
Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus

 , and
, and 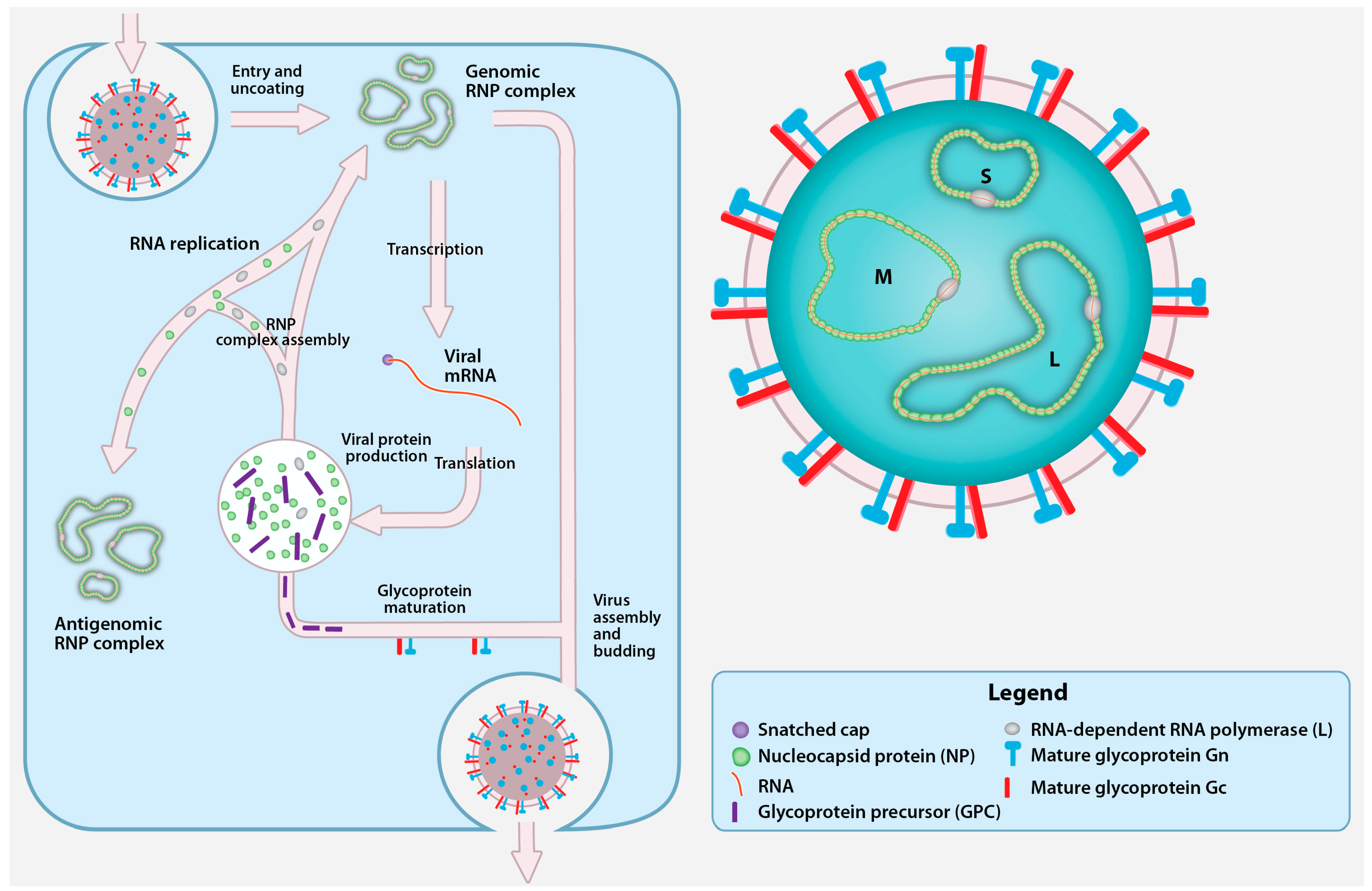{kind=link}
{kind=link}
{kind=link}
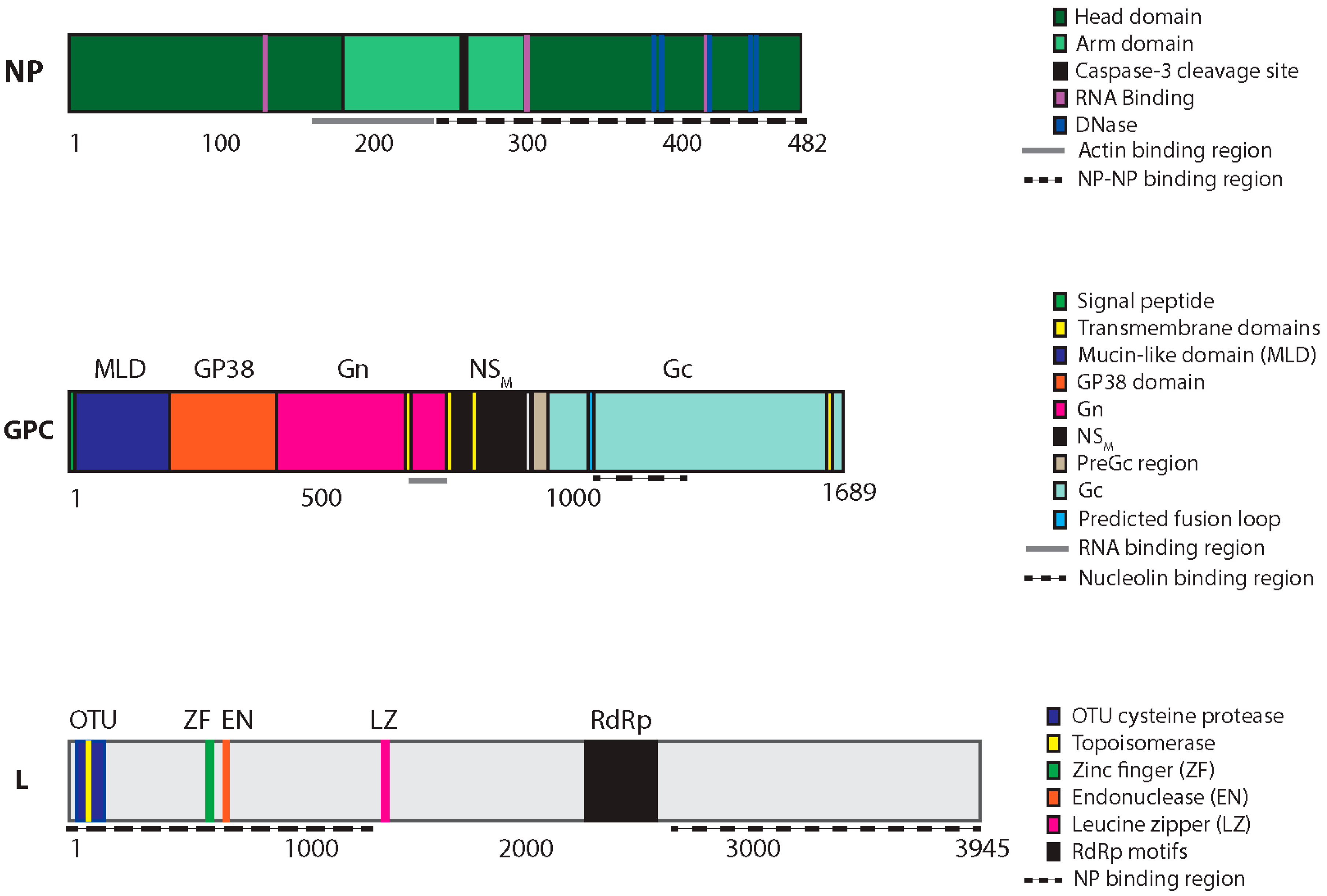{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CCHFV Genome and Replication Cycle
2.1. CCHFV Genome
2.2. Cell Entry
2.3. Transcription and Replication
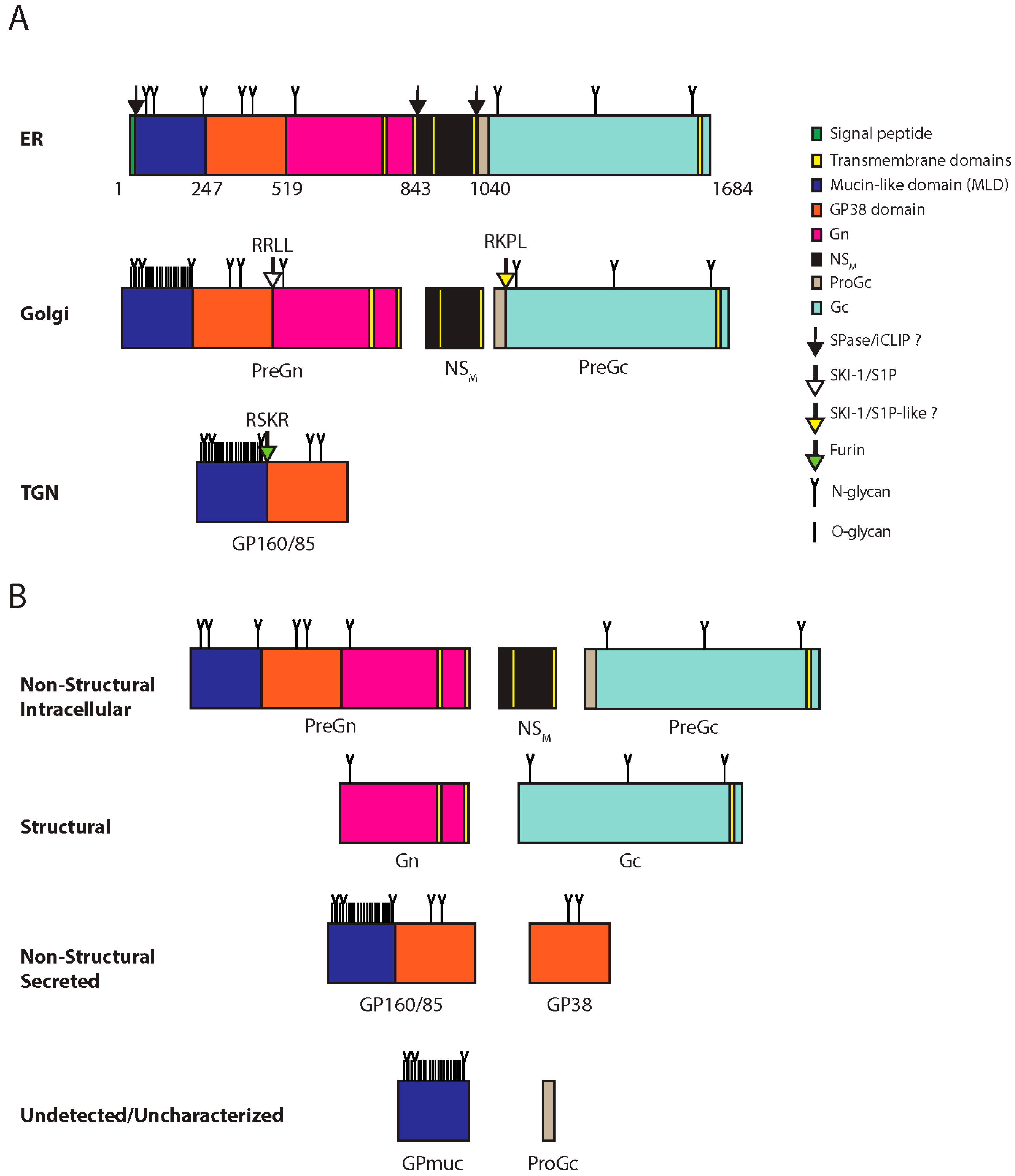
2.4. Glycoprotein Maturation, Viral Assembly, and Egress
3. Viral Protein Function
3.1. S Segment: NP and NSS
3.2. M Segment: Glycoproteins
3.3. L Segment: Ovarian Tumor Protease, Nuclease, and RdRp Activities
4. CCHFV Reverse Genetics
4.1. CCHFV Minigenome System
4.2. CCHFV Virus-like Particle System
4.3. CCHFV Infectious Clone System
5. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Ergönül, Ö. Crimean-Congo haemorrhagic fever. Lancet Infect. Dis. 2006, 6, 203–214. [Google Scholar]
- Whitehouse, C. A Crimean-Congo hemorrhagic fever. Antivir. Res. 2004, 64, 145–160. [Google Scholar] [CrossRef]
- Bente, D.A.; Forrester, N.L.; Watts, D.M.; McAuley, A.J.; Whitehouse, C.A.; Bray, M. Crimean-Congo hemorrhagic fever: History, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir. Res. 2013, 100, 159–189. [Google Scholar] [CrossRef] [PubMed]
- Vorou, R.; Pierroutsakos, I.N.; Maltezou, H.C. Crimean-Congo hemorrhagic fever. Curr. Opin. Infect. Dis. 2007, 20, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraal, H. The epidemiology of tick-borne Crimean-Congo hemorrhagic fever in Asia, Europe, and Africa. J. Med. Entomol. 1979, 15, 307–417. [Google Scholar] [CrossRef] [PubMed]
- Wolfel, R.; Paweska, J.T.; Petersen, N.; Grobbelaar, A.A.; Leman, P.A.; Hewson, R.; Georges-Courbot, M.-C.; Papa, A.; Günther, S.; Drosten, C. Virus detection and monitoring of viral load in Crimean-Congo hemorrhagic fever virus patients. Emerg. Infect. Dis. 2007, 13, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Drosten, C.; Bino, S.; Papadimitriou, E.; Panning, M.; Velo, E.; Kota, M.; Harxhi, A.; Antoniadis, A. Viral load and hemorrhagic fever. Emerg. Infect. Dis. 2007, 13, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Ergonul, O.; Tuncbilek, S.; Baykam, N.; Celikbas, A.; Dokuzoguz, B. Evaluation of serum levels of interleukin (IL)-6, IL-10, and tumor necrosis factor-alpha in patients with Crimean-Congo hemorrhagic fever. J. Infect. Dis. 2006, 193, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Ergonul, O.; Celikbas, A.; Baykam, N.; Eren, S.; Dokuzoguz, B. Analysis of risk-factors among patients with Crimean-Congo haemorrhagic fever virus infection: Severity criteria revisited. Clin. Microbiol. Infect. 2006, 12, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Duh, D.; Saksida, A.; Petrovec, M.; Ahmeti, S.; Dedushaj, I.; Panning, M.; Drosten, C.; Avsic-Zupanc, T. Viral load as predictor of Crimean-Congo hemorrhagic fever outcome. Emerg. Infect. Dis. 2007, 13, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Koksal, I.; Topbas, M.; Yilmaz, H.; Aksoy, F. The effectiveness of routine laboratory findings in determining disease severity in patients with Crimean-Congo hemorrhagic fever: Severity prediction criteria. J. Clin. Virol. 2010, 47, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Yesilyurt, M.; Gul, S.; Ozturk, B.; Kayhan, B.C.; Celik, M.; Uyar, C.; Erdogan, F. The early prediction of fatality in Crimean Congo hemorrhagic fever patients. Saudi Med. J. 2011, 32, 742–743. [Google Scholar] [PubMed]
