
A Yellow Fever 17D Virus Replicon-Based Vaccine Platform for Emerging Coronaviruses

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Construction of YF17D Replicons with Foreign Inserts
2.3. RNA Synthesis and Electroporation
2.4. Immunofluorescence Assay (IFA)
2.5. Polyacrylamide Gel Electrophoresis (PAGE) and Western Blotting
2.6. Production of Replicon Particles
2.7. Immunoperoxidase Monolayer Assay (IPMA)
2.8. Recombinant Protein Expression
2.9. Mouse Experiment
2.10. Indirect ELISA
2.11. Virus Titration
2.12. Virus Neutralization Test (VNT)
2.13. Statistical Analysis
3. Results
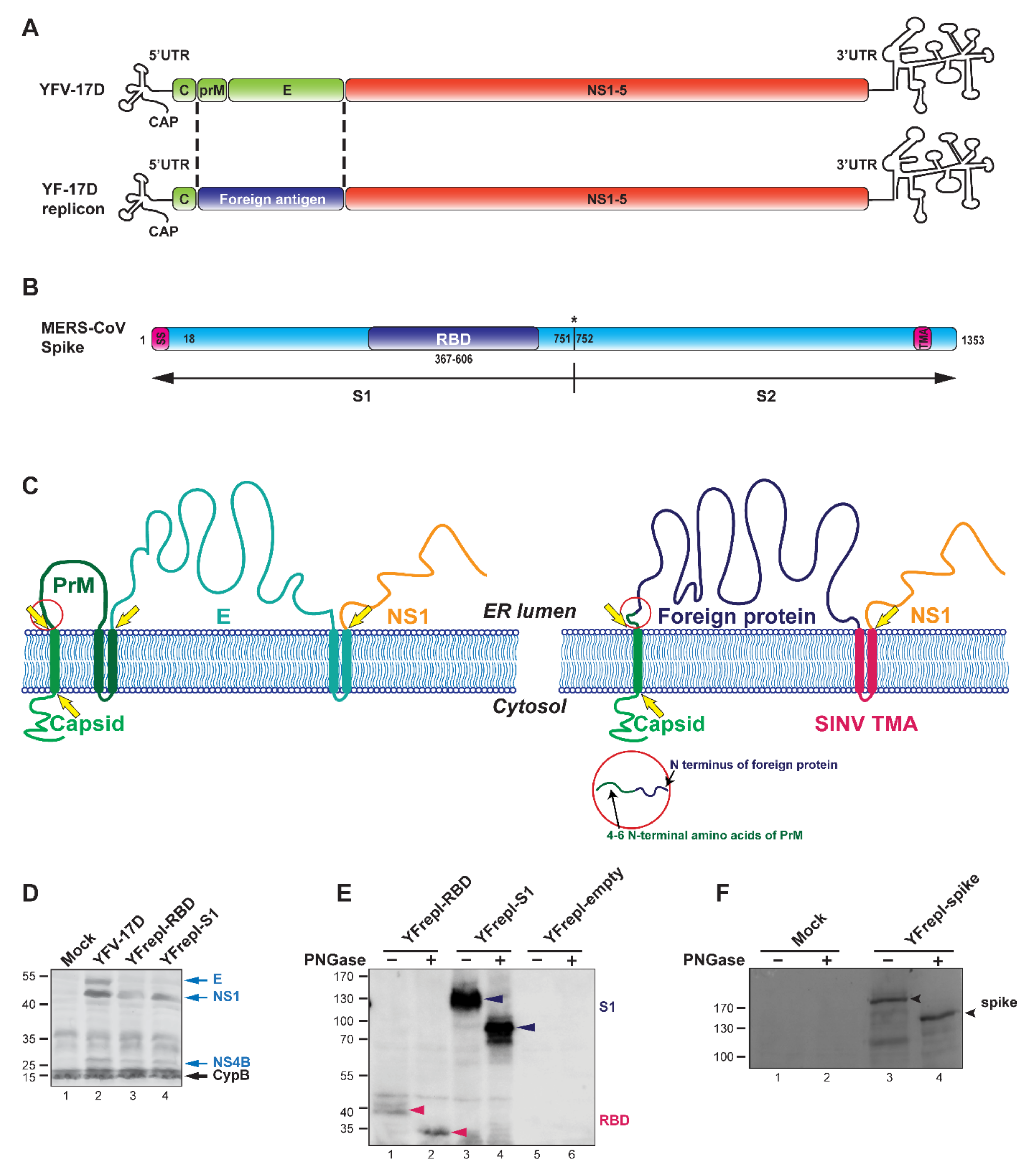
3.1. Design and Characterization of YF-Based RNA Replicons Expressing Partial or Full-Length MERS-CoV S Protein
3.2. Cellular Localization of the Foreign RBD and S1 Expression Products
3.3. The YF-Replicons Can Express Diverse Inserts in Either Cell-Associated or Secreted Form
3.4. YF-Replicons Are Packaged into Infectious Particles and Express the Encoded Foreign Protein Inserts
3.5. S Protein-Expressing YF-Replicons Induce Neutralizing Antibodies against Both MERS-CoV and SARS-CoV in Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyriakidis, N.C.; Lopez-Cortes, A.; Gonzalez, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- RAPS. Available online: https://www.raps.org/news-and-articles/news-articles/2020/3/covid-19-vaccine-tracker (accessed on 15 December 2021).
