
Replication and Inhibitors of Enteroviruses and Parechoviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Enterovirus and Parechovirus Associated Diseases
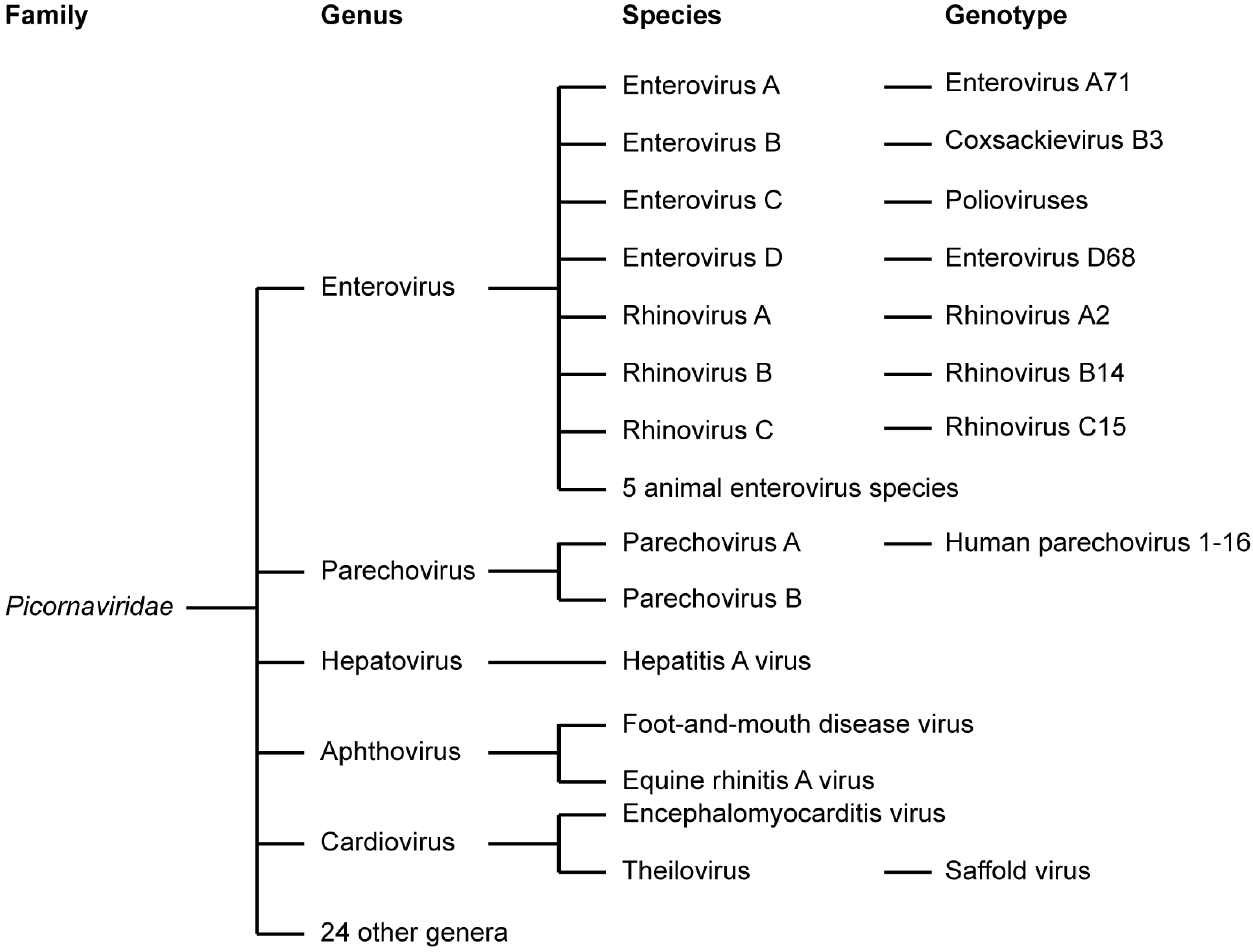
1.1. Picornaviridae

1.2. Enteroviruses
1.3. Parechoviruses
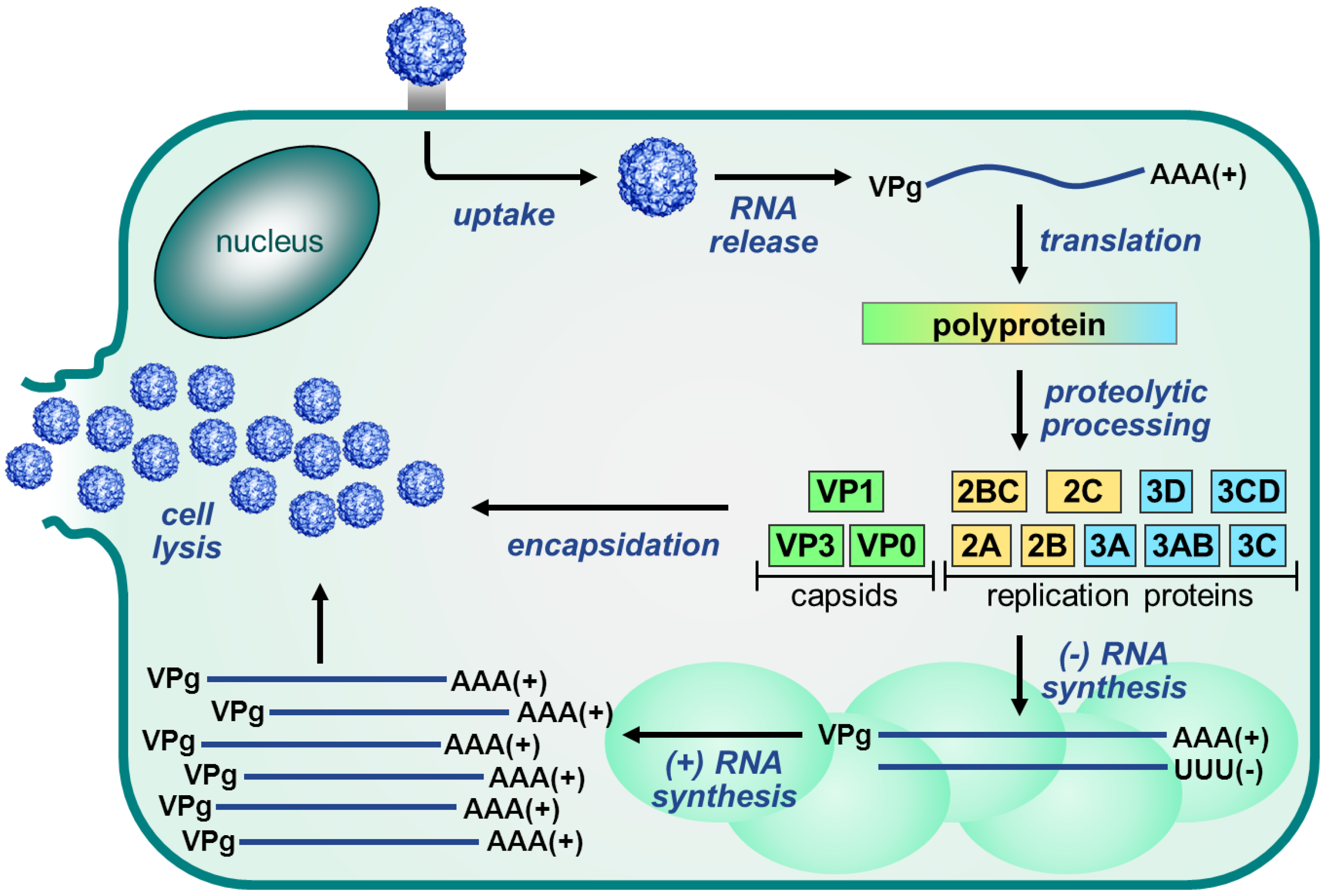
2. Enterovirus Replication Cycle
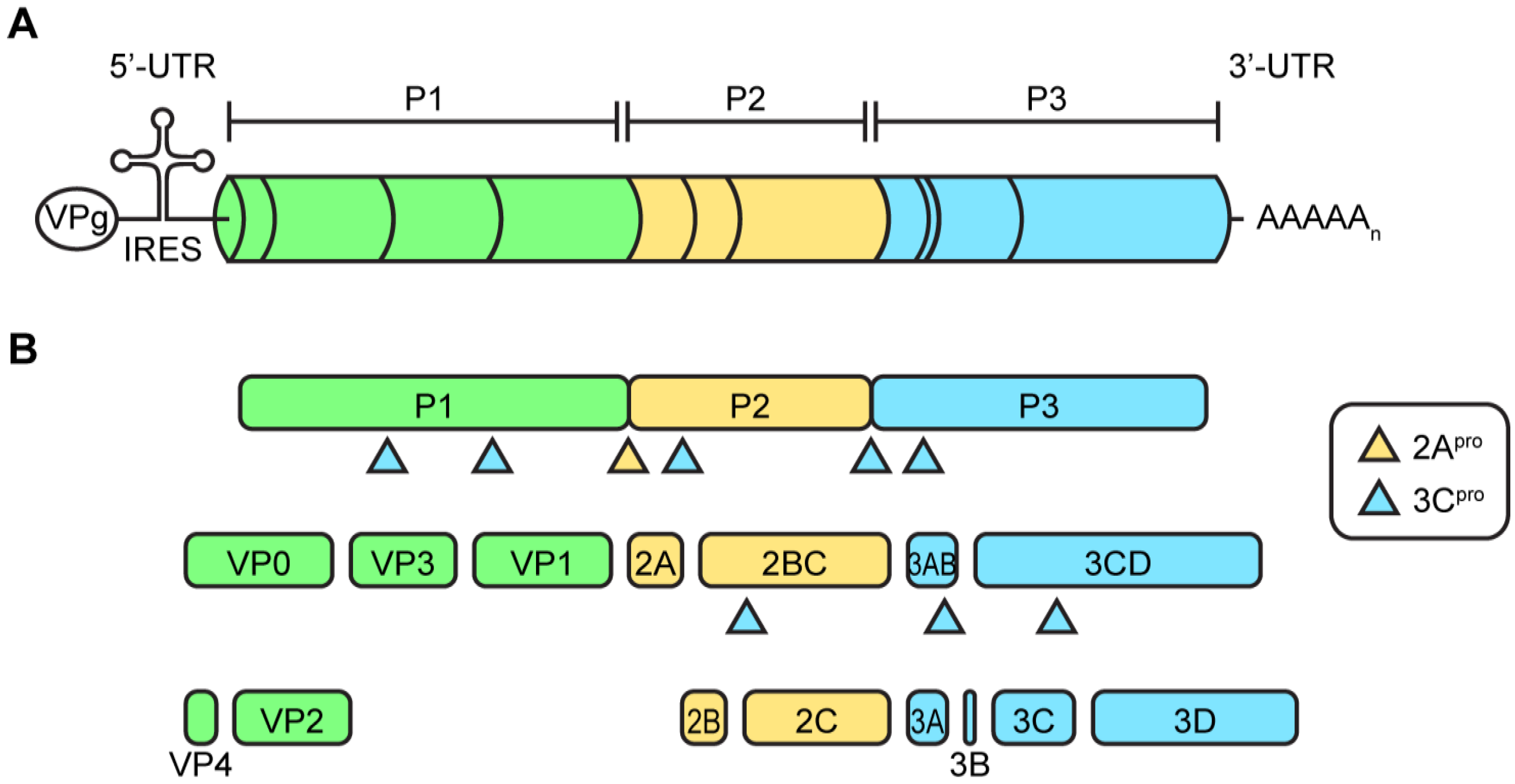
2.1. Enterovirus Virions and Viral Genome Organization

2.2. Protein Translation and Processing

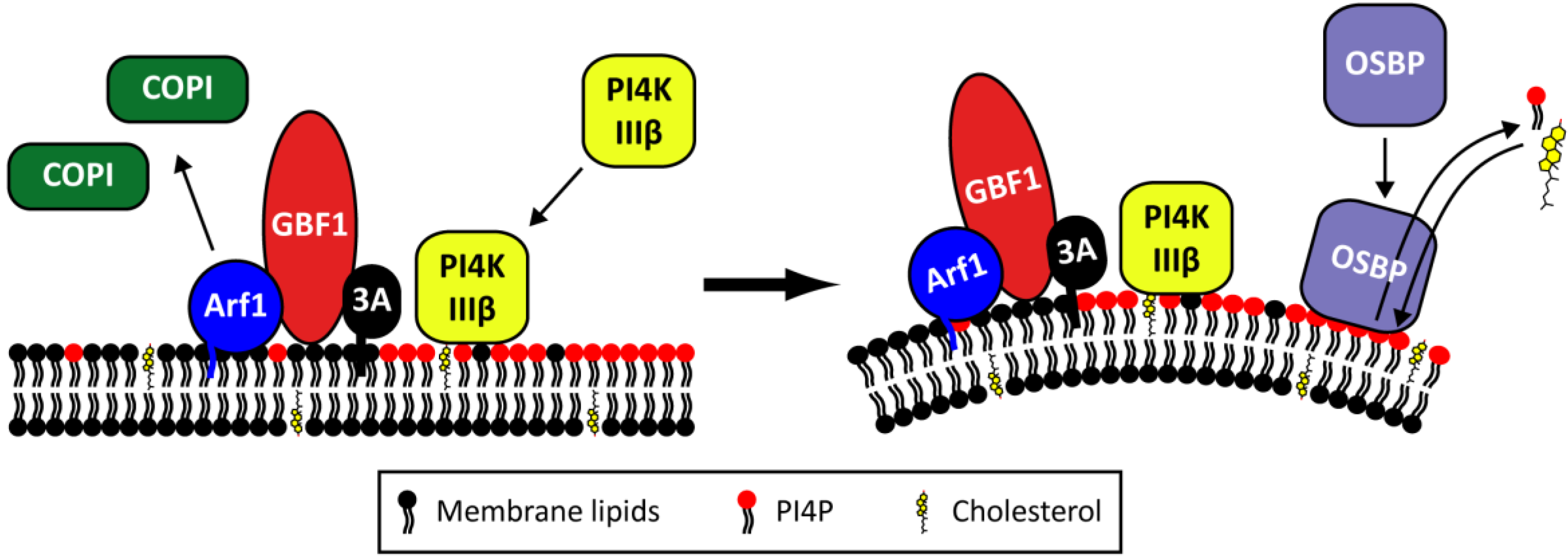
2.4. The Role of Viral Proteins and Host Factors in Membrane Rearrangements


2.5. Morphogenesis and Virus Release

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Adams, M.J.; King, A.M.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch. Virol. 2013, 158, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Knowles, N.J.; Hovi, T.; Hyypiä, T.; King, A.M.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Simmonds, P.; Skern, T.; Stanway, G.; et al. Picornaviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 855–880. [Google Scholar]
- Strikas, R.A.; Anderson, L.J.; Parker, R.A. Temporal and geographic patterns of isolates of nonpolio enterovirus in the United States, 1970–1983. J. Infect. Dis. 1986, 153, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Poliomyelitis. In Epidemiology and Prevention of Vaccine-Preventable Diseases; Atkinson, W., Hamborsky, J., Wolfe, S., Eds.; Public Health Foundation: Washington DC, 2012; Volume 12, pp. 249–261. [Google Scholar]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Rotbart, H.A. Viral meningitis. Semin. Neurol. 2000, 20, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Ooi, M.H.; Wong, S.C.; Lewthwaite, P.; Cardosa, M.J.; Solomon, T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010, 9, 1097–1105. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Messacar, K.; Clayton, A.; Yu, G.; Somasekar, S.; Federman, S.; Stryke, D.; Anderson, C.; Yagi, S.; et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012–14): A retrospective cohort study. Lancet. Infect. Dis. 2015, 15, 671–682. [Google Scholar] [CrossRef]
- Meijer, A.; Benschop, K.S.; Donker, G.A.; van der Avoort, H.G. Continued seasonal circulation of enterovirus D68 in the Netherlands, 2011–2014. Euro Surveill. 2014, 19, art 1. [Google Scholar] [CrossRef]
- Midgley, C.M.; Jackson, M.A.; Selvarangan, R.; Turabelidze, G.; Obringer, E.; Johnson, D.; Giles, B.L.; Patel, A.; Echols, F.; Oberste, M.S.; et al. Severe respiratory illness associated with enterovirus D68—Missouri and Illinois, 2014. MMWR. Morb. Mortal. Wkly. Rep. 2014, 63, 798–799. [Google Scholar] [PubMed]
- Bragstad, K.; Jakobsen, K.; Rojahn, A.E.; Skram, M.K.; Vainio, K.; Holberg-Petersen, M.; Hungnes, O.; Dudman, S.G.; Kran, A.-M.B. High frequency of enterovirus D68 in children hospitalised with respiratory illness in Norway, Autumn 2014. Influenza Other Respi. Viruses 2015, 9, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Messacar, K.; Schreiner, T.L.; Maloney, J.A.; Wallace, A.; Ludke, J.; Oberste, M.S.; Nix, W.A.; Robinson, C.C.; Glodé, M.P.; Abzug, M.J.; et al. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet 2015, 385, 1662–1671. [Google Scholar] [CrossRef]
- Fendrick, A.M.; Monto, A.S.; Nightengale, B.; Sarnes, M. The economic burden of non-influenza-related viral respiratory tract infection in the United States. Arch. Intern. Med. 2003, 163, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, G.; Lerikou, M.; Tsiodras, S.; Chranioti, A.; Perros, E.; Anagnostopoulou, U.; Armaganidis, A.; Karakitsos, P. Viral epidemiology of acute exacerbations of chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2012, 25, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Gern, J.E. The ABCs of rhinoviruses, wheezing, and asthma. J. Virol. 2010, 84, 7418–7426. [Google Scholar] [CrossRef] [PubMed]