- Weber, F.; Mirazimi, A. Interferon and cytokine responses to Crimean Congo hemorrhagic fever virus; an emerging and neglected viral zonoosis. Cytokine Growth Factor Rev. 2008, 19, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Saksida, A.; Duh, D.; Wraber, B.; Dedushaj, I.; Ahmeti, S.; Avsic-Zupanc, T. Interacting roles of immune mechanisms and viral load in the pathogenesis of Crimean-Congo hemorrhagic fever. Clin. Vaccine Immunol. 2010, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Bino, S.; Velo, E.; Harxhi, A.; Kota, M.; Antoniadis, A. Cytokine levels in Crimean-Congo hemorrhagic fever. J. Clin. Virol. 2006, 36, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Onguru, P.; Dagdas, S.; Bodur, H.; Yilmaz, M.; Akinci, E.; Eren, S.; Ozet, G. Coagulopathy parameters in patients with Crimean-Congo hemorrhagic fever and its relation with mortality. J. Clin. Lab. Anal. 2010, 24, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Cevik, M.A.; Erbay, A.; Bodur, H.; Gülderen, E.; Baştuğ, A.; Kubar, A.; Akinci, E. Clinical and laboratory features of Crimean-Congo hemorrhagic fever: Predictors of fatality. Int. J. Infect. Dis. 2008, 12, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Cevik, M.A.; Erbay, A.; Bodur, H.; Eren, S.S.; Akinci, E.; Sener, K.; Ongürü, P.; Kubar, A. Viral load as a predictor of outcome in Crimean-Congo hemorrhagic fever. Clin. Infect. Dis. 2007, 45, e96–e100. [Google Scholar] [CrossRef] [PubMed]
- Burt, F.J.; Swanepoel, R.; Shieh, W.J.; Smith, J.F.; Leman, P.A.; Greer, P.W.; Coffield, L.M.; Rollin, P.E.; Ksiazek, T.G.; Peters, C.J.; et al. Immunohistochemical and in situ localization of Crimean-Congo hemorrhagic fever (CCHF) virus in human tissues and implications for CCHF pathogenesis. Arch. Pathol. Lab. Med. 1997, 121, 839–846. [Google Scholar] [PubMed]
- Peyrefitte, C.N.; Perret, M.; Garcia, S.; Rodrigues, R.; Bagnaud, A.; Lacote, S.; Crance, J.-M.; Vernet, G.; Garin, D.; Bouloy, M.; et al. Differential activation profiles of Crimean-Congo hemorrhagic fever virus- and Dugbe virus-infected antigen-presenting cells. J. Gen. Virol. 2010, 91, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Connolly-Andersen, A.-M.; Douagi, I.; Kraus, A.A.; Mirazimi, A. Crimean Congo hemorrhagic fever virus infects human monocyte-derived dendritic cells. Virology 2009, 390, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Connolly-Andersen, A.-M.; Moll, G.; Andersson, C.; Akerström, S.; Karlberg, H.; Douagi, I.; Mirazimi, A. Crimean-Congo hemorrhagic fever virus activates endothelial cells. J. Virol. 2011, 85, 7766–7774. [Google Scholar] [CrossRef] [PubMed]
- ICTV. Virus Taxonomy 2014. Available online: http://www.ictvonline.org/virusTaxonomy.asp?bhcp=1 (accessed on 15 April 2016).
- Hewlett, M.J.; Pettersson, R.F.; Baltimore, D. Circular forms of Uukuniemi virion RNA: An electron microscopic study. J. Virol. 1977, 21, 1085–1093. [Google Scholar] [PubMed]
- Devignot, S.; Bergeron, E.; Nichol, S.; Mirazimi, A.; Weber, F. A Virus-like particle system identifies the endonuclease domain of Crimean-Congo hemorrhagic fever virus. J. Virol. 2015, 89, 5957–5967. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, E.; Albariño, C.G.; Khristova, M.L.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus-encoded ovarian tumor protease activity is dispensable for virus RNA polymerase function. J. Virol. 2010, 84, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Zivcec, M.; Metcalfe, M.G.; Albariño, C.G.; Guerrero, L.W.; Pegan, S.D.; Spiropoulou, C.F.; Bergeron, É. Assessment of inhibitors of pathogenic Crimean-Congo hemorrhagic fever virus strains using virus-like particles. PLoS Negl. Trop. Dis. 2015, 9, e0004259. [Google Scholar] [CrossRef] [PubMed]
- Barnwal, B.; Karlberg, H.; Mirazimi, A.; Tan, Y.-J. Non-structural protein of Crimean-Congo hemorrhagic fever virus disrupts mitochondrial membrane potential and induces apoptosis. J. Biol. Chem. 2015, 291, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Albariño, C.G.; Bird, B.H.; Nichol, S.T. A shared transcription termination signal on negative and ambisense RNA genome segments of Rift Valley fever, sandfly fever Sicilian, and Toscana viruses. J. Virol. 2007, 81, 5246–5256. [Google Scholar] [CrossRef] [PubMed]