- Brett, D.; Lindenbach, C.L.M.; Heinz-Jürgen, T.; Rice, C.M. Flaviviridae. In Fields Virology, 6th ed.; Knipe, D.M., Ed.; Lippincott Willims & Wilkins: Philadelphia, PA, USA, 2013; p. 712. [Google Scholar]
- WHO. Yellow Fever. 7 May 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/yellow-fever (accessed on 15 December 2021).
- Theiler, M.; Smith, H.H. The Effect Of Prolonged Cultivation In Vitro Upon The Pathogenicity Of Yellow Fever Virus. J. Exp. Med. 1937, 65, 767. [Google Scholar] [CrossRef]
- WHO. SAGE Working Group: Background Paper on Yellow Fever Vaccine. 2013. Available online: https://www.who.int/immunization/sage/meetings/2013/april/1_Background_Paper_Yellow_Fever_Vaccines.pdf (accessed on 15 December 2021).
- Gotuzzo, E.; Yactayo, S.; Cordova, E. Efficacy and duration of immunity after yellow fever vaccination: Systematic review on the need for a booster every 10 years. Am. J. Trop. Med. Hyg. 2013, 89, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Wieten, R.W.; Jonker, E.F.; van Leeuwen, E.M.; Remmerswaal, E.B.; Ten Berge, I.J.; de Visser, A.W.; van Genderen, P.J.; Goorhuis, A.; Visser, L.G.; Grobusch, M.P.; et al. A Single 17D Yellow Fever Vaccination Provides Lifelong Immunity; Characterization of Yellow-Fever-Specific Neutralizing Antibody and T-Cell Responses after Vaccination. PLoS ONE 2016, 11, e0149871. [Google Scholar] [CrossRef] [Green Version]
- Grobusch, M.P.; van Aalst, M.; Goorhuis, A. Yellow fever vaccination—Once in a lifetime? Travel Med. Infect. Dis. 2017, 15, 1–2. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Sequeira, P.C.; Galler, R. The yellow fever 17D virus as a platform for new live attenuated vaccines. Hum. Vaccin Immunother. 2014, 10, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- McAllister, A.; Arbetman, A.E.; Mandl, S.; Pena-Rossi, C.; Andino, R. Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine experimental solid tumors and pulmonary metastases. J. Virol 2000, 74, 9197–9205. [Google Scholar] [CrossRef] [Green Version]
- Tao, D.; Barba-Spaeth, G.; Rai, U.; Nussenzweig, V.; Rice, C.M.; Nussenzweig, R.S. Yellow fever 17D as a vaccine vector for microbial CTL epitopes: Protection in a rodent malaria model. J. Exp. Med. 2005, 201, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barba-Spaeth, G.; Longman, R.S.; Albert, M.L.; Rice, C.M. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J. Exp. Med. 2005, 202, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, R.T.; Nogueira, A.R.; Pereira, M.C.; Rodrigues, M.M.; Galler, R.; Bonaldo, M.C. Biological and immunological characterization of recombinant Yellow Fever 17D viruses expressing a Trypanosoma cruzi Amastigote Surface Protein-2 CD8+ T cell epitope at two distinct regions of the genome. Virol. J. 2011, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Bonaldo, M.C.; Garratt, R.C.; Freire, M.S.; Galler, R. Expression of foreign protein epitopes at the surface of recombinant yellow fever 17D viruses based on three-dimensional modeling of its envelope protein. Cell Biochem. Biophys. 2006, 44, 313–324. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Garratt, R.C.; Marchevsky, R.S.; Coutinho, E.S.; Jabor, A.V.; Almeida, L.F.; Yamamura, A.M.; Duarte, A.S.; Oliveira, P.J.; Lizeu, J.O.; et al. Attenuation of recombinant yellow fever 17D viruses expressing foreign protein epitopes at the surface. J. Virol. 2005, 79, 8602–8613. [Google Scholar] [CrossRef] [Green Version]