- Kherad, O.; Kaiser, L.; Bridevaux, P.-O.; Sarasin, F.; Thomas, Y.; Janssens, J.-P.; Rutschmann, O.T. Upper-respiratory viral infection, biomarkers, and COPD exacerbations. Chest 2010, 138, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Mallia, P.; Message, S.D.; Kebadze, T.; Parker, H.L.; Kon, O.M.; Johnston, S.L. An experimental model of rhinovirus induced chronic obstructive pulmonary disease exacerbations: A pilot study. Respir. Res. 2006, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- McManus, T.E.; Marley, A.-M.; Baxter, N.; Christie, S.N.; O’Neill, H.J.; Elborn, J.S.; Coyle, P.V.; Kidney, J.C. Respiratory viral infection in exacerbations of COPD. Respir. Med. 2008, 102, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.G.; Christodoulou, I.; Rohde, G.; Agache, I.; Almqvist, C.; Bruno, A.; Bonini, S.; Bont, L.; Bossios, A.; Bousquet, J.; et al. Viruses and bacteria in acute asthma exacerbations—A GA2 LEN-DARE systematic review. Allergy 2011, 66, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Seemungal, T.A.; Harper-Owen, R.; Bhowmik, A.; Jeffries, D.J.; Wedzicha, J.A. Detection of rhinovirus in induced sputum at exacerbation of chronic obstructive pulmonary disease. Eur. Respir. J. 2000, 16, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.L.; Emerson, J.; Kuypers, J.; Campbell, A.P.; Gibson, R.L.; McNamara, S.; Worrell, K.; Englund, J.A. Respiratory viruses in children with cystic fibrosis: viral detection and clinical findings. Influenza Other Respi. Viruses 2012, 6, 218–223. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, M.B.; Zerbinati, R.M.; Tateno, A.F.; Oliveira, C.M.; Romão, R.M.; Rodrigues, J.C.; Pannuti, C.S.; da Silva Filho, L.V.F. Rhinovirus C and respiratory exacerbations in children with cystic fibrosis. Emerg. Infect. Dis. 2010, 16, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Kieninger, E.; Singer, F.; Tapparel, C.; Alves, M.P.; Latzin, P.; Tan, H.-L.; Bossley, C.; Casaulta, C.; Bush, A.; Davies, J.C.; et al. High rhinovirus burden in lower airways of children with cystic fibrosis. Chest 2013, 143, 782–790. [Google Scholar] [PubMed]
- Wat, D.; Gelder, C.; Hibbitts, S.; Cafferty, F.; Bowler, I.; Pierrepoint, M.; Evans, R.; Doull, I. The role of respiratory viruses in cystic fibrosis. J. Cyst. Fibros. 2008, 7, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Turner, R.B. Rhinovirus genetics and virulence: looking for needles in a haystack. Am. J. Respir. Crit. Care Med. 2012, 186, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Joki-Korpela, P.; Hyypiä, T. Parechoviruses, a novel group of human picornaviruses. Ann. Med. 2001, 33, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Stanway, G.; Joki-Korpela, P.; Hyypiä, T. Human parechoviruses—Biology and clinical significance. Rev. Med. Virol. 2000, 10, 57–69. [Google Scholar] [CrossRef]
- Stanway, G.; Brown, F.; Christian, P.; Hovi, T.; Hyypiä, T.; King, A.M.Q.; Knowles, N.J.; Lemon, S.M.; Minor, P.D.; Pallansch, M.A.; et al. Family Picornaviridae. In Virus Taxonomy. Eighth Report of the International Committee on Taxonomy of Viruses; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier/Academic Press: London, UK, 2005; pp. 757–778. [Google Scholar]
- Benschop, K.; Thomas, X.; Serpenti, C.; Molenkamp, R.; Wolthers, K. High prevalence of human Parechovirus (HPeV) genotypes in the Amsterdam region and identification of specific HPeV variants by direct genotyping of stool samples. J. Clin. Microbiol. 2008, 46, 3965–3970. [Google Scholar] [CrossRef] [PubMed]
- Khatami, A.; McMullan, B.J.; Webber, M.; Stewart, P.; Francis, S.; Timmers, K.J.; Rodas, E.; Druce, J.; Mehta, B.; Sloggett, N.A.; et al. Sepsis-like disease in infants due to human parechovirus type 3 during an outbreak in Australia. Clin. Infect. Dis. 2015, 60, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Wildenbeest, J.G.; Wolthers, K.C.; Straver, B.; Pajkrt, D. Successful IVIG treatment of human parechovirus-associated dilated cardiomyopathy in an infant. Pediatrics 2013, 132, e243–e247. [Google Scholar] [CrossRef] [PubMed]
