
Nuclear Translocation of Crk Adaptor Proteins by the Influenza A Virus NS1 Protein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Recombinant Influenza A Viruses
2.3. DNA Transfections and Plasmids
2.4. Antibodies
2.5. Immunoprecipitation and Detection
2.6. Cell Fractionation
2.7. Immunofluorescence Staining and Confocal Imaging
3. Results
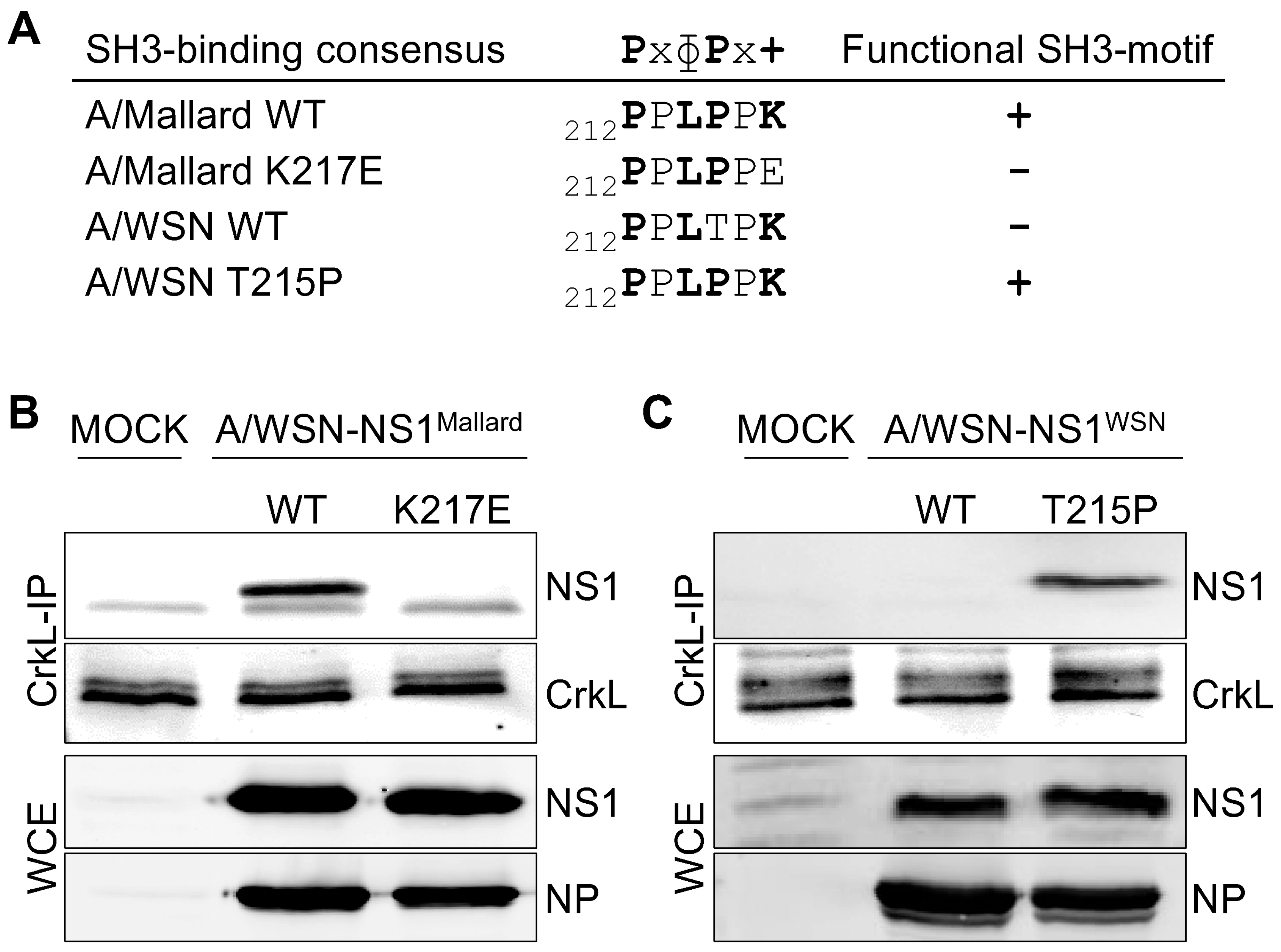
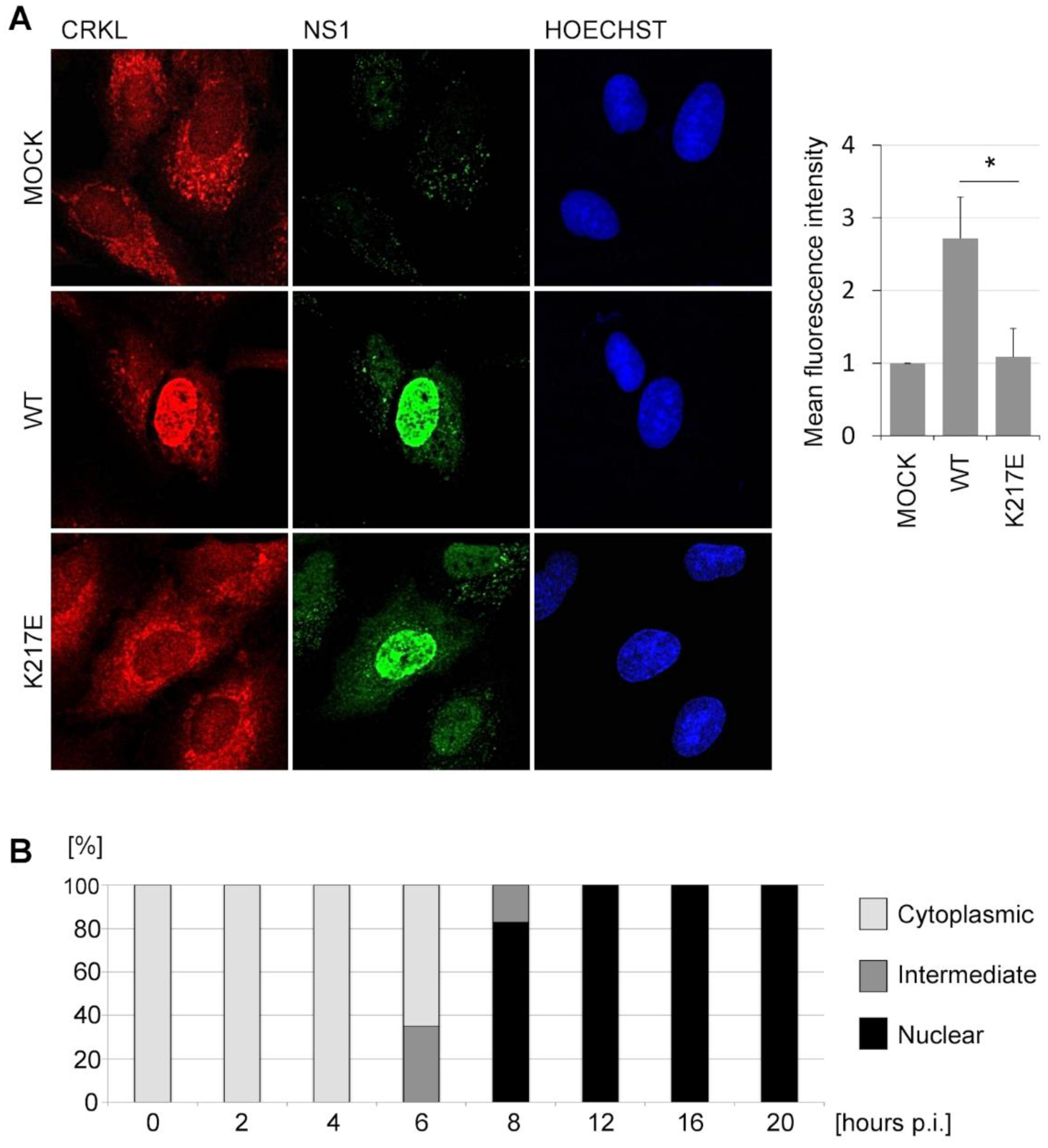
3.1. SH3 Binding-Competent NS1 Proteins Translocate Crk Proteins into the Nucleus
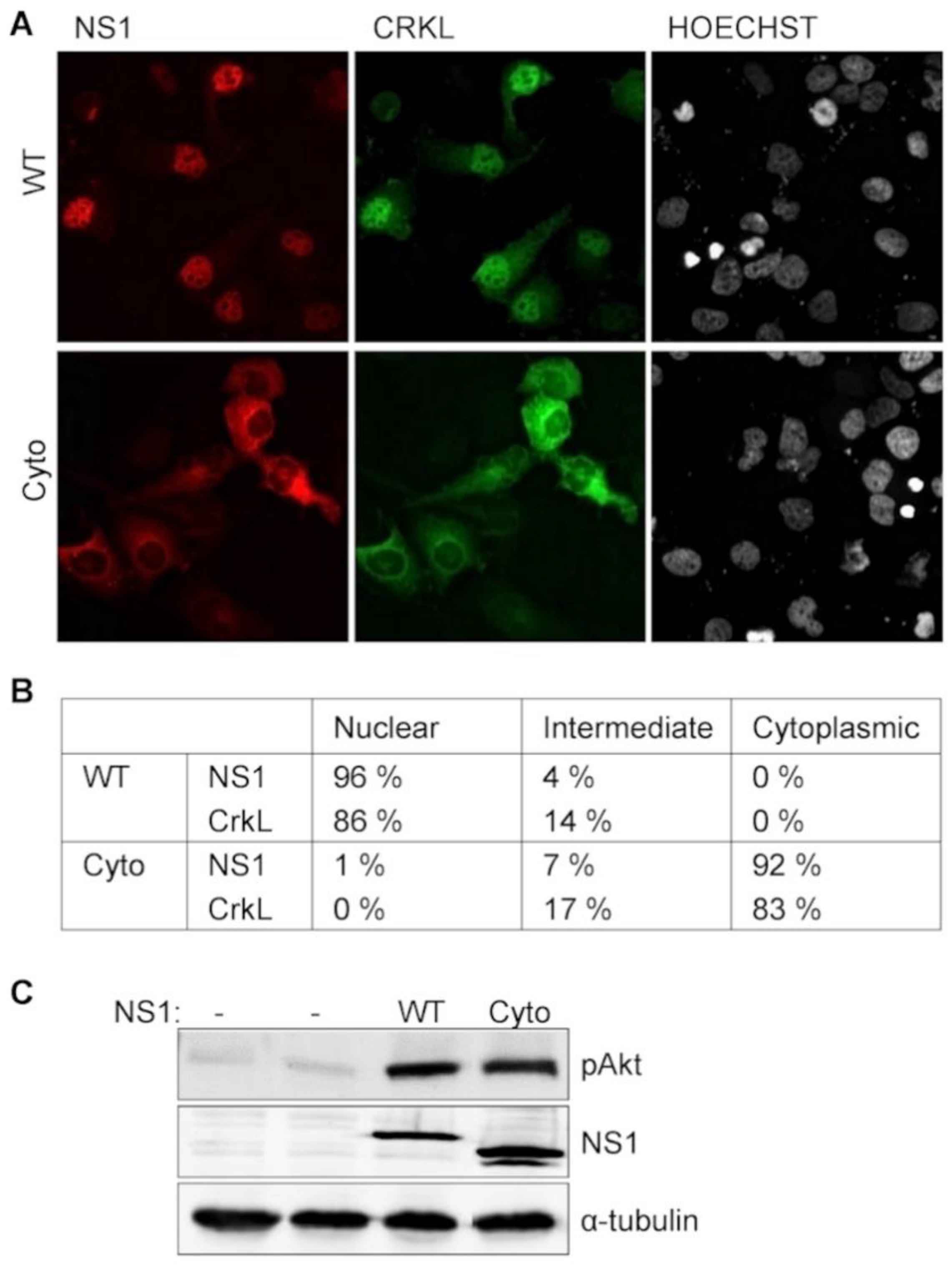
3.2 NS1-Induced PI3K-Activation does not Depend on Crk Relocalization into the Nucleus
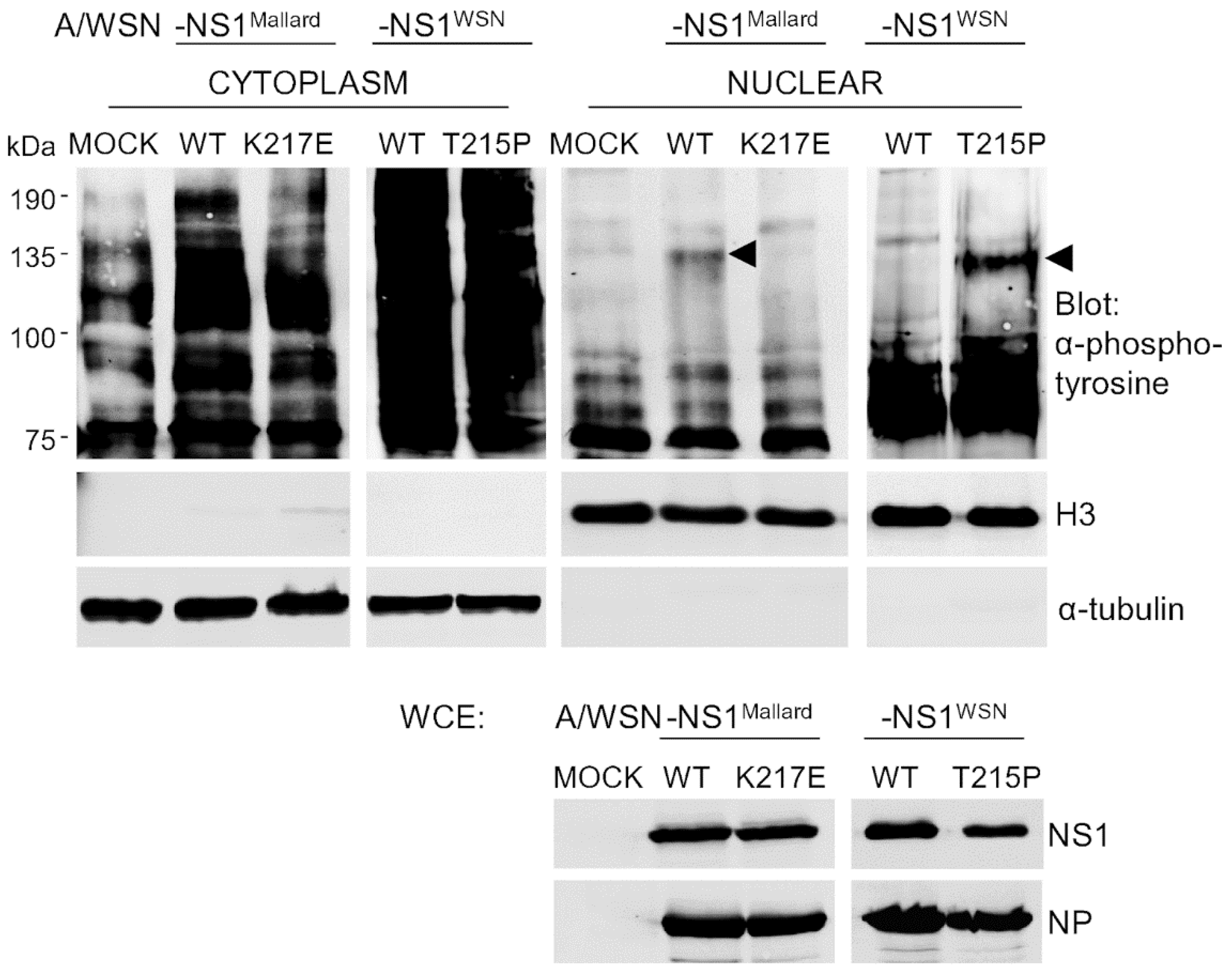
3.3. A Change in Nuclear Protein Tyrosine Phosphorylation after NS1-Mediated Nuclear Re-Localization of Crk
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Garcia-Sastre, A. The NS1 protein: A multitasking virulence factor. Curr. Top Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar] [PubMed]
- Melen, K.; Kinnunen, L.; Fagerlund, R.; Ikonen, N.; Twu, K.Y.; Krug, R.M.; Julkunen, I. Nuclear and nucleolar targeting of influenza A virus NS1 protein: Striking differences between different virus subtypes. J. Virol. 2007, 81, 5995–6006. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cui, Z.Q.; Wang, W.; Zhang, Z.P.; Wei, H.P.; Zhou, Y.F.; Zhang, X.E. New regulatory mechanisms for the intracellular localization and trafficking of influenza A virus NS1 protein revealed by comparative analysis of A/PR/8/34 and A/Sydney/5/97. J. Gener. Virol. 2010, 91, 2907–2917. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.; Selman, M.; Pelchat, M.; Jia, J.J.; Stintzi, A.; Brown, E.G. Identification of adaptive mutations in the influenza A virus non-structural 1 gene that increase cytoplasmic localization and differentially regulate host gene expression. PLoS ONE 2013, 8, e84673. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, D.; Palese, P.; Krystal, M. Two nuclear location signals in the influenza virus NS1 nonstructural protein. J. Virol. 1988, 62, 3020–3026. [Google Scholar] [PubMed]
- Melen, K.; Tynell, J.; Fagerlund, R.; Roussel, P.; Hernandez-Verdun, D.; Julkunen, I. Influenza A H3N2 subtype virus NS1 protein targets into the nucleus and binds primarily via its C-terminal NLS2/NoLS to nucleolin and fibrillarin. Virol. J. 2012, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yamakita, Y.; Krug, R.M. Regulation of a nuclear export signal by an adjacent inhibitory sequence: The effector domain of the influenza virus NS1 protein. Proc.Natl. Acad. Sci. USA 1998, 95, 4864–4869. [Google Scholar] [CrossRef] [PubMed]
- Tynell, J.; Melen, K.; Julkunen, I. Mutations within the conserved NS1 nuclear export signal lead to inhibition of influenza A virus replication. Virol. J. 2014, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Qiu, Y.; Krug, R.M. The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J. Virol. 1994, 68, 2425–2432. [Google Scholar] [PubMed]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; Schulz, O.; Pichlmair, A.; Bier, K.; Robb, N.; Vreede, F.; Barclay, W.; Fodor, E.; Reis e Sousa, C. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villan, E.; Garcia-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′–5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gener. Virol. 2007, 88, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, L.S.; Kazlauskas, A.; Melen, K.; Wagner, R.; Ziegler, T.; Julkunen, I.; Saksela, K. Avian and 1918 Spanish influenza a virus NS1 proteins bind to Crk/CrkL Src homology 3 domains to activate host cell signaling. J. Biol. Chem. 2008, 283, 5719–5727. [Google Scholar] [CrossRef] [PubMed]