- Rumyantsev, A.A.; Zhang, Z.X.; Gao, Q.S.; Moretti, N.; Brown, N.; Kleanthous, H.; Delagrave, S.; Guirakhoo, F.; Collett, M.S.; Pugachev, K.V. Direct random insertion of an influenza virus immunologic determinant into the NS1 glycoprotein of a vaccine flavivirus. Virology 2010, 396, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Bredenbeek, P.J.; Molenkamp, R.; Spaan, W.J.; Deubel, V.; Marianneau, P.; Salvato, M.S.; Moshkoff, D.; Zapata, J.; Tikhonov, I.; Patterson, J.; et al. A recombinant Yellow Fever 17D vaccine expressing Lassa virus glycoproteins. Virology 2006, 345, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Bonaldo, M.C.; Mello, S.M.; Trindade, G.F.; Rangel, A.A.; Duarte, A.S.; Oliveira, P.J.; Freire, M.S.; Kubelka, C.F.; Galler, R. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virol. J. 2007, 4, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaldo, M.C.; Martins, M.A.; Rudersdorf, R.; Mudd, P.A.; Sacha, J.B.; Piaskowski, S.M.; Costa Neves, P.C.; Veloso de Santana, M.G.; Vojnov, L.; Capuano, S., 3rd; et al. Recombinant yellow fever vaccine virus 17D expressing simian immunodeficiency virus SIVmac239 gag induces SIV-specific CD8+ T-cell responses in rhesus macaques. J. Virol. 2010, 84, 3699–3706. [Google Scholar] [CrossRef] [Green Version]
- Franco, D.; Li, W.; Qing, F.; Stoyanov, C.T.; Moran, T.; Rice, C.M.; Ho, D.D. Evaluation of yellow fever virus 17D strain as a new vector for HIV-1 vaccine development. Vaccine 2010, 28, 5676–5685. [Google Scholar] [CrossRef]
- Jiang, X.; Dalebout, T.J.; Bredenbeek, P.J.; Carrion, R., Jr.; Brasky, K.; Patterson, J.; Goicochea, M.; Bryant, J.; Salvato, M.S.; Lukashevich, I.S. Yellow fever 17D-vectored vaccines expressing Lassa virus GP1 and GP2 glycoproteins provide protection against fatal disease in guinea pigs. Vaccine 2011, 29, 1248–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, R.T.; Nogueira, A.R.; Pereira, M.C.; Rodrigues, M.M.; Neves, P.C.; Galler, R.; Bonaldo, M.C. Recombinant yellow fever viruses elicit CD8+ T cell responses and protective immunity against Trypanosoma cruzi. PLoS ONE 2013, 8, e59347. [Google Scholar] [CrossRef] [Green Version]
- Martins, M.A.; Bonaldo, M.C.; Rudersdorf, R.A.; Piaskowski, S.M.; Rakasz, E.G.; Weisgrau, K.L.; Furlott, J.R.; Eernisse, C.M.; Veloso de Santana, M.G.; Hidalgo, B.; et al. Immunogenicity of seven new recombinant yellow fever viruses 17D expressing fragments of SIVmac239 Gag, Nef, and Vif in Indian rhesus macaques. PLoS ONE 2013, 8, e54434. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Felipe, L.; Vercruysse, T.; Sharma, S.; Ma, J.; Lemmens, V.; Van Looveren, D.; Arkalagud Javarappa, M.P.; Boudewijns, R.; Malengier-Devlies, B.; Liesenborghs, L.; et al. A single-dose live-attenuated YF17D-vectored SARS-CoV-2 vaccine candidate. Nature 2021, 590, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Nestorowicz, A.; Mason, P.W.; Rice, C.M. Yellow fever/Japanese encephalitis chimeric viruses: Construction and biological properties. J. Virol. 1999, 73, 3095–3101. [Google Scholar] [CrossRef] [Green Version]
- Guirakhoo, F.; Weltzin, R.; Chambers, T.J.; Zhang, Z.X.; Soike, K.; Ratterree, M.; Arroyo, J.; Georgakopoulos, K.; Catalan, J.; Monath, T.P. Recombinant chimeric yellow fever-dengue type 2 virus is immunogenic and protective in nonhuman primates. J. Virol. 2000, 74, 5477–5485. [Google Scholar] [CrossRef] [Green Version]