- Benschop, K.S.; Schinkel, J.; Minnaar, R.P.; Pajkrt, D.; Spanjerberg, L.; Kraakman, H.C.; Berkhout, B.; Zaaijer, H.L.; Beld, M.G.H.M.; Wolthers, K.C. Human parechovirus infections in Dutch children and the association between serotype and disease severity. Clin. Infect. Dis. 2006, 42, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Harvala, H.; Robertson, I.; Chieochansin, T.; McWilliam Leitch, E.C.; Templeton, K.; Simmonds, P. Specific association of human parechovirus type 3 with sepsis and fever in young infants, as identified by direct typing of cerebrospinal fluid samples. J. Infect. Dis. 2009, 199, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Wolthers, K.C.; Benschop, K.S.M.; Schinkel, J.; Molenkamp, R.; Bergevoet, R.M.; Spijkerman, I.J.B.; Kraakman, H.C.; Pajkrt, D. Human parechoviruses as an important viral cause of sepsislike illness and meningitis in young children. Clin. Infect. Dis. 2008, 47, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Abed, Y.; Boucher, F.D. Human parechovirus 3 and neonatal infections. Emerg. Infect. Dis. 2005, 11, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Renaud, C.; Kuypers, J.; Ficken, E.; Cent, A.; Corey, L.; Englund, J.A. Introduction of a novel parechovirus RT-PCR clinical test in a regional medical center. J. Clin. Virol. 2011, 51, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Sainato, R.; Flanagan, R.; Mahlen, S.; Fairchok, M.; Braun, L. Severe human parechovirus sepsis beyond the neonatal period. J. Clin. Virol. 2011, 51, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Schuffenecker, I.; Javouhey, E.; Gillet, Y.; Kugener, B.; Billaud, G.; Floret, D.; Lina, B.; Morfin, F. Human parechovirus infections, Lyon, France, 2008-10: evidence for severe cases. J. Clin. Virol. 2012, 54, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Selvarangan, R.; Nzabi, M.; Selvaraju, S.B.; Ketter, P.; Carpenter, C.; Harrison, C.J. Human parechovirus 3 causing sepsis-like illness in children from midwestern United States. Pediatr. Infect. Dis. J. 2011, 30, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Verboon-Maciolek, M.A.; Groenendaal, F.; Hahn, C.D.; Hellmann, J.; van Loon, A.M.; Boivin, G.; de Vries, L.S. Human parechovirus causes encephalitis with white matter injury in neonates. Ann. Neurol. 2008, 64, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Walters, B.; Peñaranda, S.; Nix, W.A.; Oberste, M.S.; Todd, K.M.; Katz, B.Z.; Zheng, X. Detection of human parechovirus (HPeV)-3 in spinal fluid specimens from pediatric patients in the Chicago area. J. Clin. Virol. 2011, 52, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Gonzalez, R.; Xie, Z.; Xiao, Y.; Liu, J.; Chen, L.; Liu, C.; Zhang, J.; Ren, L.; Vernet, G.; et al. Human rhinovirus C infections mirror those of human rhinovirus A in children with community-acquired pneumonia. J. Clin. Virol. 2010, 49, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Yuzurihara, S.S.; Ao, K.; Hara, T.; Tanaka, F.; Mori, M.; Kikuchi, N.; Kai, S.; Yokota, S. Human parechovirus-3 infection in nine neonates and infants presenting symptoms of hemophagocytic lymphohistiocytosis. J. Infect. Chemother. 2013, 19, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.A.; Diop, O.M.; Burns, C.C.; Tangermann, R.H.; Wassilak, S.G.F. Tracking progress toward polio eradication—Worldwide, 2013–2014. MMWR. Morb. Mortal. Wkly. Rep. 2015, 64, 415–420. [Google Scholar] [PubMed]
- Bejing Vigoo Biological Co., Ltd. A clinical trial to assess the efficacy and safety of an inactivated vaccine (vero cell) against EV71 in Chinese children aged 6–35 months. Available online: https://clinicaltrials.gov/ct2/show/NCT01508247 (accessed on 27 March 2015).