- Pinschewer, D.D.; Perez, M.; de La Torre, J.C. Dual role of the lymphocytic choriomeningitis virus intergenic region in transcription termination and virus propagation. J. Virol. 2005, 79, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.J.; Vincent, M.J.; Nichol, S.T. Characterization of the glycoproteins of Crimean-Congo hemorrhagic fever virus. J. Virol. 2002, 76, 7263–7275. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-R.; Wang, M.-L.; Deng, F.; Li, T.-X.; Hu, Z.-H.; Wang, H.-L. Production of CCHF virus-like particle by a baculovirus-insect cell expression system. Virol. Sin. 2011, 26, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.R.; King, J.B.; Rossouw, D.J.; Cooper, R. A nosocomial outbreak of Crimean-Congo haemorrhagic fever at Tygerberg Hospital Part III. Clinical pathology and pathogenesis. S. Afr. Med. J. 1985, 68, 722–728. [Google Scholar] [PubMed]
- Hardestam, J.; Simon, M.; Hedlund, K.O.; Vaheri, A.; Klingström, J.; Lundkvist, A. Ex vivo stability of the rodent-borne Hantaan virus in comparison to that of arthropod-borne members of the Bunyaviridae family. Appl. Environ. Microbiol. 2007, 73, 2547–2551. [Google Scholar] [CrossRef] [PubMed]
- Korolev, M.B.; Donets, M.A.; Rubin, S.G.; Chumakov, M.P. Morphology and morphogenesis of Crimean hemorrhagic fever virus. Arch. Virol. 1976, 50, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Donets, M.A.; Chumakov, M.P.; Korolev, M.B.; Rubin, S.G. Physicochemical characteristics, morphology and morphogenesis of virions of the causative agent of Crimean hemorrhagic fever. Intervirology 1977, 8, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti-ciarlet, A.; Smith, J.; Strecker, K.; Paragas, J.; Altamura, L.A.; Mcfalls, J.M.; Frias-sta, N.; Schmaljohn, C.S.; Doms, R.W. Cellular localization and antigenic characterization of Crimean-Congo hemorrhagic fever virus glycoproteins. J. Virol. 2005, 79, 6152–6161. [Google Scholar] [CrossRef] [PubMed]
- Garry, C.E.; Garry, R.F. Proteomics computational analyses suggest that the carboxyl terminal glycoproteins of Bunyaviruses are class II viral fusion protein (beta-penetrenes). Theor. Biol. Med. Model. 2004, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessau, M.; Modis, Y. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc. Natl. Acad. Sci. USA 2013, 110, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Feng, Y.; Zhu, Z.; Dimitrov, D.S. Identification of a putative Crimean-Congo hemorrhagic fever virus entry factor. Biochem. Biophys. Res. Commun. 2011, 411, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Tayyari, F.; Marchant, D.; Moraes, T.J.; Duan, W.; Mastrangelo, P.; Hegele, R.G. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 2011, 17, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Thongtan, T.; Wikan, N.; Wintachai, P.; Rattanarungsan, C.; Srisomsap, C.; Cheepsunthorn, P.; Smith, D.R. Characterization of putative Japanese encephalitis virus receptor molecules on microglial cells. J. Med. Virol. 2012, 84, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Tajrishi, M.M.; Tuteja, R.; Tuteja, N. Nucleolin: The most abundant multifunctional phosphoprotein of nucleolus. Commun. Integr. Biol. 2011, 4, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Tokunaga, K.; Sawa, H.; Maeda, M.; Chiba, J.; Kojima, A.; Hasegawa, H.; Shoya, Y.; Sata, T.; Kurata, T.; et al. Nucleolin and the packaging signal, psi, promote the budding of human immunodeficiency virus type-1 (HIV-1). Microbiol. Immunol. 2004, 48, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Johansson, C.; Mirazimi, A. Crimean-Congo hemorrhagic fever virus entry and replication is clathrin-, pH- and cholesterol-dependent. J. Gen. Virol. 2009, 90, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.R.; Radoshitzky, S.R.; Kota, K.P.; Pegoraro, G.; Ruthel, G.; Kuhn, J.H.; Altamura, L.A.; Kwilas, S.A.; Bavari, S.; Haucke, V.; et al. Crimean-Congo hemorrhagic fever virus utilizes a clathrin- and early endosome-dependent entry pathway. Virology 2013, 444, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Shtanko, O.; Nikitina, R.A.; Altuntas, C.Z.; Chepurnov, A.A.; Davey, R.A. Crimean-Congo hemorrhagic fever virus entry into host cells occurs through the multivesicular body and requires ESCRT regulators. PLoS Pathog. 2014, 10, e1004390. [Google Scholar] [CrossRef] [PubMed]