- Guirakhoo, F.; Zhang, Z.X.; Chambers, T.J.; Delagrave, S.; Arroyo, J.; Barrett, A.D.; Monath, T.P. Immunogenicity, genetic stability, and protective efficacy of a recombinant, chimeric yellow fever-Japanese encephalitis virus (ChimeriVax-JE) as a live, attenuated vaccine candidate against Japanese encephalitis. Virology 1999, 257, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of YFV 17D-based chimeric vaccines against dengue, West Nile and Japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Kum, D.B.; Mishra, N.; Boudewijns, R.; Gladwyn-Ng, I.; Alfano, C.; Ma, J.; Schmid, M.A.; Marques, R.E.; Schols, D.; Kaptein, S.; et al. A yellow fever-Zika chimeric virus vaccine candidate protects against Zika infection and congenital malformations in mice. NPJ Vaccines 2018, 3, 56. [Google Scholar] [CrossRef]
- Thomas, R.E.; Lorenzetti, D.L.; Spragins, W.; Jackson, D.; Williamson, T. The safety of yellow fever vaccine 17D or 17DD in children, pregnant women, HIV+ individuals, and older persons: Systematic review. Am. J. Trop. Med. Hyg. 2012, 86, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.D.; Teuwen, D.E. Yellow fever vaccine—How does it work and why do rare cases of serious adverse events take place? Curr. Opin. Immunol. 2009, 21, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Dalebout, T.J.; Lukashevich, I.S.; Bredenbeek, P.J.; Franco, D. Molecular and immunological characterization of a DNA-launched yellow fever virus 17D infectious clone. J. Gen. Virol. 2015, 96, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Fayzulin, R.; Scholle, F.; Petrakova, O.; Frolov, I.; Mason, P.W. Evaluation of replicative capacity and genetic stability of West Nile virus replicons using highly efficient packaging cell lines. Virology 2006, 351, 196–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, P.W.; Shustov, A.V.; Frolov, I. Production and characterization of vaccines based on flaviviruses defective in replication. Virology 2006, 351, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Giel-Moloney, M.; Vaine, M.; Zhang, L.; Parrington, M.; Gajewska, B.; Vogel, T.U.; Pougatcheva, S.O.; Duan, X.; Farrell, T.; Ustyugova, I.; et al. Application of replication-defective West Nile virus vector to non-flavivirus vaccine targets. Hum. Vaccin. Immunother. 2017, 13, 2982–2986. [Google Scholar] [CrossRef] [Green Version]
- Kümmerer, B.M. Establishment and Application of Flavivirus Replicons. In Dengue and Zika: Control and Antiviral Treatment Strategies; Hilgenfeld, R., Vasudevan, S.G., Eds.; Springer Singapore: Singapore, 2018; pp. 165–173. [Google Scholar]
- Huang, Y.T.; Liao, J.T.; Yen, L.C.; Chang, Y.K.; Lin, Y.L.; Liao, C.L. Japanese encephalitis virus replicon-based vaccine expressing enterovirus-71 epitope confers dual protection from lethal challenges. J. Biomed. Sci. 2015, 22, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynard, O.; Mokhonov, V.; Mokhonova, E.; Leung, J.; Page, A.; Mateo, M.; Pyankova, O.; Georges-Courbot, M.C.; Raoul, H.; Khromykh, A.A.; et al. Kunjin virus replicon-based vaccines expressing Ebola virus glycoprotein GP protect the guinea pig against lethal Ebola virus infection. J. Infect. Dis. 2011, 204, S1060–S1065. [Google Scholar] [CrossRef] [Green Version]
