
Cellular microRNAs Repress Vesicular Stomatitis Virus but Not Theiler’s Virus Replication


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viruses
2.3. miRNA Target Sequence Predictions
2.4. Construction of a CRE Expressing Lentiviral Vector and Cell Transduction
2.5. Generation of L929 Cells Overexpressing miR-770-3p and Construction of a miR-142-3p Expressing Vector
2.6. Reporter Vectors and Luciferase Assays
2.7. RNA Extraction and Quantitative RT-PCR
2.8. Flow Cytometry
2.9. Statistical Analysis
3. Results
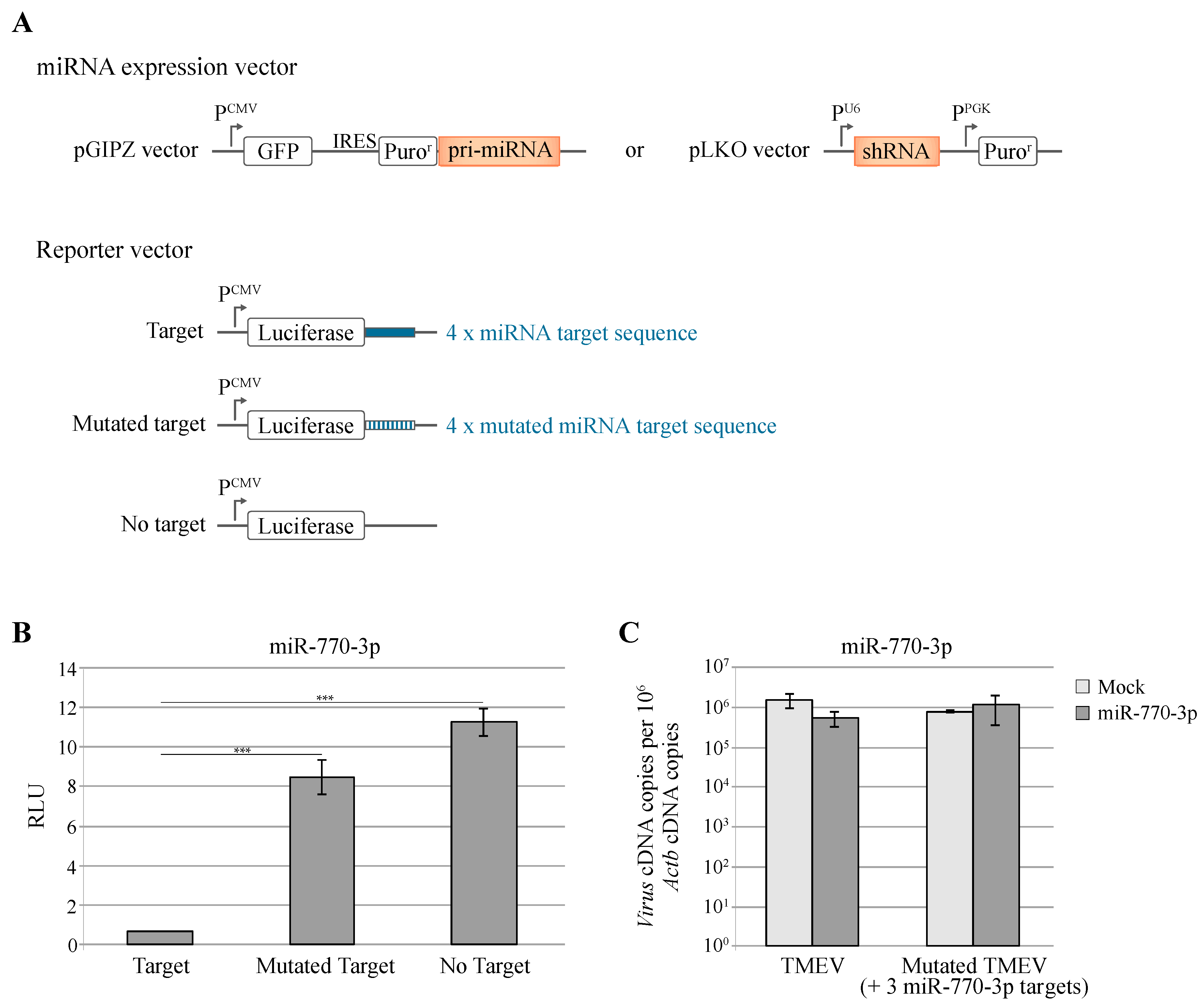
3.1. Under-Represented miRNA Target Sequences in TMEV Genome
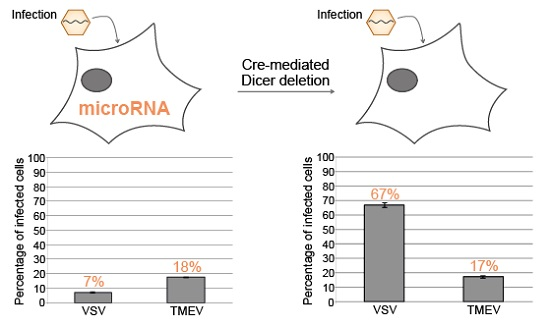
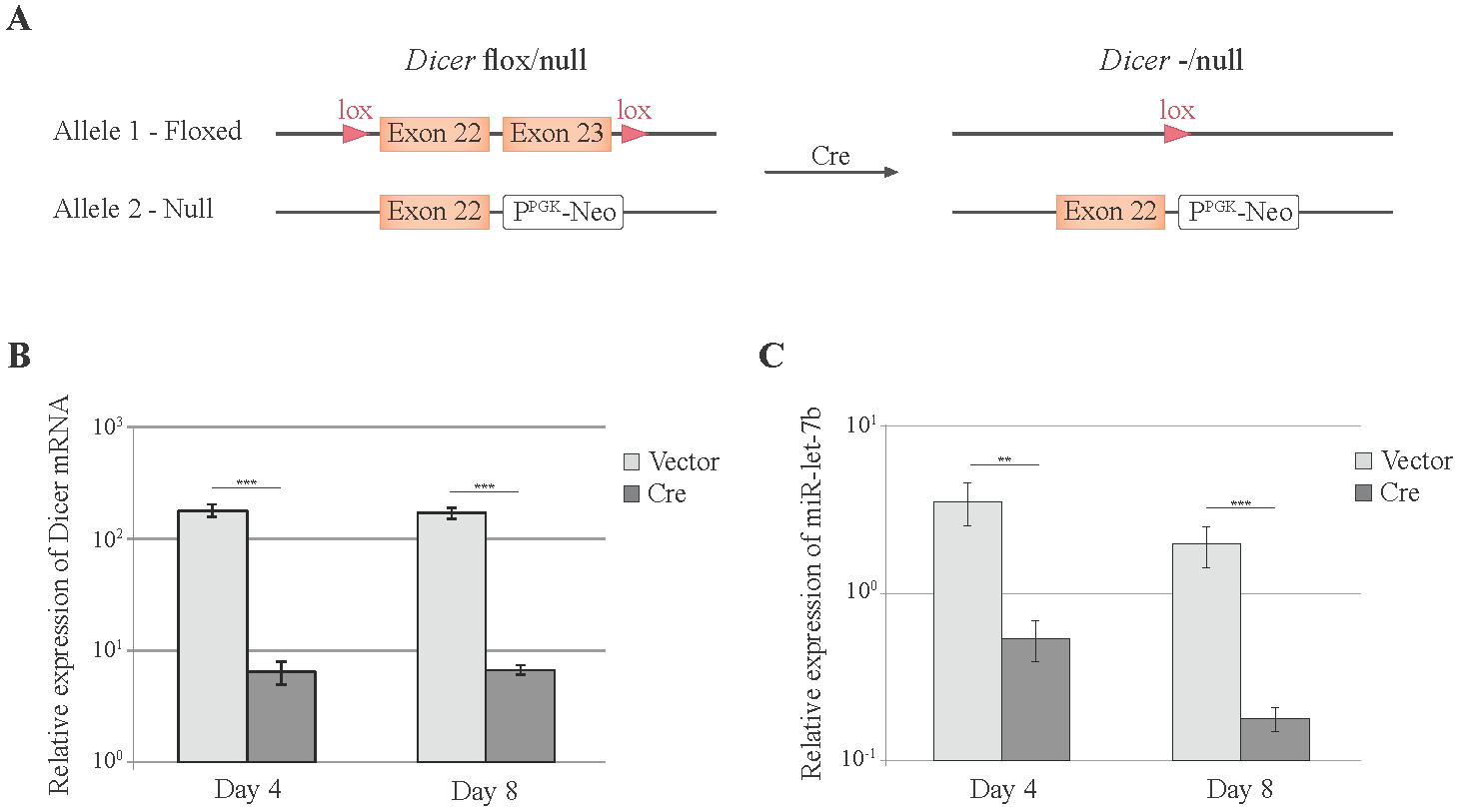
3.2. Cre-Mediated Dicer Inactivation Reduces Expression and Activity of Cellular miRNAs
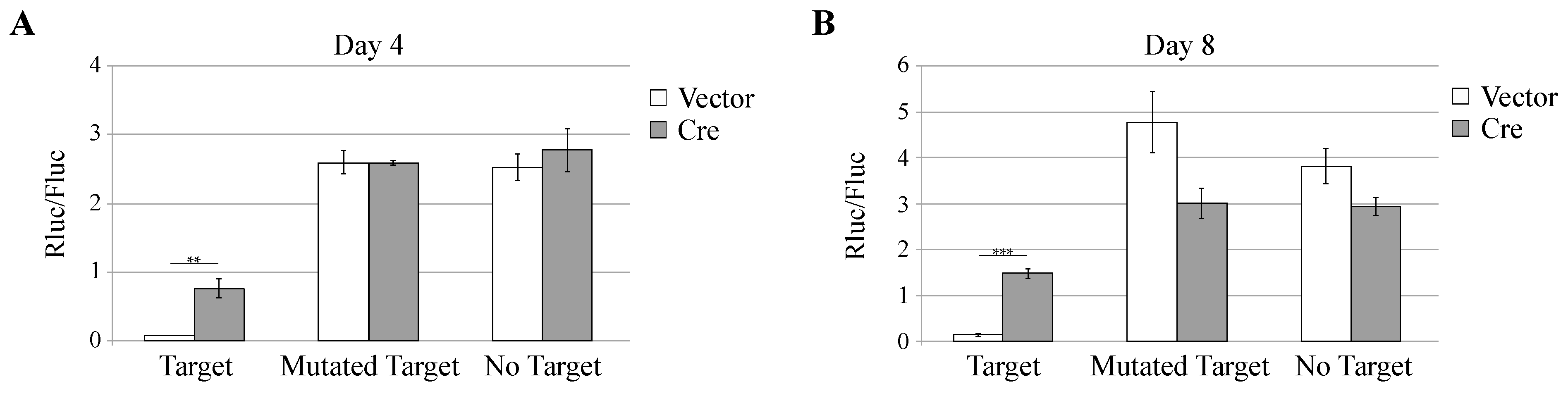
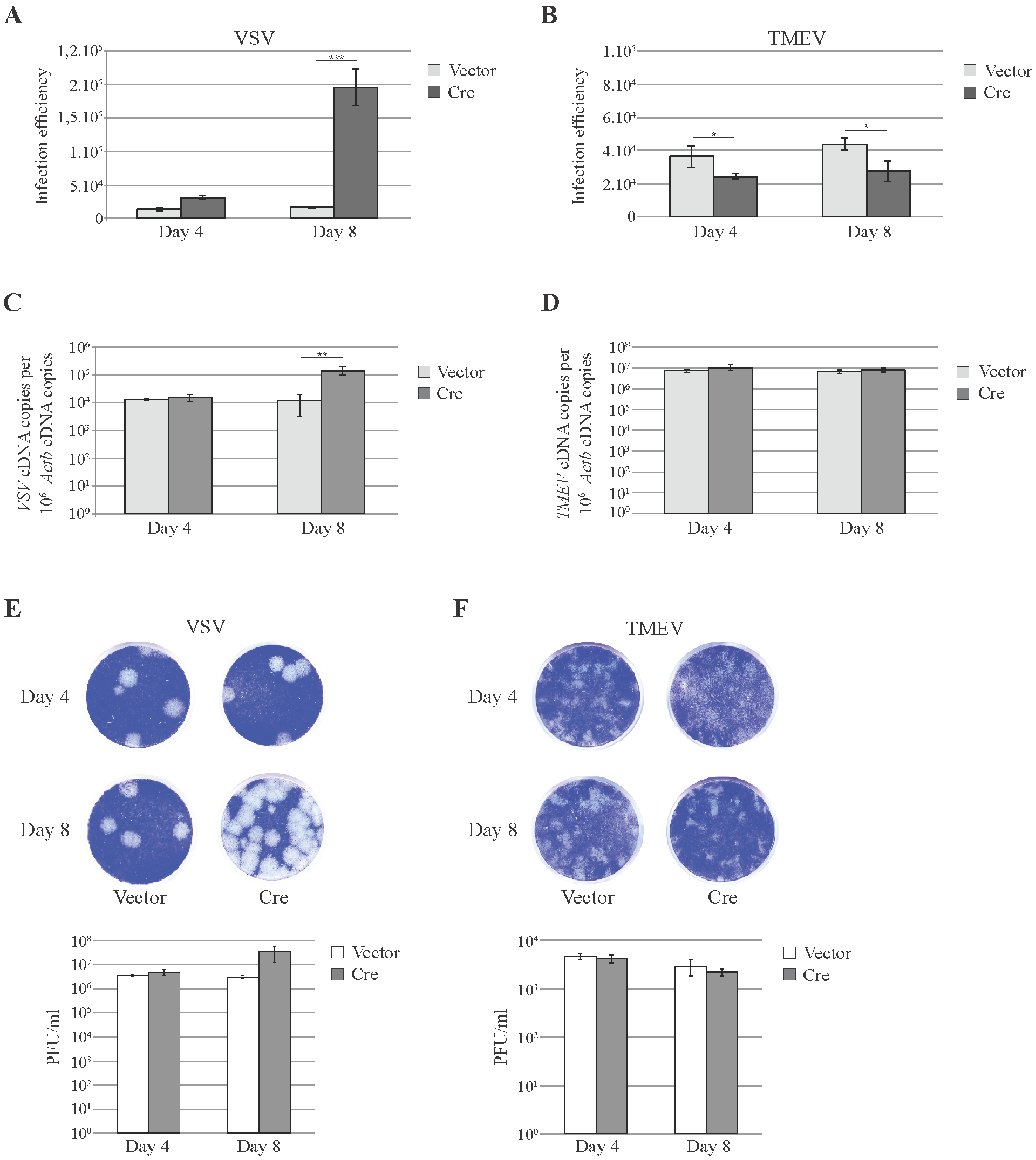
3.3. Knockdown of Cellular miRNAs Increases VSV but Not TMEV Replication
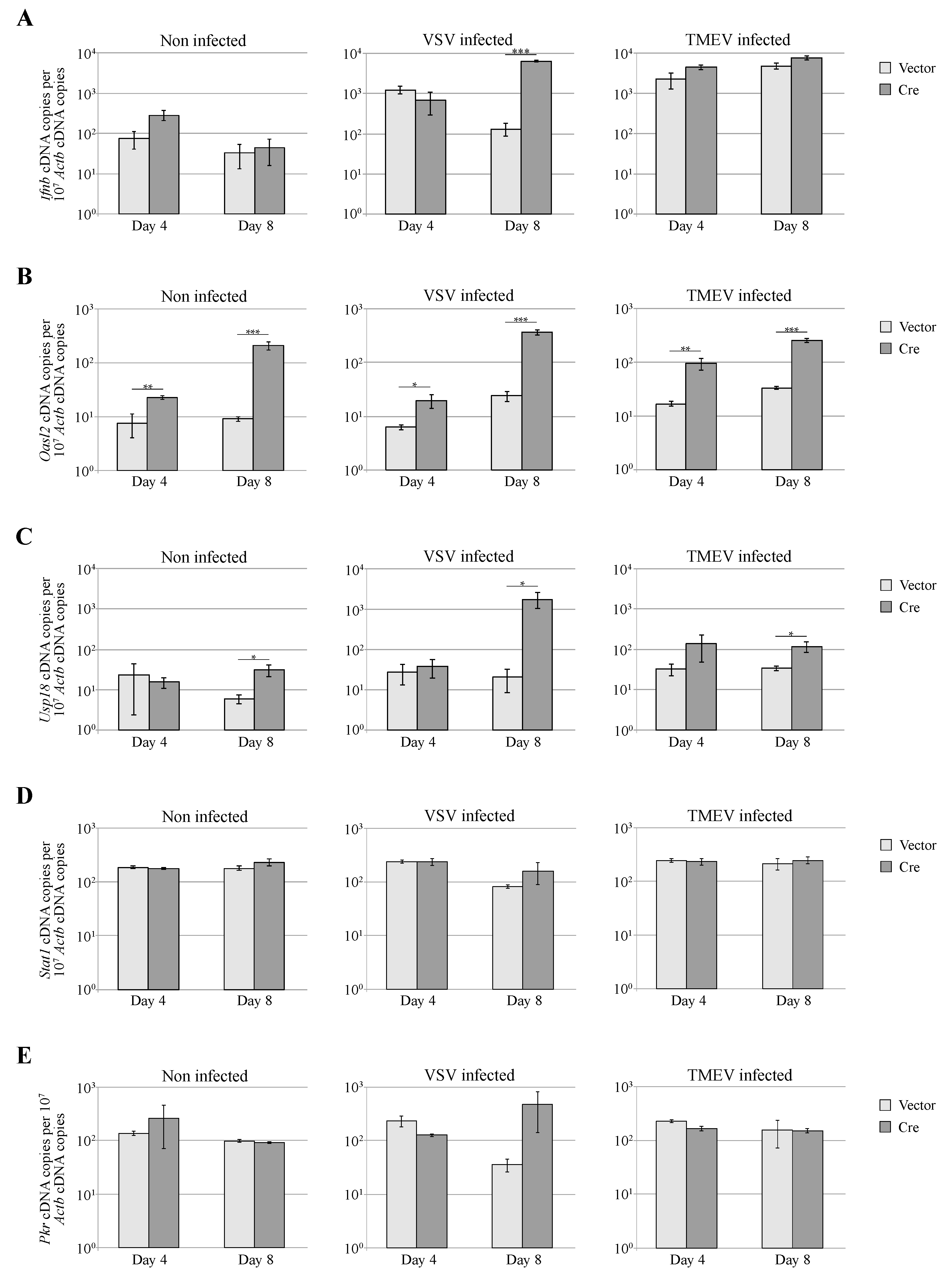
3.4. Dicer Inactivation Increases Oasl2 Expression
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nilsen, T.W. Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet. 2007, 23, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y. Principles of micro-RNA production and maturation. Oncogene 2006, 25, 6156–6162. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Plasterk, R.H. The diverse functions of microRNAs in animal development and disease. Dev. Cell 2006, 11, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.W.; Voinnet, O. Antiviral immunity directed by small RNAs. Cell 2007, 130, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Zambon, R.A.; Vakharia, V.N.; Wu, L.P. RNAi is an antiviral immune response against a dsRNA virus in Drosophila melanogaster. Cell. Microbiol. 