- Longding Liu, Chinese Academy of Medical Sciences. A protected study of inactivated EV71 vaccine (human diploid cell, KMB-17) in Chinese infants and children. Available online: https://clinicaltrials.gov/ct2/show/NCT01569581 (accessed on 10 March 2015).
- Sinovac Biotech Co., Ltd. An Efficacy Trial in inactivated enterovirus type 71 (EV71) vaccine. Available online: https://clinicaltrials.gov/ct2/show/NCT01507857 (accessed on 27 March 2015).
- Abzug, M.J.; Keyserling, H.L.; Lee, M.L.; Levin, M.J.; Rotbart, H.A. Neonatal enterovirus infection: virology, serology, and effects of intravenous immune globulin. Clin. Infect. Dis. 1995, 20, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Nagington, J. Echovirus 11 infection and prophylactic antiserum. Lancet 1982, 1, 446. [Google Scholar] [CrossRef]
- Wildenbeest, J.G.; van den Broek, P.J.; Benschop, K.S.M.; Koen, G.; Wierenga, P.C.; Vossen, A.C.; Kuijpers, T.W.; Wolthers, K.C. Pleconaril revisited: clinical course of chronic enteroviral meningoencephalitis after treatment correlates with in vitro susceptibility. Antivir. Ther. 2012, 17, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.-H.; Huang, Y.-C.; Chen, M.-C.; Liu, C.-C.; Chiu, N.-C.; Lien, R.; Chang, L.-Y.; Chiu, C.-H.; Tsao, K.-C.; Lin, T.-Y. Effect of intravenous immunoglobulin for neonates with severe enteroviral infections with emphasis on the timing of administration. J. Clin. Virol. 2015, 64, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Enserink, M. Polio endgame. Wanted: Drug for a disappearing disease. Science 2004, 303. [Google Scholar] [CrossRef] [PubMed]
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 43–89. [Google Scholar] [PubMed]
- Kräusslich, H.G.; Nicklin, M.J.; Toyoda, H.; Etchison, D.; Wimmer, E. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol. 1987, 61, 2711–2718. [Google Scholar] [PubMed]
- Lloyd, R.E.; Grubman, M.J.; Ehrenfeld, E. Relationship of p220 cleavage during picornavirus infection to 2A proteinase sequencing. J. Virol. 1988, 62, 4216–4223. [Google Scholar] [PubMed]
- Joachims, M.; Van Breugel, P.C.; Lloyd, R.E. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 1999, 73, 718–727. [Google Scholar] [PubMed]
- Badorff, C.; Lee, G.H.; Lamphear, B.J.; Martone, M.E.; Campbell, K.P.; Rhoads, R.E.; Knowlton, K.U. Enteroviral protease 2A cleaves dystrophin: Evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 1999, 5, 320–326. [Google Scholar] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Castelló, A.; Alvarez, E.; Carrasco, L. The multifaceted poliovirus 2A protease: Regulation of gene expression by picornavirus proteases. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Gerber, K.; Wimmer, E.; Paul, A.V. Biochemical and genetic studies of the initiation of human rhinovirus 2 RNA replication: Identification of a cis-replicating element in the coding sequence of 2A(pro). J. Virol. 2001, 75, 10979–10990. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V.; Rieder, E.; Kim, D.W.; van Boom, J.H.; Wimmer, E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 2000, 74, 10359–10370. [Google Scholar] [CrossRef] [PubMed]
- Rieder, E.; Paul, A.V.; Kim, D.W.; van Boom, J.H.; Wimmer, E. Genetic and biochemical studies of poliovirus cis-acting replication element cre in relation to VPg uridylylation. J. Virol. 2000, 74, 10371–10380. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rijnbrand, R.; McKnight, K.L.; Wimmer, E.; Paul, A.; Martin, A.; Lemon, S.M. Sequence requirements for viral RNA replication and VPg uridylylation directed by the internal cis-acting replication element (cre) of human rhinovirus type 14. J. Virol. 2002, 76, 7485–7494. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; van Kuppeveld, F.J. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2012, 2, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Bárcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio 2011. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Feng, Q.; Nikovics, K.; Jackson, C.L.; Ehrenfeld, E. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 2008, 4, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Gazina, E.V.; Mackenzie, J.M.; Gorrell, R.J.; Anderson, D.A. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J. Virol. 2002, 76, 11113–11122. [Google Scholar] [CrossRef] [PubMed]
- Irurzun, A.; Perez, L.; Carrasco, L. Involvement of membrane traffic in the replication of poliovirus genomes: effects of brefeldin A. Virology 1992, 191, 166–175. [Google Scholar] [CrossRef]
- Lanke, K.H.; van der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; van Kuppeveld, F.J. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef] [PubMed]
- Wessels, E.; Duijsings, D.; Niu, T.-K.; Neumann, S.; Oorschot, V.M.; de Lange, F.; Lanke, K.H.; Klumperman, J.; Henke, A.; Jackson, C.L.; et al. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell 2006, 11, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Wessels, E.; Duijsings, D.; Lanke, K.H.; van Dooren, S.H.J.; Jackson, C.L.; Melchers, W.J.; van Kuppeveld, F.J. Effects of picornavirus 3A Proteins on Protein Transport and GBF1-dependent COP-I recruitment. J. Virol. 2006, 80, 11852–11860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, N.-Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Doedens, J.R.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [PubMed]
- Wessels, E.; Duijsings, D.; Notebaart, R.A.; Melchers, W.J.; van Kuppeveld, F.J. A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-golgi transport. J. Virol. 2005, 79, 5163–5173. [Google Scholar] [CrossRef] [PubMed]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.A.; Giddings, T.H.; Kirkegaard, K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 2001, 75, 8158–8165. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Vicinanza, M.; Di Campli, A.; De Matteis, M.A. The multiple roles of PtdIns(4)P—Not just the precursor of PtdIns(4,5)P2. J. Cell Sci. 2008, 121, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Burd, C.G. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011, 21, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Tirado, F.H.; Bretscher, A. Membrane-trafficking sorting hubs: cooperation between PI4P and small GTPases at the trans-Golgi network. Trends Cell Biol. 2011, 21, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Dorobantu, C.M.; van der Schaar, H.M.; Ford, L.A.; Strating, J.R.; Ulferts, R.; Fang, Y.; Belov, G.; van Kuppeveld, F.J. Recruitment of PI4KIIIβ to coxsackievirus B3 replication organelles is independent of ACBD3, GBF1, and Arf1. J. Virol. 2014, 88, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Téoulé, F.; Brisac, C.; Pelletier, I.; Vidalain, P.-O.; Jégouic, S.; Mirabelli, C.; Bessaud, M.; Combelas, N.; Autret, A.; Tangy, F.; et al. The Golgi protein ACBD3, an interactor for poliovirus protein 3A, modulates poliovirus replication. J. Virol. 2013, 87, 11031–11046. [Google Scholar] [CrossRef] [PubMed]