- Ylosmaki, L.; Schmotz, C.; Ylosmaki, E.; Saksela, K. Reorganization of the host cell Crk(L)-PI3 kinase signaling complex by the influenza A virus NS1 protein. Virology 2015, 484, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Reichman, C.T.; Mayer, B.J.; Keshav, S.; Hanafusa, H. The product of the cellular crk gene consists primarily of SH2 and SH3 regions. Cell Growth Differ. 1992, 3, 451–460. [Google Scholar] [PubMed]
- Hoeve, J.T.; Morris, C.; Heisterkamp, N.; Groffen, J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene 1993, 8, 2469–2474. [Google Scholar] [PubMed]
- Feller, S.M. Crk family adaptors-signalling complex formation and biological roles. Oncogene 2001, 20, 6348–6371. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Kalodimos, C.; Inagaki, F.; Tanaka, S. Crk and CrkL adaptor proteins: Networks for physiological and pathological signaling. Cell Commun. Sign. 2009, 7, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Curran, T. Essential roles of Crk and CrkL in fibroblast structure and motility. Oncogene 2014, 33, 5121–5132. [Google Scholar] [CrossRef] [PubMed]
- Guris, D.L.; Fantes, J.; Tara, D.; Druker, B.J.; Imamoto, A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 2001, 27, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Boyd, K.; Curran, T. Cardiovascular and craniofacial defects in Crk-null mice. Mol. Cell. Biol. 2006, 26, 6272–6282. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y. The capable ABL: What is its biological function? Mol. Cell. Biol. 2014, 34, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Richardson, D.A.; Kopf, J.; Yoshida, M.; Hollingsworth, R.E.; Kornbluth, S. Apoptotic regulation by the Crk adapter protein mediated by interactions with Wee1 and Crm1/exportin. Mol. Cell Biol. 2002, 22, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Evans, E.K.; Murakami, M.; Moyer, M.B.; Moseley, M.A.; Vande Woude, G.; Kornbluth, S. Wee1-Regulated apoptosis mediated by the crk adaptor protein in Xenopus egg extracts. J. Cell Biol. 2000, 151, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.N.; Uddin, S.; Korkmaz, M.; Majchrzak, B.; Druker, B.J.; Platanias, L.C. Activation of a CrkL-stat5 signaling complex by type I interferons. J. Biol. Chem. 1999, 274, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; York, R.D.; Tara, D.; Tajinda, K.; Druker, B.J. CrkL functions as a nuclear adaptor and transcriptional activator in Bcr-Abl-expressing cells. Exp. Hematol. 2000, 28, 305–310. [Google Scholar] [CrossRef]
- Harkiolaki, M.; Gilbert, R.J.; Jones, E.Y.; Feller, S.M. The C-terminal SH3 domain of CRKL as a dynamic dimerization module transiently exposing a nuclear export signal. Structure 2006, 14, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Kar, B.; Reichman, C.T.; Singh, S.; O'Connor, J.P.; Birge, R.B. Proapoptotic function of the nuclear Crk II adaptor protein. Biochemistry 2007, 46, 10828–10840. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Watanabe, T.; Ito, H.; Watanabe, S.; Goto, H.; Gao, P.; Hughes, M.; Perez, D.R.; Donis, R.; Hoffmann, E.; Hobom, G.; Kawaoka, Y. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 1999, 96, 9345–9350. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; Tinevez, J.Y.; White, D.J.; Hartenstein, V.; Eliceiri, K.; Tomancak, P.; Cardona, A. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Knebel, A.; Botting, C.H.; Galloway, C.S.; Precious, B.L.; Jackson, D.; Elliott, R.M.; Randall, R.E. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology 2009, 383, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, T.Y.; Zhou, L.; Krug, R.M. Roles of the phosphorylation of specific serines and threonines in the NS1 protein of human influenza A viruses. J. Virol. 2012, 86, 10370–10376. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Gotoh, I.; Gotoh, Y.; Nishida, E. Cytoplasmic localization of mitogen-activated protein kinase kinase directed by its NH2-terminal, leucine-rich short amino acid sequence, which acts as a nuclear export signal. J. Biol. Chem. 1996, 271, 20024–20028. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.J.; Hanafusa, H. Mutagenic analysis of the v-crk oncogene: Requirement for SH2 and SH3 domains and correlation between increased cellular phosphotyrosine and transformation. J. Virol. 1990, 64, 3581–3589. [Google Scholar] [PubMed]
- Hrincius, E.R.; Wixler, V.; Wolff, T.; Wagner, R.; Ludwig, S.; Ehrhardt, C. CRK adaptor protein expression is required for efficient replication of avian influenza A viruses and controls JNK-mediated apoptotic responses. Cell 2010, 12, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Hrincius, E.R.; Liedmann, S.; Anhlan, D.; Wolff, T.; Ludwig, S.; Ehrhardt, C. Avian influenza viruses inhibit the major cellular signalling integrator c-Abl. Cell Microbiol. 2014, 16, 1854–1874. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Steel, J.; Manicassamy, B.; Medina, R.A.; Ye, J.; Hickman, D.; Lowen, A.C.; Perez, D.R.; Garcia-Sastre, A. Mutations in the NS1 C-terminal tail do not enhance replication or virulence of the 2009 pandemic H1N1 influenza A virus. J. Gen. Virol. 