- Morin, B.; Coutard, B.; Lelke, M.; Ferron, F.; Kerber, R.; Jamal, S.; Frangeul, A.; Baronti, C.; Charrel, R.; de Lamballerie, X.; et al. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog. 2010, 6, e1001038. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Weber, F.; Cusack, S. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010, 6, e10011101. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Bouvier, D.; Crépin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.J.; Vincent, M.J.; Erickson, B.R.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus glycoprotein precursor is cleaved by Furin-like and SKI-1 proteases to generate a novel 38-kilodalton glycoprotein. J. Virol. 2006, 80, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Altamura, L.A.; Bertolotti-Ciarlet, A.; Teigler, J.; Paragas, J.; Schmaljohn, C.S.; Doms, R.W. Identification of a novel C-terminal cleavage of Crimean-Congo hemorrhagic fever virus PreGN that leads to generation of an NSM protein. J. Virol. 2007, 81, 6632–6642. [Google Scholar] [CrossRef] [PubMed]
- Estrada, D.F.; De Guzman, R.N. Structural characterization of the Crimean-Congo hemorrhagic fever virus Gn tail provides insight into virus assembly. J. Biol. Chem. 2011, 286, 21678–21686. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, E.; Vincent, M.J.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus glycoprotein processing by the endoprotease SKI-1/S1P is critical for virus infectivity. J. Virol. 2007, 81, 13271–13276. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.R.; Deyde, V.; Sanchez, A.J.; Vincent, M.J.; Nichol, S.T. N-linked glycosylation of Gn (but not Gc) is important for Crimean Congo hemorrhagic fever virus glycoprotein localization and transport. Virology 2007, 361, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, É.; Zivcec, M.; Chakrabarti, A.K.; Nichol, S.T.; Albariño, C.G.; Spiropoulou, C.F. Recovery of recombinant Crimean Congo hemorrhagic fever virus reveals a function for non-structural glycoproteins cleavage by furin. PLoS Pathog. 2015, 11, e1004879. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Fernando, L.; Schwarz, T.F.; Feldmann, H.; Flick, R. Intracellular localization of Crimean-Congo hemorrhagic fever (CCHF) virus glycoproteins. Virol. J. 2005, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Sanchez, A.J.; Erickson, B.R.; Basak, A.; Chretien, M.; Seidah, N.G.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus glycoprotein proteolytic processing by subtilase SKI-1. J. Virol. 2003, 77, 8640–8649. [Google Scholar] [CrossRef] [PubMed]
- Pasquato, A.; de Palma, J.R.; Galan, C.; Seidah, N.G.; Kunz, S. Viral envelope glycoprotein processing by proprotein convertases. Antivir. Res. 2013, 99, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Lee, A.M.; Nguyen, N.; Spiropoulou, C.F.; Kunz, S. Site 1 protease is required for proteolytic processing of the glycoproteins of the South American hemorrhagic fever viruses Junin, Machupo, and Guanarito. J. Virol. 2008, 82, 6045–6051. [Google Scholar] [CrossRef] [PubMed]
- Andersson, I.; Simon, M.; Lundkvist, A.; Nilsson, M.; Holmström, A.; Elgh, F.; Mirazimi, A. Role of actin filaments in targeting of Crimean Congo hemorrhagic fever virus nucleocapsid protein to perinuclear regions of mammalian cells. J. Med. Virol. 2004, 72, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Andersson, I.; Bladh, L.; Mousavi-jazi, M.; Magnusson, K.; Lundkvist, A.; Haller, O.; Mirazimi, A. Human MxA protein inhibits the replication of Crimean-Congo hemorrhagic fever virus. J. Virol. 2004, 78, 4323–4329. [Google Scholar] [CrossRef] [PubMed]