- Mesmin, B.; Bigay, J.; Moser von Filseck, J.; Lacas-Gervais, S.; Drin, G.; Antonny, B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 2013, 155, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Strating, J.R.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanke, K.H.; Thibaut, H.J.; et al. Itraconazole Inhibits Enterovirus Replication by Targeting the Oxysterol-Binding Protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014, 58, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Roulin, P.S.; Lötzerich, M.; Torta, F.; Tanner, L.B.; van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, L.; Wubbolts, R.; van Kuppeveld, F.J.; Strating, J.R. Cholesterol shuttling is important for RNA replication of coxsackievirus B3 and encephalomyocarditis virus. Cell. Microbiol. 2015, 17, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Ilnytska, O.; Santiana, M.; Hsu, N.-Y.; Du, W.-L.; Chen, Y.-H.; Viktorova, E.G.; Belov, G.; Brinker, A.; Storch, J.; Moore, C.; et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013, 14, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T.; Giddings, T.H.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, A.L.; Jackson, W.T. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 2012, 8, e1003046. [Google Scholar] [CrossRef] [PubMed]
- Barco, A.; Carrasco, L. A human virus protein, poliovirus protein 2BC, induces membrane proliferation and blocks the exocytic pathway in the yeast Saccharomyces cerevisiae. EMBO J. 1995, 14, 3349–3364. [Google Scholar] [PubMed]
- Cho, M.W.; Teterina, N.; Egger, D.; Bienz, K.; Ehrenfeld, E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 1994, 202, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Agirre, A.; Barco, A.; Carrasco, L.; Nieva, J.L. Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. J. Biol. Chem. 2002, 277, 40434–40441. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; Wessels, E.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the Golgi complex as the target organelle. J. Biol. Chem. 2003, 278, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; Visch, H.-J.; de Mattia, F.; van Dommelen, M.M.; Swarts, H.G.; Luyten, T.; Callewaert, G.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. The Coxsackievirus 2B Protein Increases Efflux of Ions from the Endoplasmic Reticulum and Golgi, thereby Inhibiting Protein Trafficking through the Golgi. J. Biol. Chem. 2006, 281, 14144–14150. [Google Scholar] [CrossRef] [PubMed]
- Van Kuppeveld, F.J.; Hoenderop, J.G.; Smeets, R.L.; Willems, P.H.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 1997, 16, 3519–3532. [Google Scholar] [CrossRef] [PubMed]
- Aldabe, R.; Carrasco, L. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem. Biophys. Res. Commun. 1995, 206, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Van Kuppeveld, F.J.; Melchers, W.J.; Kirkegaard, K.; Doedens, J.R. Structure-function analysis of coxsackie B3 virus protein 2B. Virology 1997, 227, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Cornell, C.T.; Kiosses, W.B.; Harkins, S.; Whitton, J.L. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J. Virol. 2007, 81, 6785–6797. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.-F.; Yang, S.-Y.; Wu, B.-W.; Jheng, J.-R.; Chen, Y.-L.; Shih, C.-H.; Lin, K.-H.; Lai, H.-C.; Tang, P.; Horng, J.-T. Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. J. Biol. Chem. 2007, 282, 5888–5898. [Google Scholar] [CrossRef] [PubMed]
- Molla, A.; Paul, A.V.; Wimmer, E. Cell-free, de novo synthesis of poliovirus. Science 1991, 254, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Nugent, C.I.; Johnson, K.L.; Sarnow, P.; Kirkegaard, K. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 1999, 73, 427–435. [Google Scholar] [PubMed]
- Liu, Y.; Wang, C.; Mueller, S.; Paul, A.V.; Wimmer, E.; Jiang, P. Direct interaction between two viral proteins, the nonstructural protein 2C and the capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog. 2010, 6, e1001066. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.; Vignuzzi, M.; Andino, R.; Frydman, J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007, 21, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Ypma-Wong, M.F.; Dewalt, P.G.; Johnson, V.H.; Lamb, J.G.; Semler, B.L. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 1988, 166, 265–270. [Google Scholar] [CrossRef]
- Mikami, T.; Satoh, N.; Hatayama, I.; Nakane, A. Buthionine sulfoximine inhibits cytopathic effect and apoptosis induced by infection with human echovirus 9. Arch. Virol. 2004, 149, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Dawson, H. Glutathione is required for efficient production of infectious picornavirus virions. Virology 2006, 353, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Thibaut, H.J.; van der Linden, L.; Jiang, P.; Thys, B.; Canela, M.-D.; Aguado, L.; Rombaut, B.; Wimmer, E.; Paul, A.; Pérez-Pérez, M.-J.; et al. Binding of glutathione to enterovirus capsids is essential for virion morphogenesis. PLoS Pathog. 2014, 10, e1004039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.-C.; Liu, Y.; Wang, C.; Strauss, M.; Rehage, N.; Chen, Y.-H.; Altan-Bonnet, N.; Hogle, J.; Wimmer, E.; Mueller, S.; et al. An interaction between glutathione and the capsid is required for the morphogenesis of C-cluster enteroviruses. PLoS Pathog. 2014, 10, e1004052. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.-C.; et al. Phosphatidylserine Vesicles Enable Efficient En Bloc Transmission of Enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.-H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Abzug, M.J.; Cloud, G.; Bradley, J.; Sánchez, P.J.; Romero, J.; Powell, D.; Lepow, M.; Mani, C.; Capparelli, E.V.; Blount, S.; et al. Double blind placebo-controlled trial of pleconaril in infants with enterovirus meningitis. Pediatr. Infect. Dis. J. 2003, 22, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Desmond, R.A.; Accortt, N.A.; Talley, L.; Villano, S.A.; Soong, S.-J.; Whitley, R.J. Enteroviral meningitis: Natural history and outcome of pleconaril therapy. Antimicrob. Agents Chemother. 2006, 50, 2409–2414. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Coats, T.; Kim, K.; Hassman, H.A.; Blatter, M.M.; Zhang, B.; Liu, S. Oral pleconaril treatment of picornavirus-associated viral respiratory illness in adults: Efficacy and tolerability in phase II clinical trials. Antivir. Ther. 2002, 7, 53–65. [Google Scholar] [PubMed]
- Hayden, F.G.; Herrington, D.T.; Coats, T.L.; Kim, K.; Cooper, E.C.; Villano, S.A.; Liu, S.; Hudson, S.; Pevear, D.C.; Collett, M.; et al. Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: Results of 2 double-blind, randomized, placebo-controlled trials. Clin. Infect. Dis. 2003, 36, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Rotbart, H.A.; Webster, A.D. Treatment of potentially life-threatening enterovirus infections with pleconaril. Clin. Infect. Dis. 2001, 32, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Schiff, G.M.; Sherwood, J.R. Clinical activity of pleconaril in an experimentally induced coxsackievirus A21 respiratory infection. J. Infect. Dis. 2000, 181, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Senior, K. FDA panel rejects common cold treatment. Lancet. Infect. Dis. 2002, 2, 264. [Google Scholar] [CrossRef]
- Merck Sharp & Dohme Corp. Effects of Pleconaril Nasal Spray on Common Cold Symptoms and Asthma Exacerbations Following Rhinovirus Exposure. Available online: https://clinicaltrials.gov/ct2/show/NCT00394914 (accessed on 23 June 2015).
- Pleconaril Enteroviral Sepsis Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT00031512 (accessed on 28 February 2013).
- Biota Pharmaceuticals HRV Phase IIb Study Achieves Primary Endpoint. Available online: http://www.biota.com.au/uploaded/154/1021819_20hrvphaseiibstudyachieve.pdf (accessed on 28 March 2015).
- Biota Pharmaceuticals Biota Commences Dosing in Vapendavir SPIRITUS Phase 2b Trial. Available online: http://investors.biotapharma.com/releasedetail.cfm?releaseid=899451 (accessed on 28 March 2015).
