A. Role of ATM and the damage response mediator proteins 53bp1 and mdc1 in the maintenance of G2/M checkpoint arrest. Mol. Cell. Biol. 2010, 30, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Jung, E.J.; Flemington, E.K. Cell cycle analysis of epstein-barr virus-infected cells following treatment with lytic cycle-inducing agents. J. Virol. 2001, 75, 4482–4489. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Tang, Q.Q.; Chen, H.; ApRhys, C.; Farrell, C.; Chen, J.; Fujimuro, M.; Lane, M.D.; Hayward, G.S. Lytic replication-associated protein (RAP) encoded by kaposi sarcoma-associated herpesvirus causes p21CIP-1-mediated G1 cell cycle arrest through ccaat/enhancer-binding protein-alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 10683–10688. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Lin, S.F.; Ellison, T.J.; Levy, A.M.; Mayeur, G.L.; Izumiya, C.; Kung, H.J. Cell cycle regulation by kaposi’s sarcoma-associated herpesvirus k-bzip: Direct interaction with cyclin-CDK2 and induction of G1 growth arrest. J. Virol. 2003, 77, 9652–9661. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wood, C. Kaposi’s sarcoma-associated herpesvirus transactivator rta induces cell cycle arrest in G0/G1 phase by stabilizing and promoting nuclear localization of p27kip. J. Virol. 2013, 87, 13226–13238. [Google Scholar] [CrossRef] [PubMed]
- Jarviluoma, A.; Koopal, S.; Rasanen, S.; Makela, T.P.; Ojala, P.M. Kshv viral cyclin binds to p27kip1 in primary effusion lymphomas. Blood 2004, 104, 3349–3354. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. Kshv 2.0: A comprehensive annotation of the kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Koopal, S.; Furuhjelm, J.H.; Jarviluoma, A.; Jaamaa, S.; Pyakurel, P.; Pussinen, C.; Wirzenius, M.; Biberfeld, P.; Alitalo, K.; Laiho, M.; et al. Viral oncogene-induced DNA damage response is activated in kaposi sarcoma tumorigenesis. PLoS Pathog. 2007, 3, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; Dyson, O.F.; Akula, S.M. Identifying cellular genes crucial for the reactivation of kaposi’s sarcoma-associated herpesvirus latency. J. Gen. Virol. 2006, 87, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an atr-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [PubMed]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced g2-m checkpoint activation by histone h2ax and 53bp1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.G.; Verrall, E.; Schelcher, C.; Rhie, A.; Doherty, A.J.; Sinclair, A.J. Functional interaction between epstein-barr virus replication protein zta and host DNA damage response protein 53bp1. J. Virol. 2009, 83, 11116–11122. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollingworth, R.; Skalka, G.L.; Stewart, G.S.; Hislop, A.D.; Blackbourn, D.J.; Grand, R.J. Activation of DNA Damage Response Pathways during Lytic Replication of KSHV. Viruses 2015, 7, 2908-2927. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062752
Hollingworth R, Skalka GL, Stewart GS, Hislop AD, Blackbourn DJ, Grand RJ. Activation of DNA Damage Response Pathways during Lytic Replication of KSHV. Viruses. 2015; 7(6):2908-2927. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062752
Chicago/Turabian StyleHollingworth, Robert, George L. Skalka, Grant S. Stewart, Andrew D. Hislop, David J. Blackbourn, and Roger J. Grand. 2015. "Activation of DNA Damage Response Pathways during Lytic Replication of KSHV" Viruses 7, no. 6: 2908-2927. https://0-doi-org.brum.beds.ac.uk/10.3390/v7062752