2006, 8, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Aliyari, R.; Wu, Q.; Li, H.W.; Wang, X.H.; Li, F.; Green, L.D.; Han, C.S.; Li, W.X.; Ding, S.W. Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell Host Microbe 2008, 4, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Wu, Q.; Ito, T.; Cillo, F.; Li, W.X.; Chen, X.; Yu, J.L.; Ding, S.W. RNAi-mediated viral immunity requires amplification of virus-derived siRNAs in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, L.; Csorba, T.; Pantaleo, V.; Chapman, E.J.; Carrington, J.C.; Liu, Y.P.; Dolja, V.V.; Calvino, L.F.; Lopez-Moya, J.J.; Burgyan, J. Small RNA binding is a common strategy to suppress RNA silencing by several viral suppressors. EMBO J. 2006, 25, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, W.X.; Ding, S.W. Induction and suppression of RNA silencing by an animal virus. Science 2002, 296, 1319–1321. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.A.; Lee, J.H.; Chapados, B.R.; Debler, E.W.; Schneemann, A.; Williamson, J.R. Dual modes of RNA-silencing suppression by flock house virus protein B2. Nat. Struct. Mol. Biol. 2005, 12, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Galiana-Arnoux, D.; Dostert, C.; Schneemann, A.; Hoffmann, J.A.; Imler, J.L. Essential function in vivo for Dicer-2 in host defense against RNA viruses in Drosophila. Nat. Immunol. 2006, 7, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Bouche, N.; Lauressergues, D.; Gasciolli, V.; Vaucheret, H. An antagonistic function for Arabidopsis DCL2 in development and a new function for DCL4 in generating viral siRNAs. EMBO J. 2006, 25, 3347–3356. [Google Scholar] [CrossRef] [PubMed]