- Oberste, M.S.; Moore, D.; Anderson, B.; Pallansch, M.A.; Pevear, D.C.; Collett, M.S. In vitro antiviral activity of V-073 against polioviruses. Antimicrob. Agents Chemother. 2009, 53, 4501–4503. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, P.J.; Cox, S.; Wright-Minogue, J.; DeMartino, J.L.; Skelton, A.M.; Ferrari, E.; Albin, R.; Rozhon, E.J.; Girijavallabhan, V.; Modlin, J.F.; et al. SCH 48973: A potent, broad-spectrum, antienterovirus compound. Antimicrob. Agents Chemother. 1997, 41, 1220–1225. [Google Scholar] [PubMed]
- Torres-Torres, S.; Myers, A.L.; Klatte, J.M.; Rhoden, E.E.; Oberste, M.S.; Collett, M.S.; McCulloh, R.J. First use of investigational antiviral drug pocapavir (v-073) for treating neonatal enteroviral sepsis. Pediatr. Infect. Dis. J. 2015, 34, 52–54. [Google Scholar] [CrossRef] [PubMed]
- De Palma, A.M.; Vliegen, I.; De Clercq, E.; Neyts, J. Selective inhibitors of picornavirus replication. Med. Res. Rev. 2008, 28, 823–884. [Google Scholar] [CrossRef] [PubMed]
- Pevear, D.C.; Hayden, F.G.; Demenczuk, T.M.; Barone, L.R.; McKinlay, M.A.; Collett, M.S. Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses. Antimicrob. Agents Chemother. 2005, 49, 4492–4499. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, L.; Crump, C.E.; Hayden, F.G. In vitro activity of pleconaril and AG7088 against selected serotypes and clinical isolates of human rhinoviruses. Antivir. Res. 2000, 47, 215–220. [Google Scholar] [CrossRef]
- Ledford, R.M.; Patel, N.R.; Demenczuk, T.M.; Watanyar, A.; Herbertz, T.; Collett, M.S.; Pevear, D.C. VP1 sequencing of all human rhinovirus serotypes: Insights into genus phylogeny and susceptibility to antiviral capsid-binding compounds. J. Virol. 2004, 78, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Pevear, D.C.; Tull, T.M.; Seipel, M.E.; Groarke, J.M. Activity of pleconaril against enteroviruses. Antimicrob. Agents Chemother. 1999, 43, 2109–2115. [Google Scholar] [PubMed]
- Benschop, K.S.; Wildenbeest, J.G.; Koen, G.; Minnaar, R.P.; van Hemert, F.J.; Westerhuis, B.M.; Pajkrt, D.; van den Broek, P.J.; Vossen, A.C.; Wolthers, K.C. Genetic and antigenic structural characterization for resistance of echovirus 11 to pleconaril in an immunocompromised patient. J. Gen. Virol. 2015, 96, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Webber, S.E.; Marakovits, J.T.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Lee, C.A.; Ford, C.E.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements. J. Med. Chem. 1999, 42, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- De Palma, A.M.; Pürstinger, G.; Wimmer, E.; Patick, A.K.; Andries, K.; Rombaut, B.; De Clercq, E.; Neyts, J. Potential use of antiviral agents in polio eradication. Emerg. Infect. Dis. 2008, 14, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Shih, S.-R.; Chang, T.-Y.; Tseng, H.-Y.; Shih, Y.-F.; Yen, K.-J.; Chen, W.-C.; Shie, J.-J.; Fang, J.-M.; Liang, P.-H.; et al. A mammalian cell-based reverse two-hybrid system for functional analysis of 3C viral protease of human enterovirus 71. Anal. Biochem. 2008, 375, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.K.; Binford, S.L.; Brothers, M.A.; Jackson, R.L.; Ford, C.E.; Diem, M.D.; Maldonado, F.; Dragovich, P.S.; Zhou, R.; Prins, T.J.; et al. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 1999, 43, 2444–2450. [Google Scholar] [PubMed]
- Tsai, M.-T.; Cheng, Y.-H.; Liu, Y.-N.; Liao, N.-C.; Lu, W.-W.; Kung, S.-H. Real-time monitoring of human enterovirus (HEV)-infected cells and anti-HEV 3C protease potency by fluorescence resonance energy transfer. Antimicrob. Agents Chemother. 2009, 53, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.K.; Brothers, M.A.; Maldonado, F.; Binford, S.; Maldonado, O.; Fuhrman, S.; Petersen, A.; Smith, G.J.; Zalman, L.S.; Burns-Naas, L.A.; et al. In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 2005, 49, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Johnson, T.O.; Hua, Y.; Luu, H.T.; Sakata, S.K.; Brown, E.L.; Maldonado, F.C.; Tuntland, T.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 8. Pharmacological optimization of orally bioavailable 2-pyridone-containing peptidomimetics. J. Med. Chem. 2003, 46, 4572–4585. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [PubMed]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 2001, 98, 6895–6900. [Google Scholar] [CrossRef] [PubMed]
- Gazina, E.V.; Smidansky, E.D.; Holien, J.K.; Harrison, D.N.; Cromer, B.A.; Arnold, J.J.; Parker, W.W.; Cameron, C.E.; Petrou, S. Amiloride is a competitive inhibitor of coxsackievirus B3 RNA polymerase. J. Virol. 2011, 85, 10364–10374. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, L.; Vives-Adrián, L.; Selisko, B.; Ferrer-Orta, C.; Liu, X.; Lanke, K.; Ulferts, R.; De Palma, A.M.; Tanchis, F.; Goris, N.; et al. The RNA template channel of the RNA-dependent RNA polymerase as a target for development of antiviral therapy of multiple genera within a virus family. PLoS Pathog. 2015, 11, e1004733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, H.-C.; Chen, T.-C.; Fang, M.-Y.; Yen, K.-J.; Shih, S.-R.; Hsu, J.T.-A.; Tseng, C.-P. Inhibition of enterovirus 71 replication and the viral 3D polymerase by aurintricarboxylic acid. J. Antimicrob. Chemother. 2010, 65, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.A.; Milstrey, K.P.; Trown, P.W. Specific inhibition of viral ribonucleic acid replication by Gliotoxin. Science 1968, 159, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.L.; Carrasco, L. Gliotoxin: inhibitor of poliovirus RNA synthesis that blocks the viral RNA polymerase 3Dpol. J. Virol. 1992, 66, 1971–1976. [Google Scholar] [PubMed]
- Velu, A.B.; Chen, G.-W.; Hsieh, P.-T.; Horng, J.-T.; Hsu, J.T.-A.; Hsieh, H.-P.; Chen, T.-C.; Weng, K.-F.; Shih, S.-R. BPR-3P0128 inhibits RNA-dependent RNA polymerase elongation and VPg uridylylation activities of Enterovirus 71. Antivir. Res. 2014, 112, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-C.; Chang, H.-Y.; Lin, P.-F.; Chern, J.-H.; Hsu, J.T.-A.; Chang, C.-Y.; Shih, S.-R. Novel antiviral agent DTriP-22 targets RNA-dependent RNA polymerase of enterovirus 71. Antimicrob. Agents Chemother. 2009, 53, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.J.; Flanegan, J.B. Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J. Virol. 1997, 71, 8482–8489. [Google Scholar] [PubMed]
- Li, J.P.; Baltimore, D. Isolation of poliovirus 2C mutants defective in viral RNA synthesis. J. Virol. 1988, 62, 4016–4021. [Google Scholar] [PubMed]
- Pfister, T.; Jones, K.W.; Wimmer, E. A cysteine-rich motif in poliovirus protein 2C(ATPase) is involved in RNA replication and binds zinc in vitro. J. Virol. 2000, 74, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Teterina, N.L.; Kean, K.M.; Gorbalenya, A.E.; Agol, V.I.; Girard, M. Analysis of the functional significance of amino acid residues in the putative NTP-binding pattern of the poliovirus 2C protein. J. Gen. Virol. 1992, 73 (Pt 8), 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Teterina, N.L.; Levenson, E.; Rinaudo, M.S.; Egger, D.; Bienz, K.; Gorbalenya, A.E.; Ehrenfeld, E. Evidence for functional protein interactions required for poliovirus RNA replication. J. Virol. 2006, 80, 5327–5337. [Google Scholar] [CrossRef] [PubMed]
- Tolskaya, E.A.; Romanova, L.I.; Kolesnikova, M.S.; Gmyl, A.P.; Gorbalenya, A.E.; Agol, V.I. Genetic studies on the poliovirus 2C protein, an NTPase. A plausible mechanism of guanidine effect on the 2C function and evidence for the importance of 2C oligomerization. J. Mol. Biol. 1994, 236, 1310–1323. [Google Scholar] [CrossRef]