2010, 91, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Hrincius, E.R.; Liedmann, S.; Finkelstein, D.; Vogel, P.; Gansebom, S.; Ehrhardt, C.; Ludwig, S.; Hains, D.S.; Webby, R.; McCullers, J.A. Nonstructural protein 1 (NS1)-mediated inhibition of c-Abl results in acute lung injury and priming for bacterial co-infections: Insights into 1918 H1N1 pandemic? J. Infect. Dis. 2015, 211, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Oda, A.; Wakao, H.; Rhodes, J.; Druker, B.J.; Ishida, A.; Wakui, M.; Okamoto, S.; Morita, K.; Handa, M.; Komatsu, N.; Ohashi, H.; Miyajima, A.; Ikeda, Y. Thrombopoietin induces association of Crkl with STAT5 but not STAT3 in human platelets. Blood 1998, 92, 4652–4662. [Google Scholar] [PubMed]
- Uddin, S.; Lekmine, F.; Sassano, A.; Rui, H.; Fish, E.N.; Platanias, L.C. Role of Stat5 in type I interferon-signaling and transcriptional regulation. Biochem. Biophys. Res. Commun. 2003, 308, 325–330. [Google Scholar] [CrossRef]
- Lekmine, F.; Sassano, A.; Uddin, S.; Majchrzak, B.; Miura, O.; Druker, B.J.; Fish, E.N.; Imamoto, A.; Platanias, L.C. The CrkL adapter protein is required for type I interferon-dependent gene transcription and activation of the small G-protein Rap1. Biochem. Biophys. Res. Commun. 2002, 291, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Iwamatsu, A.; Hirano, N.; Ogawa, S.; Tanaka, T.; Mano, H.; Yazaki, Y.; Hirai, H. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. Embo J. 1994, 13, 3748–3756. [Google Scholar] [PubMed]
- Birge, R.B.; Fajardo, J.E.; Mayer, B.J.; Hanafusa, H. Tyrosine-phosphorylated epidermal growth factor receptor and cellular p130 provide high affinity binding substrates to analyze Crk-phosphotyrosine-dependent interactions in vitro. J. Biol. Chem. 1992, 267, 10588–10595. [Google Scholar] [PubMed]