- Connolly-Andersen, A.M.; Magnusson, K.E.; Mirazimi, A. Basolateral entry and release of Crimean-Congo hemorrhagic fever virus in polarized MDCK-1 cells. J. Virol. 2007, 81, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Macleod, J.M.L.; Marmor, H.; García-Sastre, A.; Frias-Staheli, N. Mapping of the interaction domains of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J. Gen. Virol. 2015, 96, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.D.; Surtees, R.; Walter, C.T.; Ariza, A.; Bergeron, É.; Nichol, S.T.; Hiscox, J.A.; Edwards, T.A.; Barr, J.N. Structure, function, and evolution of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J. Virol. 2012, 86, 10914–10923. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, X.; Wang, X.; Dong, H.; Ma, C.; Wang, J.; Liu, B.; Mao, Y.; Wang, Y.; Li, T.; et al. Structural and functional diversity of nairovirus-encoded nucleoproteins. J. Virol. 2015, 89, 11740–11749. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, W.; Ji, W.; Deng, M.; Sun, Y.; Zhou, H.; Yang, C.; Deng, F.; Wang, H.; Hu, Z.; et al. Crimean-Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, 5046–5051. [Google Scholar] [CrossRef] [PubMed]
- Hastie, K.M.; Liu, T.; Li, S.; King, L.B.; Ngo, N.; Zandonatti, M.A.; Woods, V.L.; de la Torre, J.C.; Saphire, E.O. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc. Natl. Acad. Sci. USA 2011, 108, 19365–19370. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.D.; Piper, M.E.; Gerrard, S.R.; Smith, J.L. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc. Natl. Acad. Sci. USA 2010, 107, 11769–11774. [Google Scholar] [CrossRef] [PubMed]
- Olal, D.; Dick, A.; Woods, V.L.; Liu, T.; Li, S.; Devignot, S.; Weber, F.; Saphire, E.O.; Daumke, O. Structural insights into RNA encapsidation and helical assembly of the Toscana virus nucleoprotein. Nucleic Acids Res. 2014, 42, 6025–6037. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Malet, H.; Weber, F.; Cusack, S. Structural basis for encapsidation of genomic RNA by La Crosse Orthobunyavirus nucleoprotein. Proc. Natl. Acad. Sci. USA 2013, 110, 7246–7251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dutta, S.; Karlberg, H.; Devignot, S.; Weber, F.; Hao, Q.; Tan, Y.J.; Mirazimi, A.; Kotaka, M. Structure of Crimean-Congo hemorrhagic fever virus nucleoprotein: Superhelical homo-oligomers and the role of caspase-3 cleavage. J. Virol. 2012, 86, 12294–12303. [Google Scholar] [CrossRef] [PubMed]
- Burt, F.J.; Swanepoel, R.; Braack, L.E.O. Enzyme-linked immunosorbent assays for the detection of antibody to Crimean-Congo haemorrhagic fever virus in the sera of livestock and wild vertebrates. Epidemiol. Infect. 2009, 111, 547–558. [Google Scholar] [CrossRef]
- Burt, F.J.; Samudzi, R.R.; Randall, C.; Pieters, D.; Vermeulen, J.; Knox, C.M. Human defined antigenic region on the nucleoprotein of Crimean-Congo hemorrhagic fever virus identified using truncated proteins and a bioinformatics approach. J. Virol. Methods 2013, 193, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Luo, Y.; Li, T.; Wang, H.; Hu, Z.; Zhang, F.; Zhang, Y.; Deng, F.; Sun, S. Serial expression of the truncated fragments of the nucleocapsid protein of CCHFV and identification of the epitope region. Virol. Sin. 2010, 25, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Mousavi-Jazi, M.; Karlberg, H.; Papa, A.; Christova, I.; Mirazimi, A. Healthy individuals’ immune response to the Bulgarian Crimean-Congo hemorrhagic fever virus vaccine. Vaccine 2012, 30, 6225–6229. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.R.; Prescott, J.; Brown, K.S.; Best, S.M.; Ebihara, H.; Feldmann, H. Antagonism of type I interferon responses by new world hantaviruses. J. Virol. 2010, 84, 11790–11801. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, H.; Tan, Y.J.; Mirazimi, A. Crimean-Congo haemorrhagic fever replication interplays with regulation mechanisms of apoptosis. J. Gen. Virol. 2015, 96, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, H.; Tan, Y.-J.; Mirazimi, A. Induction of caspase activation and cleavage of the viral nucleocapsid protein in different cell types during Crimean-Congo hemorrhagic fever virus infection. J. Biol. Chem. 2011, 286, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Becker, S.; Groseth, A. Cleavage of the Junin virus nucleoprotein serves a decoy function to inhibit the induction of apoptosis during infection. J. Virol. 2013, 87, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Volchkova, V.A.; Ströher, U.; Becker, S.; Dolnik, O.; Cieplik, M.; Garten, W.; Klenk, H.D.; Feldmann, H. Proteolytic processing of Marburg virus glycoprotein. Virology 2000, 268, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fusco, M.L.; Hashiguchi, T.; Cassan, R.; Biggins, J.E.; Murin, C.D.; Warfield, K.L.; Li, S.; Holtsberg, F.W.; Shulenin, S.; Vu, H.; et al. Protective mAbs and cross-reactive mAbs raised by immunization with engineered marburg virus GPs. PLoS Pathog. 2015, 11, e1005016. [Google Scholar]
- Honig, J.E.; Osborne, J.C.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus genome L RNA segment and encoded protein. Virology 2004, 321, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, E.; Martin, S.G.; Grolla, A.; Czub, M.; Feldmann, H.; Flick, R. Sequence determination of the Crimean-Congo hemorrhagic fever virus L segment. Virology 2004, 321, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingström, J.; Schümann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Mühlberger, E.; et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 2008, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Spengler, J.R.; Patel, J.R.; Chakrabarti, A.K.; Zivcec, M.; García-Sastre, A.; Spiropoulou, C.F.; Bergeron, E. RIG-I mediates an antiviral response to Crimean-Congo hemorrhagic fever virus. J. Virol. 2015, 89, 10119–10229. [Google Scholar] [CrossRef] [PubMed]
- Garcin, D.; Lezzi, M.; Dobbs, M.; Elliott, R.M.; Schmaljohn, C.; Kang, C.Y.; Kolakofsky, D. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J. Virol. 1995, 69, 5754–5762. [Google Scholar] [PubMed]
- Yao, H.; Dittmann, M.; Peisley, A.; Hoffmann, H.H.; Gilmore, R.H.; Schmidt, T.; Schmid-Burgk, J.L.; Hornung, V.; Rice, C.M.; Hur, S. ATP-Dependent effector-like functions of RIG-I-like receptors. Mol. Cell 2015, 58, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Lalwani, P.; Raftery, M.J.; Matthaei, M.; Lutteke, N.; Kirsanovs, S.; Binder, M.; Ulrich, R.G.; Giese, T.; Wolff, T.; et al. RNA helicase retinoic acid-inducible gene I (RIG-I) as a sensor of Hantaan virus (HTNV) replication. J. Gen. Virol. 2011, 92, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Frias-Staheli, N.; Giannakopoulos, N.V.; Kikkert, M.; Taylor, S.L.; Bridgen, A.; Paragas, J.; Richt, J.A.; Rowland, R.R.; Schmaljohn, C.S.; Lenschow, D.J.; et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2007, 2, 404–416. [Google Scholar] [CrossRef] [PubMed]
- James, T.W.; Frias-Staheli, N.; Bacik, J.-P.; Levingston Macleod, J.M.; Khajehpour, M.; García-Sastre, A.; Mark, B.L. Structural basis for the removal of ubiquitin and interferon-stimulated gene 15 by a viral ovarian tumor domain-containing protease. Proc. Natl. Acad. Sci. USA 2011, 108, 2222–2227. [Google Scholar] [CrossRef] [PubMed]
- Van Kasteren, P.B.; Beugeling, C.; Ninaber, D.K.; Frias-Staheli, N.; van Boheemen, S.; García-Sastre, A.; Snijder, E.J.; Kikkert, M. Arterivirus and nairovirus ovarian tumor domain-containing Deubiquitinases target activated RIG-I to control innate immune signaling. J. Virol. 2012, 86, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem. Sci. 2000, 25, 50–52. [Google Scholar] [CrossRef]