- Pyankov, O.V.; Bodnev, S.A.; Pyankova, O.G.; Solodkyi, V.V.; Pyankov, S.A.; Setoh, Y.X.; Volchkova, V.A.; Suhrbier, A.; Volchkov, V.V.; Agafonov, A.A.; et al. A Kunjin Replicon Virus-like Particle Vaccine Provides Protection Against Ebola Virus Infection in Nonhuman Primates. J. Infect. Dis. 2015, 212, S368–S371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corver, J.; Lenches, E.; Smith, K.; Robison, R.A.; Sando, T.; Strauss, E.G.; Strauss, J.H. Fine mapping of a cis-acting sequence element in yellow fever virus RNA that is required for RNA replication and cyclization. J. Virol. 2003, 77, 2265–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khromykh, A.A.; Westaway, E.G. Subgenomic replicons of the flavivirus Kunjin: Construction and applications. J. Virol. 1997, 71, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Schindewolf, C.; Menachery, V.D. Middle East Respiratory Syndrome Vaccine Candidates: Cautious Optimism. Viruses 2019, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Abdulla, Z.A.; Al-Bashir, S.M.; Al-Salih, N.S.; Aldamen, A.A.; Abdulazeez, M.Z. A Summary of the SARS-CoV-2 Vaccines and Technologies Available or under Development. Pathogens 2021, 10, 788. [Google Scholar] [CrossRef]
- Xia, X. Domains and Functions of Spike Protein in Sars-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Bredenbeek, P.J.; Kooi, E.A.; Lindenbach, B.; Huijkman, N.; Rice, C.M.; Spaan, W.J. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J. Gen. Virol. 2003, 84, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Touret, F.; Gilles, M.; Klitting, R.; Aubry, F.; de Lamballerie, X.; Nougairede, A. Live Zika virus chimeric vaccine candidate based on a yellow fever 17-D attenuated backbone. Emerg. Microbes Infect. 2018, 7, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spearman, C. The Method Of ‘Right And Wrong Cases’ (‘Constant Stimuli’) Without Gauss’s Formulae. Br. J. Psychol. 1908, 2, 227–242. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedebergs Arch. Für Exp. Pathol. Und Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.; Boudewijns, R.; Schmid, M.A.; Marques, R.E.; Sharma, S.; Neyts, J.; Dallmeier, K. A Chimeric Japanese Encephalitis Vaccine Protects against Lethal Yellow Fever Virus Infection without Inducing Neutralizing Antibodies. mBio 2020, 11, e02494-19. [Google Scholar] [CrossRef] [Green Version]
- Op De Beeck, A.; Rouille, Y.; Caron, M.; Duvet, S.; Dubuisson, J. The transmembrane domains of the prM and E proteins of yellow fever virus are endoplasmic reticulum localization signals. J. Virol.. 2004, 78, 12591–12602. [Google Scholar] [CrossRef] [Green Version]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Mou, H.; Raj, V.S.; van Kuppeveld, F.J.; Rottier, P.J.; Haagmans, B.L.; Bosch, B.J. The receptor binding domain of the new Middle East respiratory syndrome coronavirus maps to a 231-residue region in the spike protein that efficiently elicits neutralizing antibodies. J. Virol. 2013, 87, 9379–9383. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, M.; Nagahama, M.; Kim, W.S.; Watanabe, T.; Hatsuzawa, K.; Ikemizu, J.; Murakami, K.; Nakayama, K. Arg-X-Lys/Arg-Arg motif as a signal for precursor cleavage catalyzed by furin within the constitutive secretory pathway. J. Biol. Chem. 1991, 266, 12127–12130. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Available online. Available online: https://services.healthtech.dtu.dk/service.php?NetNGlyc-1.0 (accessed on 3 January 2020).