- Van Etten, R.A.; Jackson, P.; Baltimore, D. The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell 1989, 58, 669–678. [Google Scholar] [CrossRef]
- Shishido, T.; Akagi, T.; Chalmers, A.; Maeda, M.; Terada, T.; Georgescu, M.M.; Hanafusa, H. Crk family adaptor proteins trans-activate c-Abl kinase. Genes Cells 2001, 6, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Brasher, B.B.; Van Etten, R.A. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 2000, 275, 35631–356317. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.M.; Ren, R.; Hanafusa, H.; Baltimore, D. SH2 and SH3 domains as molecular adhesives: The interactions of Crk and Abl. Trends Biochem. Sci. 1994, 19, 453–458. [Google Scholar] [CrossRef]
- Wurzer, W.J.; Planz, O.; Ehrhardt, C.; Giner, M.; Silberzahn, T.; Pleschka, S.; Ludwig, S. Caspase 3 activation is essential for efficient influenza virus propagation. Embo J. 2003, 22, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Ylösmäki, L.; Virtanen, J.; Vihinen-Ranta, M.; Saksela, K. (unpublished observations).





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ylösmäki, L.; Fagerlund, R.; Kuisma, I.; Julkunen, I.; Saksela, K. Nuclear Translocation of Crk Adaptor Proteins by the Influenza A Virus NS1 Protein. Viruses 2016, 8, 101. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040101
Ylösmäki L, Fagerlund R, Kuisma I, Julkunen I, Saksela K. Nuclear Translocation of Crk Adaptor Proteins by the Influenza A Virus NS1 Protein. Viruses. 2016; 8(4):101. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040101
Chicago/Turabian StyleYlösmäki, Leena, Riku Fagerlund, Inka Kuisma, Ilkka Julkunen, and Kalle Saksela. 2016. "Nuclear Translocation of Crk Adaptor Proteins by the Influenza A Virus NS1 Protein" Viruses 8, no. 4: 101. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040101