- Li, H.W.; Ding, S.W. Antiviral silencing in animals. FEBS Lett. 2005, 579, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, J.; de Vries, W.; Geutjes, E.J.; Prins, M.; de Haan, P.; Berkhout, B. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007, 3, e86. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Li, H.; Lu, R.; Li, F.; Dus, M.; Atkinson, P.; Brydon, E.W.; Johnson, K.L.; Garcia-Sastre, A.; Ball, L.A.; et al. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. USA 2004, 101, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Jing, Q.; Georgel, P.; New, L.; Chen, J.; Mols, J.; Kang, Y.J.; Jiang, Z.; Du, X.; Cook, R.; et al. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 2007, 27, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, V.S.; Drake, A.; Chen, J. Virus-specific host miRNAs: Antiviral defenses or promoters of persistent infection? Trends Immunol. 2009, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Skalsky, R.L.; Kennedy, E.M.; Furuse, Y.; Whisnant, A.W.; Flores, O.; Schultz, K.L.; Putnam, N.; Barrows, N.J.; Sherry, B.; et al. Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. J. Virol. 2014, 88, 8065–8076. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Langlois, R.A.; Schmid, S.; Varble, A.; Shim, J.V.; Sachs, D.; tenOever, B.R. The Mammalian response to virus infection is independent of small RNA silencing. Cell Rep. 2014, 8, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.; Kunitomi, M.; Vignuzzi, M.; Saksela, K.; Andino, R. Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe 2008, 4, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.J.; Hadac, E.M.; Cullen, B.R.; Russell, S.J. MicroRNA antagonism of the picornaviral life cycle: Alternative mechanisms of interference. PLoS Pathog. 2010, 6, e1000820. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Dou, Y.; Bao, H.; Luo, X.; Liu, X.; Mu, K.; Liu, Z.; Liu, X.; Cai, X. Multiple microRNAs targeted to internal ribosome entry site against foot-and-mouth disease virus infection in vitro and in vivo. Virol. J. 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Karaa, Z.S.; Iacovoni, J.S.; Bastide, A.; Lacazette, E.; Touriol, C.; Prats, H. The VEGF IRESes are differentially susceptible to translation inhibition by miR-16. RNA 2009, 15, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Ke, X.; Wang, M.; He, S.; Li, Q.; Zheng, C.; Zhang, Z.; Liu, Y.; Wang, H. Human microRNA hsa-miR-296–5p suppresses enterovirus 71 replication by targeting the viral genome. J. Virol. 2013, 87, 5645–5656. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qin, Y.; Tong, L.; Wu, S.; Wang, Q.; Jiao, Q.; Guo, Z.; Lin, L.; Wang, R.; Zhao, W.; et al. MiR-342–5p suppresses coxsackievirus B3 biosynthesis by targeting the 2C-coding region. Antiviral Res. 2012, 93, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Lin, L.; Wu, S.; Guo, Z.; Wang, T.; Qin, Y.; Wang, R.; Zhong, X.; Wu, X.; Wang, Y.; et al. MiR-10a* up-regulates coxsackievirus B3 biosynthesis by targeting the 3D-coding sequence. Nucleic Acids Res. 2013, 41, 3760–3771. [Google Scholar] [CrossRef] [PubMed]