- Thibodeaux, B.A.; Garbino, N.C.; Liss, N.M.; Piper, J.; Blair, C.D.; Roehrig, J.T. A small animal peripheral challenge model of yellow fever using interferon-receptor deficient mice and the 17D-204 vaccine strain. Vaccine 2012, 30, 3180–3187. [Google Scholar] [CrossRef] [Green Version]
- Meier, K.C.; Gardner, C.L.; Khoretonenko, M.V.; Klimstra, W.B.; Ryman, K.D. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009, 5, e1000614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kum, D.B.; Mishra, N.; Vrancken, B.; Thibaut, H.J.; Wilder-Smith, A.; Lemey, P.; Neyts, J.; Dallmeier, K. Limited evolution of the yellow fever virus 17d in a mouse infection model. Emerg. Microbes Infect. 2019, 8, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- Kum, D.B.; Boudewijns, R.; Ma, J.; Mishra, N.; Schols, D.; Neyts, J.; Dallmeier, K. A chimeric yellow fever-Zika virus vaccine candidate fully protects against yellow fever virus infection in mice. Emerg. Microbes Infect. 2020, 9, 520–533. [Google Scholar] [CrossRef]
- Teo, S.P. Review of COVID-19 mRNA Vaccines: BNT162b2 and mRNA-1273. J. Pharm. Pract. 2021, 8971900211009650. [Google Scholar] [CrossRef] [PubMed]
- EMA. AstraZeneca’s COVID-19 Vaccine: Benefits and Risks in Context. 2021. Available online: https://www.ema.europa.eu/en/news/astrazenecas-covid-19-vaccine-benefits-risks-context (accessed on 15 December 2021).
- EMA. COVID-19 Vaccine Janssen. 2021. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/covid-19-vaccine-janssen (accessed on 15 December 2021).
- Cattaneo, M. Thrombosis with Thrombocytopenia Syndrome associated with viral vector COVID-19 vaccines. Eur. J. Intern. Med. 2021, 89, 22–24. [Google Scholar] [CrossRef]
- Long, B.; Bridwell, R.; Gottlieb, M. Thrombosis with thrombocytopenia syndrome associated with COVID-19 vaccines. Am. J. Emerg. Med. 2021, 49, 58–61. [Google Scholar] [CrossRef]
- Shay, D.K.; Shimabukuro, T.T.; DeStefano, F. Myocarditis Occurring after Immunization with mRNA-Based COVID-19 Vaccines. JAMA Cardiol. 2021, 6, 1115–1117. [Google Scholar] [CrossRef]
- Querec, T.; Bennouna, S.; Alkan, S.; Laouar, Y.; Gorden, K.; Flavell, R.; Akira, S.; Ahmed, R.; Pulendran, B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 2006, 203, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Querec, T.D.; Akondy, R.S.; Lee, E.K.; Cao, W.; Nakaya, H.I.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Gaucher, D.; Therrien, R.; Kettaf, N.; Angermann, B.R.; Boucher, G.; Filali-Mouhim, A.; Moser, J.M.; Mehta, R.S.; Drake, D.R., 3rd; Castro, E.; et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 2008, 205, 3119–3131. [Google Scholar] [CrossRef]
- Pulendran, B. Learning immunology from the yellow fever vaccine: Innate immunity to systems vaccinology. Nat. Rev. Immunol. 2009, 9, 741–747. [Google Scholar] [CrossRef]
- Monath, T.P.; Vasconcelos, P.F. Yellow fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef]
- Santos, A.P.; Matos, D.C.S.; Bertho, A.L.; Mendonca, S.C.F.; Marcovistz, R. Detection of Th1/Th2 cytokine signatures in yellow fever 17DD first-time vaccinees through ELISpot assay. Cytokine 2008, 42, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.M.; Klimstra, W.B. T Cell-Mediated Immunity towards Yellow Fever Virus and Useful Animal Models. Viruses 2017, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Monson, N.D.; Miller, J.D.; Edupuganti, S.; Teuwen, D.; Wu, H.; Quyyumi, F.; Garg, S.; Altman, J.D.; Del Rio, C.; et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J. Immunol. 2009, 183, 7919–7930. [Google Scholar] [CrossRef] [Green Version]
- Cottin, P.; Niedrig, M.; Domingo, C. Safety profile of the yellow fever vaccine Stamaril(R): A 17-year review. Expert Rev. Vaccines 2013, 12, 1351–1368. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Rabe, I.B.; Miller, E.R.; Fischer, M.; Staples, J.E. Adverse event reports following yellow fever vaccination, 2007–2013. J. Travel Med. 2016, 23, taw045. [Google Scholar] [CrossRef] [Green Version]
- Monath, T.P. Review of the risks and benefits of yellow fever vaccination including some new analyses. Expert Rev. Vaccines 2012, 11, 427–448. [Google Scholar] [CrossRef] [PubMed]
- WHO. 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/329432/WHO-WHE-IHM-2019.11-eng.pdf?ua=1 (accessed on 15 December 2021).
