
HACE1 Negatively Regulates Virus-Triggered Type I IFN Signaling by Impeding the Formation of the MAVS-TRAF3 Complex

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Plasmids
2.3. RNA Interference
2.4. Luciferase Assays
2.5. RT-PCR and Real-Time PCR
2.6. Co-Immunoprecipitation and Immunoblot Analysis
2.7. Statistical Analysis
3. Results
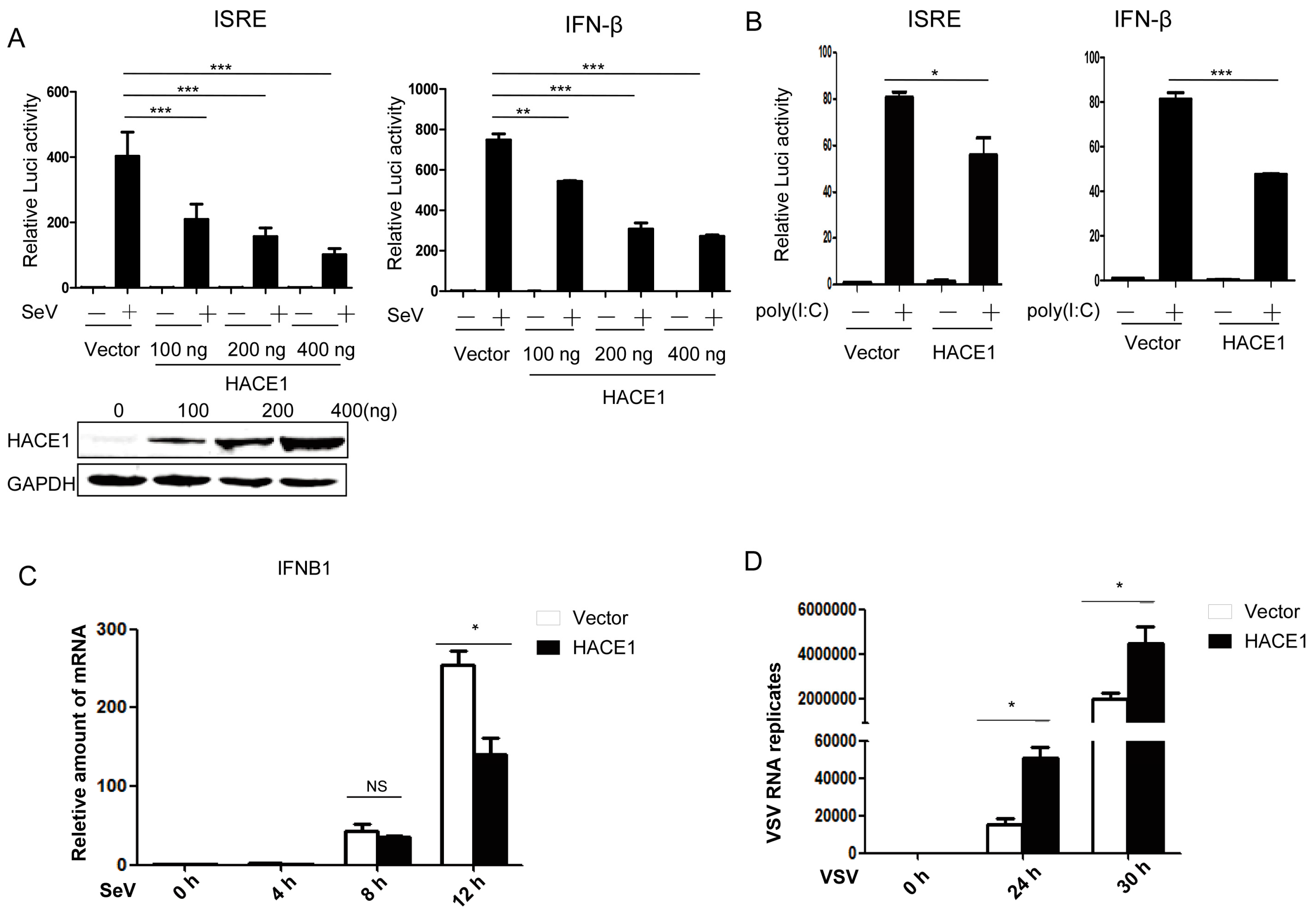
3.1. HACE1 Negatively Regulates Virus-Induced Type I IFN Signaling
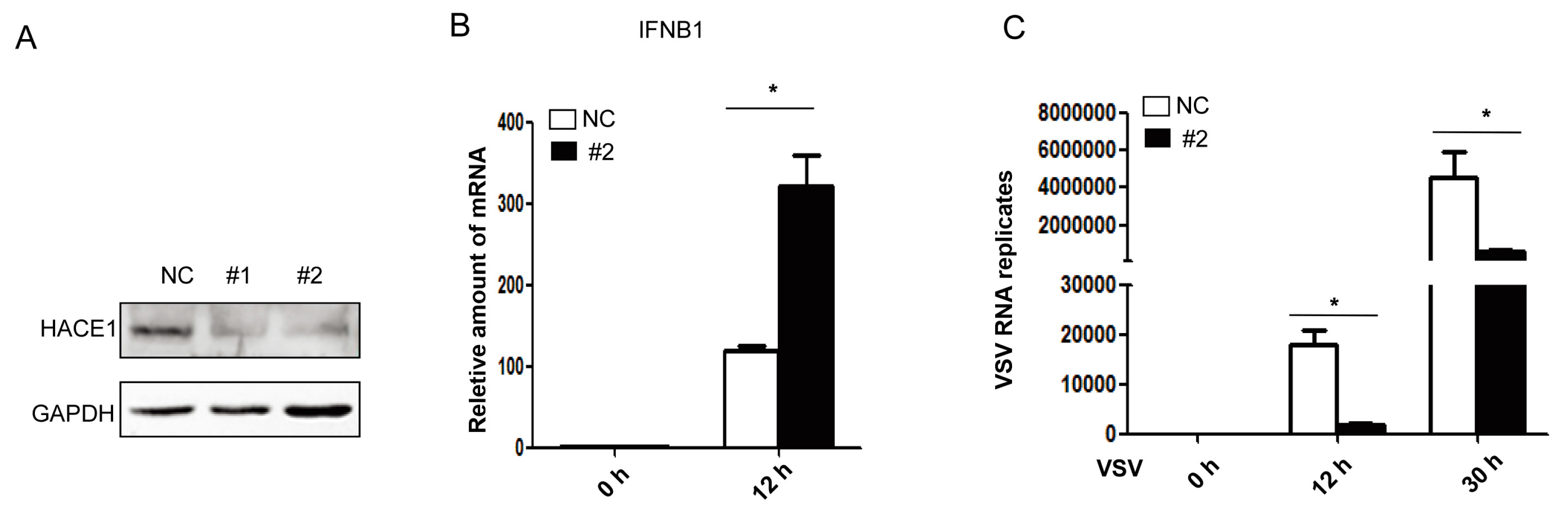
3.2. Knockdown of HACE1 Augments Virus-Induced Type I IFN Signaling
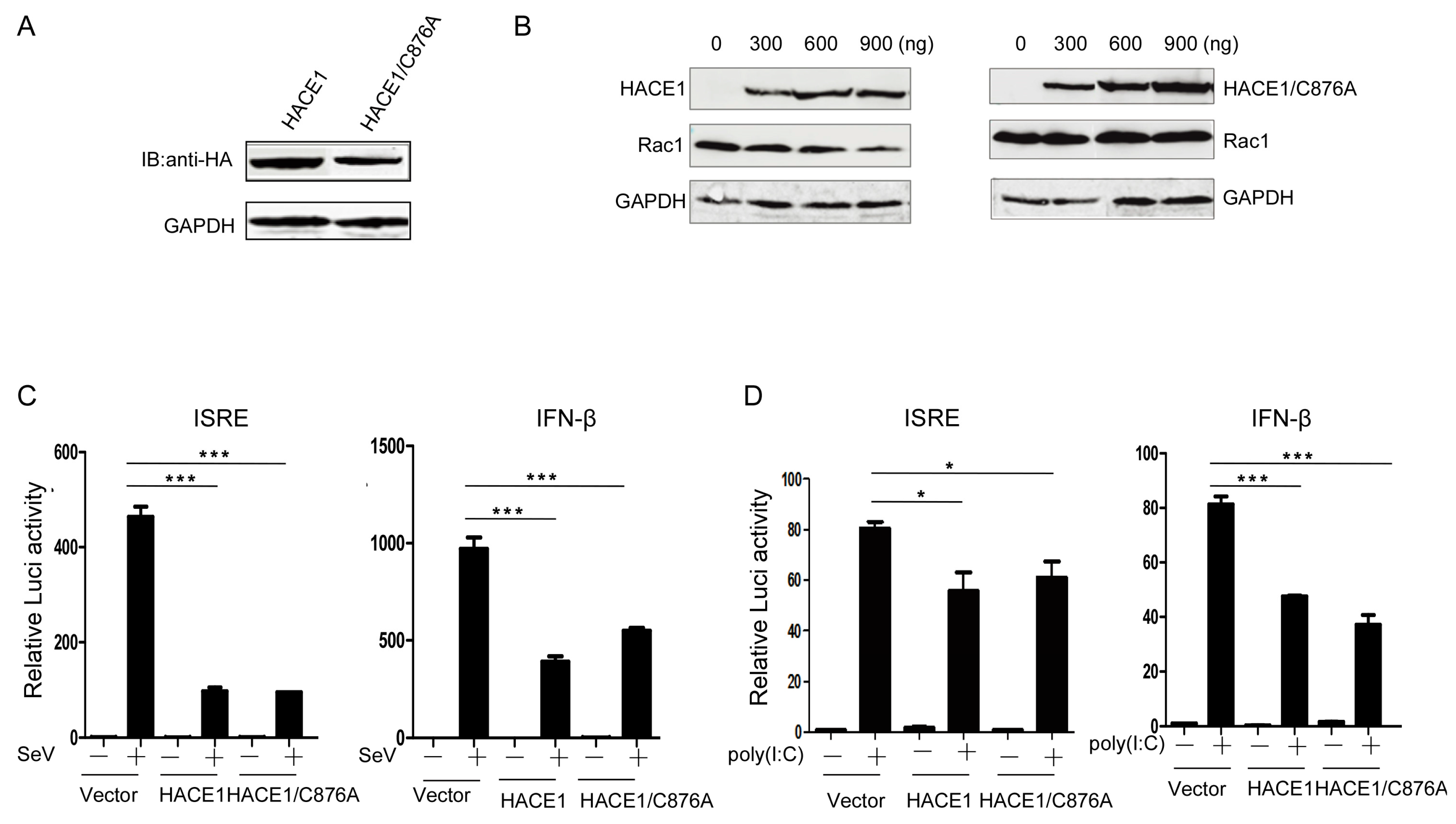
3.3. The Suppressive Function of HACE1 Is Independent of Its E3 Ligase Activity
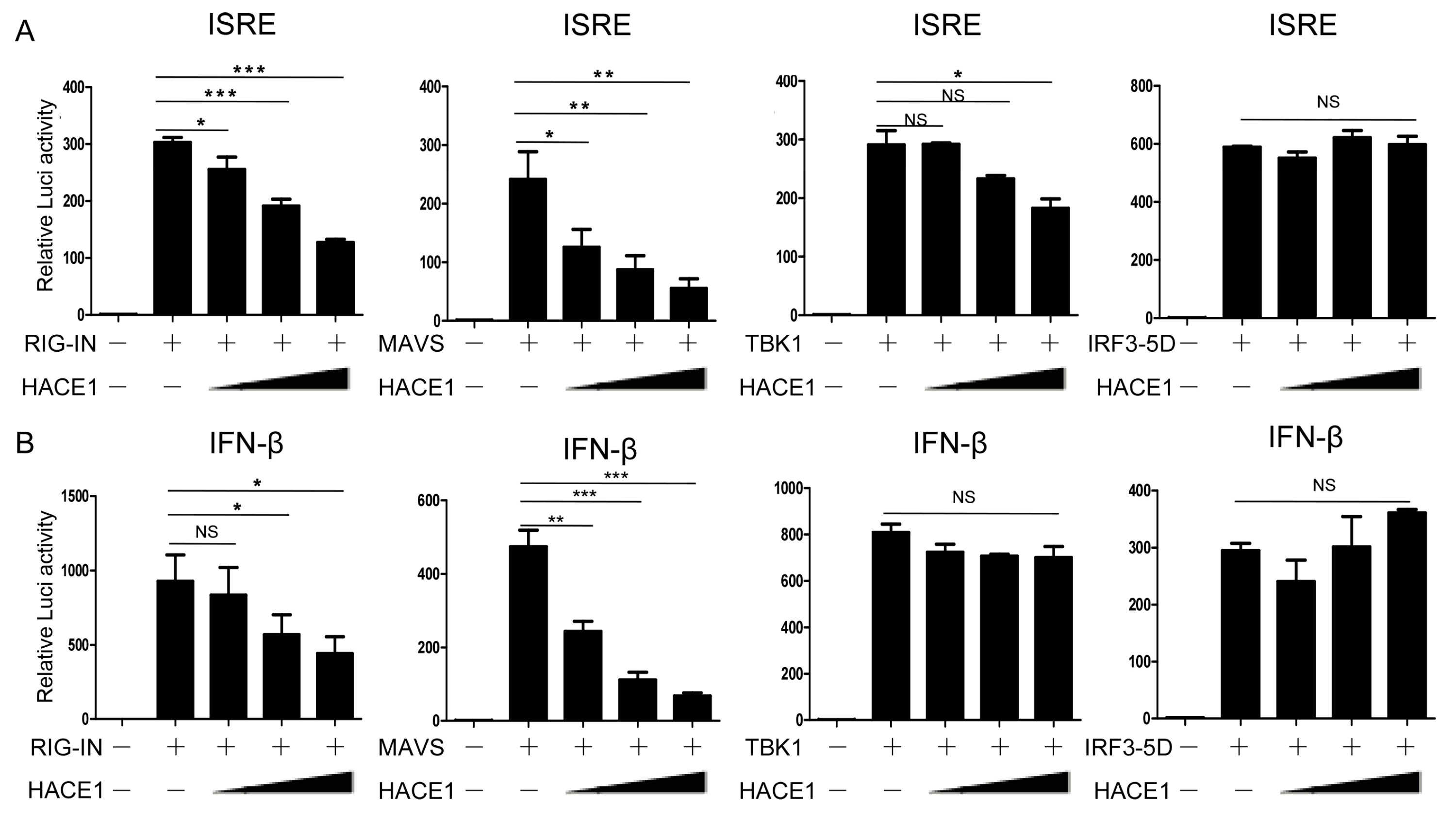
3.4. HACE1 Negatively Regulates Virus-Triggered Signaling Downstream of MAVS and Upstream of TBK1
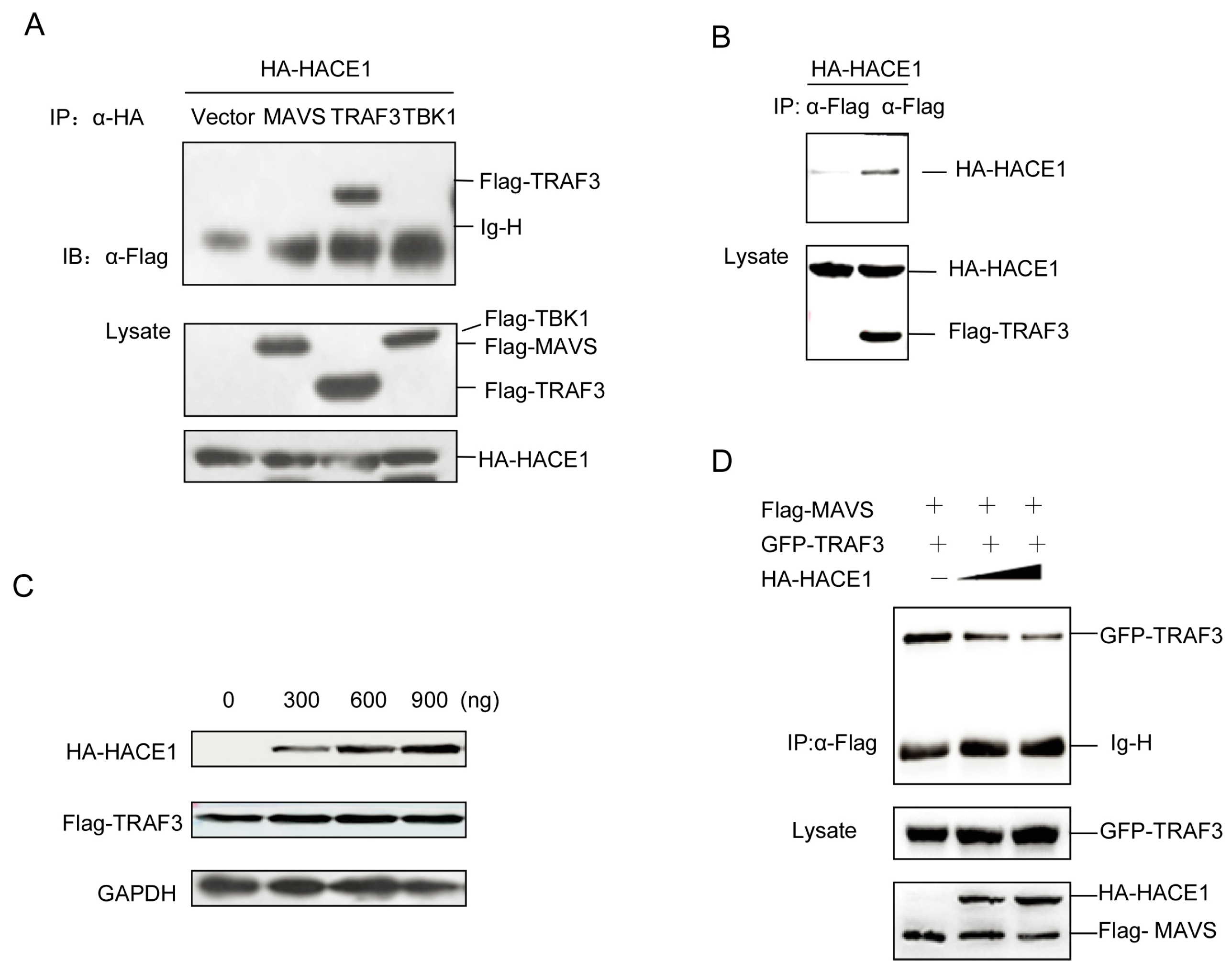
3.5. HACE1 Suppresses Virus-Induced Signaling by Disrupting the MAVS-TRAF3 Complex
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbiol. 2010, 13, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S. Innate recognition of viruses. Immunol. Lett. 2010, 128, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Nan, G.; Zhang, Y.J. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 2014, 6, 4999–5027. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.J.; Davis, M.E.; Gack, M.U. Regulation of RIG-I-like receptor signaling by host and viral proteins. Cytokine Growth Factor Rev. 2014, 25, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Garcia-Sastre, A. Activation and regulation of pathogen sensor RIG-I. Cytokine Growth Factor Rev. 2014, 25, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 2010, 20, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Weber, F. RIG-I-like receptors and negative-strand RNA viruses: RLRly bird catches some worms. Cytokine Growth Factor Rev. 2014, 25, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Pietras, E.M.; He, J.Q.; Kang, J.R.; Liu, S.Y.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.L.; Wang, Y. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006, 25, 3257–3263. [Google Scholar] [CrossRef] [PubMed]
- Paz, S.; Vilasco, M.; Werden, S.J.; Arguello, M.; Joseph-Pillai, D.; Zhao, T.; Nguyen, T.L.; Sun, Q.; Meurs, E.F.; Lin, R.; et al. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. 2011, 21, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Torrino, S.; Visvikis, O.; Doye, A.; Boyer, L.; Stefani, C.; Munro, P.; Bertoglio, J.; Gacon, G.; Mettouchi, A.; Lemichez, E. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 2011, 21, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Mettouchi, A.; Lemichez, E. Ubiquitylation of active Rac1 by the E3 ubiquitin-ligase HACE1. Small GTPases 2012, 3, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, P.; Gao, H.; Gu, Y.; Yang, J.; Peng, H.; Xu, X.; Wang, H.; Yang, M.; Liu, X.; et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell 2014, 26, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Lachance, V.; Degrandmaison, J.; Marois, S.; Robitaille, M.; Genier, S.; Nadeau, S.; Angers, S.; Parent, J.L. Ubiquitylation and activation of a Rab GTPase is promoted by a β2AR-HACE1 complex. J. Cell Sci. 2014, 127, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Anglesio, M.S.; O’Sullivan, M.; Zhang, F.; Yang, G.; Sarao, R.; Mai, P.N.; Cronin, S.; Hara, H.; Melnyk, N.; et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat. Med. 2007, 13, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, Z.; Vucetic, Z.; Soprano, K.J.; Soprano, D.R. HACE1: A novel repressor of RAR transcriptional activity. J. Cell. Biochem. 2009, 107, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Slade, I.; Stephens, P.; Douglas, J.; Barker, K.; Stebbings, L.; Abbaszadeh, F.; Pritchard-Jones, K.; Cole, R.; Pizer, B.; et al. Constitutional translocation breakpoint mapping by genome-wide paired-end sequencing identifies HACE1 as a putative Wilms tumour susceptibility gene. J. Med. Genet. 