- Murchison, E.P.; Partridge, J.F.; Tam, O.H.; Cheloufi, S.; Hannon, G.J. Characterization of Dicer-deficient murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 2005, 102, 12135–12140. [Google Scholar] [CrossRef] [PubMed]
- Hermant, P.; Francius, C.; Clotman, F.; Michiels, T. IFN-epsilon is constitutively expressed by cells of the reproductive tract and is inefficiently secreted by fibroblasts and cell lines. PLoS ONE 2013, 8, e71320. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.B.; Pappenheimer, A.M.; Richardson, S. Observations on encephalomyelitis of mice (DA strain). J. Exp. Med. 1952, 96, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Jnaoui, K.; Michiels, T. Adaptation of Theiler’s virus to L929 cells: Mutations in the putative receptor binding site on the capsid map to neutralization sites and modulate viral persistence. Virology 1998, 244, 397–404. [Google Scholar] [CrossRef] [PubMed]
- McAllister, A.; Tangy, F.; Aubert, C.; Brahic, M. Molecular cloning of the complete genome of Theiler's virus, strain DA, and production of infectious transcripts. Microb. Pathogen. 1989, 7, 381–388. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Follenzi, A.; Ailles, L.E.; Bakovic, S.; Geuna, M.; Naldini, L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 2000, 25, 217–222. [Google Scholar] [PubMed]
- Kreit, M.; Paul, S.; Knoops, L.; De Cock, A.; Sorgeloos, F.; Michiels, T. Inefficient type I interferon-mediated antiviral protection of primary mouse neurons is associated with the lack of apolipoprotein l9 expression. J. Virol. 2014, 88, 3874–3884. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.; Grueneberg, D.A.; Yang, X.; Kim, S.Y.; Kloepfer, A.M.; Hinkle, G.; Piqani, B.; Eisenhaure, T.M.; Luo, B.; Grenier, J.K.; et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006, 124, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Michiels, T. Cardiovirus leader proteins are functionally interchangeable and have evolved to adapt to virus replication fitness. J. General Virol. 2006, 87, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Prevot, P.P.; Augereau, C.; Simion, A.; Van den Steen, G.; Dauguet, N.; Lemaigre, F.P.; Jacquemin, P. Let-7b and miR-495 stimulate differentiation and prevent metaplasia of pancreatic acinar cells by repressing HNF6. Gastroenterology 2013, 145, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Stone, J.K.; Andino, R. Poliovirus escape from RNA interference: Short interfering RNA-target recognition and implications for therapeutic approaches. J. Virol. 2005, 79, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Kincaid, R.P.; Phanaksri, T.; Burke, J.M.; Pare, J.M.; Cox, J.E.; Hsiang, T.Y.; Krug, R.M.; Sullivan, C.S. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 2013, 14, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wu, Y.; Dang, Y.; Choi, J.G.; Zhang, J.; Wu, H. Pol III promoters to express small RNAs: Delineation of transcription initiation. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Sorgeloos, F.; Jha, B.K.; Silverman, R.H.; Michiels, T. Evasion of antiviral innate immunity by Theiler’s virus L* protein through direct inhibition of RNase L. PLoS Pathog. 2013, 9, e1003474. [Google Scholar] [CrossRef] [PubMed]