- Banerjee, R.; Echeverri, A.; Dasgupta, A. Poliovirus-encoded 2C polypeptide specifically binds to the 3’-terminal sequences of viral negative-strand RNA. J. Virol. 1997, 71, 9570–9578. [Google Scholar] [PubMed]
- Banerjee, R.; Tsai, W.; Kim, W.; Dasgupta, A. Interaction of poliovirus-encoded 2C/2BC polypeptides with the 3’ terminus negative-strand cloverleaf requires an intact stem-loop B. Virology 2001, 280, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Dasgupta, A. Interaction of picornavirus 2C polypeptide with the viral negative-strand RNA. J. Gen. Virol. 2001, 82, 2621–2627. [Google Scholar] [PubMed]
- Teterina, N.L.; Gorbalenya, A.E.; Egger, D.; Bienz, K.; Ehrenfeld, E. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J. Virol. 1997, 71, 8962–8972. [Google Scholar] [PubMed]
- Vance, L.M.; Moscufo, N.; Chow, M.; Heinz, B.A. Poliovirus 2C region functions during encapsidation of viral RNA. J. Virol. 1997, 71, 8759–8765. [Google Scholar] [PubMed]
- Verlinden, Y.; Cuconati, A.; Wimmer, E.; Rombaut, B. The antiviral compound 5-(3,4-dichlorophenyl) methylhydantoin inhibits the post-synthetic cleavages and the assembly of poliovirus in a cell-free system. Antivir. Res. 2000, 48, 61–69. [Google Scholar] [CrossRef]
- Li, J.P.; Baltimore, D. An intragenic revertant of a poliovirus 2C mutant has an uncoating defect. J. Virol. 1990, 64, 1102–1107. [Google Scholar] [PubMed]
- Gorbalenya, A.E.; Koonin, E.V. Viral proteins containing the purine NTP-binding sequence pattern. Nucleic Acids Res. 1989, 17, 8413–8440. [Google Scholar] [CrossRef] [PubMed]
- Mirzayan, C.; Wimmer, E. Biochemical studies on poliovirus polypeptide 2C: Evidence for ATPase activity. Virology 1994, 199, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, P.L.; Carrasco, L. Poliovirus protein 2C has ATPase and GTPase activities. J. Biol. Chem. 1993, 268, 8105–8110. [Google Scholar] [PubMed]
- De Palma, A.M.; Heggermont, W.; Lanke, K.; Coutard, B.; Bergmann, M.; Monforte, A.-M.; Canard, B.; De Clercq, E.; Chimirri, A.; Pürstinger, G.; et al. The thiazolobenzimidazole TBZE-029 inhibits enterovirus replication by targeting a short region immediately downstream from motif C in the nonstructural protein 2C. J. Virol. 2008, 82, 4720–4730. [Google Scholar] [CrossRef] [PubMed]
- Hadaschik, D.; Klein, M.; Zimmermann, H.; Eggers, H.J.; Nelsen-Salz, B. Dependence of echovirus 9 on the enterovirus RNA replication inhibitor 2-(α-Hydroxybenzyl)-benzimidazole maps to nonstructural protein 2C. J. Virol. 1999, 73, 10536–10539. [Google Scholar] [PubMed]
- Pincus, S.E.; Diamond, D.C.; Emini, E.A.; Wimmer, E. Guanidine-selected mutants of poliovirus: Mapping of point mutations to polypeptide 2C. J. Virol. 1986, 57, 638–646. [Google Scholar] [PubMed]
- Sadeghipour, S.; Bek, E.J.; McMinn, P.C. Selection and characterisation of guanidine-resistant mutants of human enterovirus 71. Virus Res. 2012, 169, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Agoh, M.; Agoh, Y.; Yoshida, H.; Yoshii, K.; Yoneyama, T.; Hagiwara, A.; Miyamura, T. Mutations in the 2C region of poliovirus responsible for altered sensitivity to benzimidazole derivatives. J. Virol. 2000, 74, 4146–4154. [Google Scholar] [CrossRef] [PubMed]
- Ulferts, R.; van der Linden, L.; Thibaut, H.J.; Lanke, K.H.; Leyssen, P.; Coutard, B.; De Palma, A.M.; Canard, B.; Neyts, J.; van Kuppeveld, F.J. Selective serotonin reuptake inhibitor fluoxetine inhibits replication of human enteroviruses B and D by targeting viral protein 2C. Antimicrob. Agents Chemother. 2013, 57, 1952–1956. [Google Scholar] [CrossRef] [PubMed]
- Wikel, J.H.; Paget, C.J.; DeLong, D.C.; Nelson, J.D.; Wu, C.Y.; Paschal, J.W.; Dinner, A.; Templeton, R.J.; Chaney, M.O.; Jones, N.D.; et al. Synthesis of syn and anti isomers of 6-[[(hydroxyimino)phenyl]methyl]-1-[(1-methylethyl)sulfonyl]-1H-benzimidazol-2-amine. Inhibitors of rhinovirus multiplication. J. Med. Chem. 1980, 23, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaar, H.M.; van der Linden, L.; Lanke, K.H.; Strating, J.R.; Pürstinger, G.; de Vries, E.; de Haan, C.A.M.; Neyts, J.; van Kuppeveld, F.J. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 2012, 22, 1576–1592. [Google Scholar] [CrossRef] [PubMed]
- Brown-Augsburger, P.; Vance, L.M.; Malcolm, S.K.; Hsiung, H.; Smith, D.P.; Heinz, B.A. Evidence that enviroxime targets multiple components of the rhinovirus 14 replication complex. Arch. Virol. 1999, 144, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Heinz, B.A.; Vance, L.M. The antiviral compound enviroxime targets the 3A coding region of rhinovirus and poliovirus. J. Virol. 1995, 69, 4189–4197. [Google Scholar] [PubMed]
- Heinz, B.A.; Vance, L.M. Sequence determinants of 3A-mediated resistance to enviroxime in rhinoviruses and enteroviruses. J. Virol. 1996, 70, 4854–4857. [Google Scholar] [PubMed]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-kinase III β is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaar, H.M.; Leyssen, P.; Thibaut, H.J.; de Palma, A.; van der Linden, L.; Lanke, K.H.; Lacroix, C.; Verbeken, E.; Conrath, K.; Macleod, A.M.; et al. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIβ. Antimicrob. Agents Chemother. 2013, 57, 4971–4981. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Gwaltney, J.M. Prophylactic activity of intranasal enviroxime against experimentally induced rhinovirus type 39 infection. Antimicrob. Agents Chemother. 1982, 21, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Barrow, G.I.; al-Nakib, W.; Tyrrell, D.A.; DeLong, D.C.; Lenox-Smith, I. Failure to demonstrate synergy between interferon-alpha and a synthetic antiviral, enviroxime, in rhinovirus infections in volunteers. Antivir. Res. 1988, 10, 141–149. [Google Scholar] [CrossRef]
- Levandowski, R.A.; Pachucki, C.T.; Rubenis, M.; Jackson, G.G. Topical enviroxime against rhinovirus infection. Antimicrob. Agents Chemother. 1982, 22, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.D.; Monto, A.S.; DeLong, D.C.; Exelby, A.; Bryan, E.R.; Srivastava, S. Controlled trial of enviroxime against natural rhinovirus infections in a community. Antimicrob. Agents Chemother. 1985, 27, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Phillpotts, R.J.; Jones, R.W.; Delong, D.C.; Reed, S.E.; Wallace, J.; Tyrrell, D.A. The activity of enviroxime against rhinovirus infection in man. Lancet 1981, 1, 1342–1344. [Google Scholar] [CrossRef]
- Phillpotts, R.J.; Wallace, J.; Tyrrell, D.A.; Tagart, V.B. Therapeutic activity of enviroxime against rhinovirus infection in volunteers. Antimicrob. Agents Chemother. 1983, 23, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, M.J.; Borawski, J.; Bose, A.; Capacci-Daniel, C.; Colvin, R.; Dennehy, M.; Ding, J.; Dobler, M.; Drumm, J.; Gaither, L.A.; et al. Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob. Agents Chemother. 2012, 56, 5149–5156. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Wakita, T.; Shimizu, H. Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J. Gen. Virol. 2008, 89, 2518–2530. [Google Scholar] [CrossRef] [PubMed]