2010, 47, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Evdokimova, V.; Melnyk, N.; Zhang, L.; Fernandez, C.V.; Grundy, P.E.; Leach, S.; Marra, M.A.; Brooks-Wilson, A.R.; Penninger, J.; et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, HACE1, in sporadic Wilms’ tumor vs. normal kidney. Hum. Mol. Genet. 2004, 13, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Kucuk, C.; Hu, X.; Iqbal, J.; Gaulard, P.; Klinkebiel, D.; Cornish, A.; Dave, B.J.; Chan, W.C. HACE1 is a tumor suppressor gene candidate in natural killer cell neoplasms. Am. J. Pathol. 2013, 182, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Sako, N.; Dessirier, V.; Bagot, M.; Bensussan, A.; Schmitt, C. HACE1, a potential tumor suppressor gene on 6q21, is not involved in extranodal natural killer/T-cell lymphoma pathophysiology. Am. J. Pathol. 2014, 184, 2899–2907. [Google Scholar] [CrossRef] [PubMed]
- Hibi, K.; Sakata, M.; Sakuraba, K.; Shirahata, A.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; et al. Aberrant methylation of the HACE1 gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2008, 28, 1581–1584. [Google Scholar] [PubMed]
- Sakata, M.; Yokomizo, K.; Kitamura, Y.; Sakuraba, K.; Shirahata, A.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G. Methylation of the HACE1 gene is frequently detected in hepatocellular carcinoma. Hepato Gastroenterol. 2013, 60, 781–783. [Google Scholar]
- Sakata, M.; Kitamura, Y.-H.; Sakuraba, K.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y. Methylation of HACE1 in gastric carcinoma. Anticancer Res. 2009, 29, 2231–2233. [Google Scholar] [PubMed]
- Goka, E.T.; Lippman, M.E. Loss of the E3 ubiquitin ligase HACE1 results in enhanced Rac1 signaling contributing to breast cancer progression. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Lluva, S.; Tan, C.T.; Daugaard, M.; Sorensen, P.H.; Malliri, A. The tumour suppressor HACE1 controls cell migration by regulating Rac1 degradation. Oncogene 2013, 32, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Rotblat, B.; Southwell, A.L.; Ehrnhoefer, D.E.; Skotte, N.H.; Metzler, M.; Franciosi, S.; Leprivier, G.; Somasekharan, S.P.; Barokas, A.; Deng, Y.; et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. USA 2014, 111, 3032–3037. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Xiang, Y.; de Renzis, S.; Rink, J.; Zheng, G.; Zerial, M.; Wang, Y. The ubiquitin ligase HACE1 regulates Golgi membrane dynamics during the cell cycle. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, X.; Sharma, P.; Moon, M.; Sheftel, A.D.; Dawood, F.; Nghiem, M.P.; Wu, J.; Li, R.K.; Gramolini, A.O.; et al. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, R.; Meng, J.L.; Mao, H.T.; Zhang, Y.; Zhang, J. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J. Immunol. 2014, 192, 4758–4764. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liao, B.; Wang, S.; Yan, B.; Jin, Y.; Shu, H.B.; Wang, Y.Y. E3 ligase WWP2 negatively regulates TLR3-mediated innate immune response by targeting TRIF for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2013, 110, 5115–5120. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, X.; Zheng, Y.; Yang, Y.; Xing, Y.; Chen, Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 2014, 5, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Kok, K.H.; Ng, M.H.; Poon, V.K.; Yuen, K.Y.; Zheng, B.J.; Jin, D.Y. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 2009, 284, 16202–16209. [Google Scholar] [CrossRef] [PubMed]
- Belgnaoui, S.M.; Paz, S.; Samuel, S.; Goulet, M.L.; Sun, Q.; Kikkert, M.; Iwai, K.; Dikic, I.; Hiscott, J.; Lin, R. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe 2012, 12, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.H.; Ho, T.H.; Kok, K.H.; Siu, K.L.; Li, J.; Jin, D.Y. MIP-T3 is a negative regulator of innate type I IFN response. J. Immunol. 