- Van Pesch, V.; van Eyll, O.; Michiels, T. The leader protein of Theiler’s virus inhibits immediate-early α/β interferon production. J. Virol. 2001, 75, 7811–7817. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vector/Virus 1 | Parental 1 | Characteristics |
|---|---|---|
| (p)KJ6 | TMEV (DA1) | Capsid adapted to infect L929 cells |
| (p)KJ7 | (p)KJ6 | GFP-coding sequence replacing codons 5–67 of L protein |
| (p)ADC43 | (p)KJ6 | TMEV carrying 3 extra target sequences for miR-770-3p |
| VSV-GFP | VSV | VSV carrying a GFP-coding sequence |
| (p)TM945 | pCCLsin 2 | Lentiviral vector carrying PCMV-MCS-IRES-mCherry 2 |
| (p)ADC82 | (p)TM945 | Lentiviral vector carrying PCMV-Cre recombinase-IRES-mCherry 2 |
| (p)ADC48 | pLKO.1 | Lentiviral vector expressing shRNA corresponding to miR-770-3p |
| (p)ADC38 | pGIPZ | Lentiviral vector expressing genomic miR-142-3p |
| (p)ADC1 | (p)TM897 | Lentiviral vector carrying PPGK-Renilla luciferase 2 |
| (p)ADC30 | (p)ADC1 | Luciferase reporter with 4 tandem miR-770-3p target sequences |
| (p)ADC39 | (p)ADC1 | Luciferase reporter with 4 tandem miR-142-3p target sequences |
| (p)ADC62 | (p)ADC1 | Luciferase reporter with 4 tandem mutated miR-770-3p target sequences |
| (p)ADC64 | (p)ADC1 | Luciferase reporter with 4 tandem copies of mutated miR-142-3p target sequences |
| pADC13 | pcDNA3 | Photinus luciferase (firefly) from pGL-2 basic (Promega, Leiden, The Netherlands) |
| Vector | miRNA Binding Site | Nucleotide Sequence of miRNA Binding Sites |
|---|---|---|
| (p)ADC30 | Target sequences for miR-770-3p | CCAGCTCCACGTCAGGCCCACG |
| (p)ADC39 | Target sequence for miR-142-3p | TCCATAAAGTAGGAAACACTACA |