- Holzer, B.; Bakshi, S.; Bridgen, A.; Baron, M.D. Inhibition of interferon induction and action by the nairovirus Nairobi sheep disease virus/Ganjam virus. PLoS ONE 2011, 6, e28594. [Google Scholar] [CrossRef] [PubMed]
- Mielech, A.M.; Kilianski, A.; Baez-Santos, Y.M.; Mesecar, A.D.; Baker, S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 2014, 450–451, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Chen, G.; Guo, B.; Cheng, G.; Tang, H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008, 18, 1105–1113. [Google Scholar] [PubMed]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Yang, J.; Sun, H.-R.; Xin, X.; Wang, H.-D.; Chen, J.-P.; Adams, M.J. Genomic analysis of rice stripe virus Zhejiang isolate shows the presence of an OTU-like domain in the RNA1 protein and a novel sequence motif conserved within the intergenic regions of ambisense segments of tenuiviruses. Arch. Virol. 2007, 152, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, C.; Ayach, M.; Beaurepaire, L.; Chenon, M.; Andreani, J.; Guerois, R.; Jupin, I.; Bressanelli, S. A compact viral processing proteinase/ubiquitin hydrolase from the OTU family. PLoS Pathog. 2013, 9, e1003560. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A deubiquitinase that regulates type I interferon production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 973–959. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Okumura, A.J.; Uematsu, K.; Hatakeyama, S.; Zhang, D.E.; Kamura, T. Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 2013, 288, 2839–2847. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. TRIM26 negatively regulates interferon-β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 2015, 11, e1004726. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.-X.; Yang, K.; Liu, X.; Liu, X.-Y.; Wei, B.; Shan, Y.-F.; Zhu, L.-H.; Wang, C. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell. Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, N.V.; Luo, J.-K.; Papov, V.; Zou, W.; Lenschow, D.J.; Jacobs, B.S.; Borden, E.C.; Li, J.; Virgin, H.W.; Zhang, D.-E. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem. Biophys. Res. Commun. 2005, 336, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Ye, Y.; Virdee, S.; Chin, J.W.; Komander, D. Molecular basis for ubiquitin and ISG15 cross-reactivity in viral ovarian tumor domains. Proc. Natl. Acad. Sci. USA 2011, 108, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Capodagli, G.C.; McKercher, M.A.; Baker, E.A.; Masters, E.M.; Brunzelle, J.S.; Pegan, S.D. Structural analysis of a viral ovarian tumor domain protease from the Crimean-Congo hemorrhagic fever virus in complex with covalently bonded ubiquitin. J. Virol. 2011, 85, 3621–3630. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Hwang, S.-Y.; Imaizumi, T.; Yoo, J.-Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Kohl, A.; Dunn, E.F.; Lowen, A.C.; Elliott, R.M. Complementarity, sequence and structural elements within the 3′ and 5′ non-coding regions of the Bunyamwera orthobunyavirus S segment determine promoter strength. J. Gen. Virol. 2004, 85, 3269–3278. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.N.; Wertz, G.W. Role of the conserved nucleotide mismatch within 3′- and 5′-terminal regions of Bunyamwera virus in signaling transcription. J. Virol. 2005, 79, 3586–3594. [Google Scholar] [CrossRef] [PubMed]