2011, 187, 6473–6482. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Mesplede, T.; Solis, M.; Sun, Q.; Zhao, T.; Yang, L.; Chuang, T.H.; Ware, C.F.; Lin, R.; Hiscott, J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009, 5, e1000650. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, H.-T.; Wang, Y.; Cai, J.; Meng, J.-L.; Zhou, Y.; Pan, Y.; Qian, X.-P.; Zhang, Y.; Zhang, J. HACE1 Negatively Regulates Virus-Triggered Type I IFN Signaling by Impeding the Formation of the MAVS-TRAF3 Complex. Viruses 2016, 8, 146. https://0-doi-org.brum.beds.ac.uk/10.3390/v8050146
Mao H-T, Wang Y, Cai J, Meng J-L, Zhou Y, Pan Y, Qian X-P, Zhang Y, Zhang J. HACE1 Negatively Regulates Virus-Triggered Type I IFN Signaling by Impeding the Formation of the MAVS-TRAF3 Complex. Viruses. 2016; 8(5):146. https://0-doi-org.brum.beds.ac.uk/10.3390/v8050146
Chicago/Turabian StyleMao, He-Ting, Yan Wang, Juan Cai, Jun-Ling Meng, Yu Zhou, Yu Pan, Xiao-Ping Qian, Yu Zhang, and Jun Zhang. 2016. "HACE1 Negatively Regulates Virus-Triggered Type I IFN Signaling by Impeding the Formation of the MAVS-TRAF3 Complex" Viruses 8, no. 5: 146. https://0-doi-org.brum.beds.ac.uk/10.3390/v8050146