| (p)ADC62 | Mutated target sequence for miR-770-3p | CCCGCACCCCGACACGCTCATG |
| (p)ADC64 | Mutated target sequence for miR-142-3p | TCTATTAAATAAGAGACTCTTCA |
| Gene | Name * | Sequence 5′–3′ | Plasmid |
|---|---|---|---|
| Actb | TM421 (Fw) | AGAGGGAAATCGTGCGTGAC | pTM793 |
| TM422 (Rev) | CAATAGTGATGACCTGGCCGT | ||
| TMEV | TM348 (Fw) | GAACGTCAGCATTTTCCGGC | pTM410 |
| TM349 (Rev) | GGTGTGAAGAGCGGCAAGTG | ||
| VSV-G | TM846 (Fw) | TTGCTGCTCCAATCCTCTCA | pVSV-G |
| TM847 (Rev) | TCGAACACCTGAGCCTTTGA | ||
| Ifnb | TM642 (Fw) | ATGAACAACAGGTGGATCCTCC | pcDNA3-IFN-β |
| TM643 (Rev) | AGGAGCTCCTGACATTTCCGAA | ||
| Oasl2 | TM638 (Fw) | GGATGCCTGGGAGAGAATCG | pCS40 |
| TM639 (Rev) | TCGCCTGCTCTTCGAAACTG | ||
| Usp18 | TM1116 (Fw) | TGCAGGGTCTGTTCACCATC | pMIP01 |
| TM1117 (Rev) | GCACATGTCGGAGCTTGCTA | ||
| Stat1 | TM881 (Fw) | GAGGGGCCTCTCATTGTCAC | pSPA31 |
| TM882 (Rev) | GATTCCTGGGCTCTGTCACC | ||
| Pkr | TM887 (Fw) | CGCTGGCAGAACTCAATCAC | pSPA33 |
| TM888 (Rev) | CTCCGGTCACGATTTGTTCA |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Cock, A.; Michiels, T. Cellular microRNAs Repress Vesicular Stomatitis Virus but Not Theiler’s Virus Replication. Viruses 2016, 8, 75. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030075
De Cock A, Michiels T. Cellular microRNAs Repress Vesicular Stomatitis Virus but Not Theiler’s Virus Replication. Viruses. 2016; 8(3):75. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030075
Chicago/Turabian StyleDe Cock, Aurélie, and Thomas Michiels. 2016. "Cellular microRNAs Repress Vesicular Stomatitis Virus but Not Theiler’s Virus Replication" Viruses 8, no. 3: 75. https://0-doi-org.brum.beds.ac.uk/10.3390/v8030075