- Duygu, F.; Kaya, T.; Baysan, P. Re-evaluation of 400 Crimean-Congo hemorrhagic fever cases in an endemic area: Is ribavirin treatment suitable? Vector Borne Zoonotic Dis. 2012, 12, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Koksal, I.; Yilmaz, G.; Aksoy, F.; Aydin, H.; Yavuz, I.; Iskender, S.; Akcay, K.; Erensoy, S.; Caylan, R.; Aydin, K. The efficacy of ribavirin in the treatment of Crimean-Congo hemorrhagic fever in Eastern Black Sea region in Turkey. J. Clin. Virol. 2010, 47, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Soares-Weiser, K.; Thomas, S.; Thomson, G.; Garner, P. Ribavirin for Crimean-Congo hemorrhagic fever: Systematic review and meta-analysis. BMC Infect. Dis. 2010, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Bell-Sakyi, L.; Kohl, A.; Bente, D.A.; Fazakerley, J.K. Tick cell lines for study of Crimean-Congo hemorrhagic fever virus and other arboviruses. Vector Borne Zoonotic Dis. 2012, 12, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Gargili, A.; Thangamani, S.; Bente, D. Influence of laboratory animal hosts on the life cycle of Hyalomma marginatum and implications for an in vivo transmission model for Crimean-Congo hemorrhagic fever virus. Front. Cell. Infect. Microbiol. 2013, 3, 39. [Google Scholar] [CrossRef] [PubMed]
- Bente, D.A.; Alimonti, J.B.; Shieh, W.-J.; Camus, G.; Ströher, U.; Zaki, S.; Jones, S.M. Pathogenesis and immune response of Crimean-Congo hemorrhagic fever virus in a STAT-1 knockout mouse model. J. Virol. 2010, 84, 11089–11100. [Google Scholar] [CrossRef] [PubMed]
- Oestereich, L.; Rieger, T.; Neumann, M.; Bernreuther, C.; Lehmann, M.; Krasemann, S.; Wurr, S.; Emmerich, P.; de Lamballerie, X.; Ölschläger, S.; et al. Evaluation of antiviral efficacy of ribavirin, arbidol, and T-705 (favipiravir) in a mouse model for Crimean-Congo hemorrhagic fever. PLoS Negl. Trop. Dis. 2014, 8, e2804. [Google Scholar] [CrossRef] [PubMed]
- Dowall, S.D.; Buttigieg, K.R.; Findlay-Wilson, S.J.D.; Rayner, E.; Pearson, G.; Miloszewska, A.; Graham, V.A.; Carroll, M.; Hewson, R. A Crimean-Congo Haemorrhagic Fever (CCHF) viral vaccine expressing nucleoprotein is immunogenic but fails to confer protection against lethal disease. Hum. Vaccin. Immunother. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, J.; Vloet, R.P.M.; McAuley, A.J.; Shen, X.; Bosch, B.J.; de Vries, L.; Moormann, R.J.M.; Bente, D.A. Crimean-Congo hemorrhagic fever virus subunit vaccines induce high levels of neutralizing Antibodies but no protection in STAT1 knockout mice. Vector Borne Zoonotic Dis. 2015, 15, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Canakoglu, N.; Berber, E.; Tonbak, S.; Ertek, M.; Sozdutmaz, I.; Aktas, M.; Kalkan, A.; Ozdarendeli, A. Immunization of knock-out α/β interferon receptor mice against high lethal dose of Crimean-Congo hemorrhagic fever virus with a cell culture based vaccine. PLoS Negl. Trop. Dis. 2015, 11, e0003579. [Google Scholar] [CrossRef] [PubMed]
- Buttigieg, K.R.; Dowall, S.D.; Findlay-Wilson, S.; Miloszewska, A.; Rayner, E.; Hewson, R.; Carroll, M.W. A novel vaccine against Crimean-Congo haemorrhagic fever protects 100% of animals against lethal challenge in a mouse model. PLoS ONE 2014, 9, e91596. [Google Scholar] [CrossRef] [PubMed]
- Andersson, I.; Lundkvist, Å.; Haller, O.; Mirazimi, A. Type I Interferon inhibits Crimean-Congo hemorrhagic fever virus in human target cells. J. Med. Virol. 2006, 222, 216–222. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zivcec, M.; Scholte, F.E.M.; Spiropoulou, C.F.; Spengler, J.R.; Bergeron, É. Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus. Viruses 2016, 8, 106. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040106
Zivcec M, Scholte FEM, Spiropoulou CF, Spengler JR, Bergeron É. Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus. Viruses. 2016; 8(4):106. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040106
Chicago/Turabian StyleZivcec, Marko, Florine E. M. Scholte, Christina F. Spiropoulou, Jessica R. Spengler, and Éric Bergeron. 2016. "Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus" Viruses 8, no. 4: 106. https://0-doi-org.brum.beds.ac.uk/10.3390/v8040106