- Spickler, C.; Lippens, J.; Laberge, M.-K.; Desmeules, S.; Bellavance, É.; Garneau, M.; Guo, T.; Hucke, O.; Leyssen, P.; Neyts, J.; et al. Phosphatidylinositol 4-kinase III beta is essential for replication of human rhinovirus and its inhibition causes a lethal phenotype in vivo. Antimicrob. Agents Chemother. 2013, 57, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J. Virol. 2013, 87, 4252–4260. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, L.; Strating, J.R.; Thibaut, H.J.; van der Linden, L.; Shair, M.D.; Neyts, J.; van Kuppeveld, F.J. Broad-range inhibition of enterovirus replication by OSW-1, a natural compound targeting OSBP. Antivir. Res. 2015, 117, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.-L.; Lin, Y.-W.; Chang, H.-W.; Lin, H.-Y.; Shao, H.-Y.; Yu, S.-L.; Liu, C.-C.; Chitra, E.; Sia, C.; Chow, Y.-H. Heat shock protein 90: Role in enterovirus 71 entry and assembly and potential target for therapy. PLoS ONE 2013, 8, e77133. [Google Scholar] [CrossRef] [PubMed]
- Bailey, H.H.; Mulcahy, R.T.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Tombes, M.B.; Wilding, G.; Pomplun, M.; Spriggs, D.R. Phase I clinical trial of intravenous l-buthionine sulfoximine and melphalan: an attempt at modulation of glutathione. J. Clin. Oncol. 1994, 12, 194–205. [Google Scholar] [PubMed]
- Bailey, H.H.; Ripple, G.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Feierabend, C.; Mahvi, D.; Schink, J.; Pomplun, M.; Mulcahy, R.T.; et al. Phase I study of continuous-infusion l-S,R-buthionine sulfoximine with intravenous melphalan. J. Natl. Cancer Inst. 1997, 89, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, P.J.; Hamilton, T.C.; LaCreta, F.P.; Gallo, J.M.; Kilpatrick, D.; Halbherr, T.; Brennan, J.; Bookman, M.A.; Hoffman, J.; Young, R.C.; et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J. Clin. Oncol. 1996, 14, 249–256. [Google Scholar] [PubMed]
- Van de Ven, A.A.; Douma, J.W.; Rademaker, C.; van Loon, A.M.; Wensing, A.M.; Boelens, J.-J.; Sanders, E.A.M.; van Montfrans, J.M. Pleconaril-resistant chronic parechovirus-associated enteropathy in agammaglobulinaemia. Antivir. Ther. 2011, 16, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Wildenbeest, J.G.; Harvala, H.; Pajkrt, D.; Wolthers, K.C. The need for treatment against human parechoviruses: How, why and when? Expert Rev. Anti. Infect. Ther. 2010, 8, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Seitsonen, J.; Susi, P.; Heikkilä, O.; Sinkovits, R.S.; Laurinmäki, P.; Hyypiä, T.; Butcher, S.J. Interaction of αVβ3 and αVβ6 integrins with human parechovirus 1. J. Virol. 2010, 84, 8509–8519. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; George, S.; Kusov, Y.; Perbandt, M.; Anemüller, S.; Mesters, J.R.; Norder, H.; Coutard, B.; Lacroix, C.; Leyssen, P.; et al. 3C protease of enterovirus 68: Structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol. 2013, 87, 4339–4351. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, L.; Ulferts, R.; Nabuurs, S.B.; Kusov, Y.; Liu, H.; George, S.; Lacroix, C.; Goris, N.; Lefebvre, D.; Lanke, K.H.; et al. Application of a cell-based protease assay for testing inhibitors of picornavirus 3C proteases. Antivir. Res. 2014, 103, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.A.; Dragovich, P.S.; Webber, S.E.; Fuhrman, S.A.; Patick, A.K.; Zalman, L.S.; Hendrickson, T.F.; Love, R.A.; Prins, T.J.; Marakovits, J.T.; et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA 1999, 96, 11000–11007. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Hansen, J.; Blaas, D.; Brunak, S. Cleavage site analysis in picornaviral polyproteins: Discovering cellular targets by neural networks. Protein Sci. 1996, 5, 2203–2216. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.H.; Panayiotou, M.; Girling, G.D.; Peard, C.I.; Oikarinen, S.; Hyöty, H.; Stanway, G. Evolution and conservation in human parechovirus genomes. J. Gen. Virol. 2009, 90, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, T.; Emerson, S.U.; Purcell, R.H.; Gauss-Müller, V. Polyprotein processing in echovirus 22: A first assessment. Biochem. Biophys. Res. Commun. 1995, 217, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Coller, B.A.; Chapman, N.M.; Beck, M.A.; Pallansch, M.A.; Gauntt, C.J.; Tracy, S.M. Echovirus 22 is an atypical enterovirus. J. Virol. 1990, 64, 2692–2701. [Google Scholar] [PubMed]
- Samuilova, O.; Krogerus, C.; Pöyry, T.; Hyypiä, T. Specific interaction between human parechovirus nonstructural 2A protein and viral RNA. J. Biol. Chem. 2004, 279, 37822–37831. [Google Scholar] [CrossRef] [PubMed]
- Hyypiä, T.; Horsnell, C.; Maaronen, M.; Khan, M.; Kalkkinen, N.; Auvinen, P.; Kinnunen, L.; Stanway, G. A distinct picornavirus group identified by sequence analysis. Proc. Natl. Acad. Sci. USA 1992, 89, 8847–8851. [Google Scholar] [CrossRef] [PubMed]
- Samuilova, O.; Krogerus, C.; Fabrichniy, I.; Hyypiä, T. ATP hydrolysis and AMP kinase activities of nonstructural protein 2C of human parechovirus 1. J. Virol. 2006, 80, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Eggers, H.J. Differences in the selective virus inhibitory action of 2-(α-hydroxybenzyl)-benzimidazole and guanidine HCl. Virology 1962, 18, 439–447. [Google Scholar] [CrossRef]
- Eggers, H.J.; Tamm, I. Spectrum and characteristics of the virus inhibitory action of 2-(α-hydroxybenzyl)-benzimidazole. J. Exp. Med. 1961, 113, 657–682. [Google Scholar] [CrossRef] [PubMed]
- Krogerus, C.; Egger, D.; Samuilova, O.; Hyypiä, T.; Bienz, K. Replication complex of human parechovirus 1. J. Virol. 2003, 77, 8512–8523. [Google Scholar] [CrossRef] [PubMed]
- Snell, N.J. Ribavirin--current status of a broad spectrum antiviral agent. Expert Opin. Pharmacother. 2001, 2, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Saleh, M.-C.; Gitlin, L.; Beske, O.; Andino, R. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J. Virol. 2004, 78, 3378–3386. [Google Scholar] [CrossRef] [PubMed]
- Koletsky, A.J.; Harding, M.W.; Handschumacher, R.E. Cyclophilin: distribution and variant properties in normal and neoplastic tissues. J. Immunol. 1986, 137, 1054–1059. [Google Scholar] [PubMed]
- Lin, K.; Gallay, P. Curing a viral infection by targeting the host: the example of cyclophilin inhibitors. Antivir. Res. 2013, 99, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Giffin, M.J.; Muller, R.; Savage, J.; Lin, Y.C.; Hong, S.; Jin, W.; Whitby, L.R.; Elder, J.H.; Boger, D.L.; et al. Identification of broad-based HIV-1 protease inhibitors from combinatorial libraries. Biochem. J. 2010, 429, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Nkeze, J.; Zhao, R.Y. Effects of HIV-1 protease on cellular functions and their potential applications in antiretroviral therapy. Cell Biosci. 2012, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Su, H.; Zhang, T.; Du, J.; Cui, S.; Yang, F.; Jin, Q. Inhibition of Enterovirus 71 replication by 7-hydroxyflavone and diisopropyl-flavon7-yl Phosphate. PLoS ONE 2014, 9, e92565. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, T.; Du, J.; Cui, S.; Yang, F.; Jin, Q. Anti-enterovirus 71 effects of chrysin and its phosphate ester. PLoS ONE 2014, 9, e89668. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L. Rationale for the evaluation of fluoxetine in the treatment of enterovirus D68-associated acute flaccid myelitis. JAMA Neurol. 2015, 72, 493–494. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linden, L.V.d.; Wolthers, K.C.; Van Kuppeveld, F.J.M. Replication and Inhibitors of Enteroviruses and Parechoviruses. Viruses 2015, 7, 4529-4562. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082832
Linden LVd, Wolthers KC, Van Kuppeveld FJM. Replication and Inhibitors of Enteroviruses and Parechoviruses. Viruses. 2015; 7(8):4529-4562. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082832
Chicago/Turabian StyleLinden, Lonneke Van der, Katja C. Wolthers, and Frank J.M. Van Kuppeveld. 2015. "Replication and Inhibitors of Enteroviruses and Parechoviruses" Viruses 7, no. 8: 4529-4562. https://0-doi-org.brum.beds.ac.uk/10.3390/v7082832