- Piras-Douce, F.; Raynal, F.; Raquin, A.; Girerd-Chambaz, Y.; Gautheron, S.; Sanchez, M.E.N.; Vangelisti, M.; Mantel, N. Next generation live-attenuated yellow fever vaccine candidate: Safety and immuno-efficacy in small animal models. Vaccine 2021, 39, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, E.C.; Tschismarov, R.; Beubler, E.; Wiedermann, U.; Firbas, C.; Loebermann, M.; Pfeiffer, A.; Muellner, M.; Tauber, E.; Ramsauer, K. Immunogenicity, safety, and tolerability of the measles-vectored chikungunya virus vaccine MV-CHIK: A double-blind, randomised, placebo-controlled and active-controlled phase 2 trial. Lancet 2019, 392, 2718–2727. [Google Scholar] [CrossRef]
- Saxena, M.; Van, T.T.H.; Baird, F.J.; Coloe, P.J.; Smooker, P.M. Pre-existing immunity against vaccine vectors—Friend or foe? Microbiology 2013, 159, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uematsu, Y.; Vajdy, M.; Lian, Y.; Perri, S.; Greer, C.E.; Legg, H.S.; Galli, G.; Saletti, G.; Otten, G.R.; Rappuoli, R.; et al. Lack of interference with immunogenicity of a chimeric alphavirus replicon particle-based influenza vaccine by preexisting antivector immunity. Clin. Vaccine Immunol. 2012, 19, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Brandriss, M.W.; Schlesinger, J.J.; Walsh, E.E. Immunogenicity of a purified fragment of 17D yellow fever envelope protein. J. Infect. Dis. 1990, 161, 1134–1139. [Google Scholar] [CrossRef]
- Lobigs, M.; Dalgarno, L.; Schlesinger, J.J.; Weir, R.C. Location of a neutralization determinant in the E protein of yellow fever virus (17D vaccine strain). Virology 1987, 161, 474–478. [Google Scholar] [CrossRef]
- Leborgne, C.; Barbon, E.; Alexander, J.M.; Hanby, H.; Delignat, S.; Cohen, D.M.; Collaud, F.; Muraleetharan, S.; Lupo, D.; Silverberg, J.; et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020, 26, 1096–1101. [Google Scholar] [CrossRef]
- Enjuanes, L.; Dediego, M.L.; Alvarez, E.; Deming, D.; Sheahan, T.; Baric, R. Vaccines to prevent severe acute respiratory syndrome coronavirus-induced disease. Virus Res. 2008, 133, 45–62. [Google Scholar] [CrossRef]
- Okba, N.M.; Raj, V.S.; Haagmans, B.L. Middle East respiratory syndrome coronavirus vaccines: Current status and novel approaches. Curr. Opin. Virol. 2017, 23, 49–58. [Google Scholar] [CrossRef]
- He, X.; Chandrashekar, A.; Zahn, R.; Wegmann, F.; Yu, J.; Mercado, N.B.; McMahan, K.; Martinot, A.J.; Piedra-Mora, C.; Beecy, S.; et al. Low-dose Ad26.COV2.S protection against SARS-CoV-2 challenge in rhesus macaques. Cell 2021, 184, 3467–3473.e11. [Google Scholar] [CrossRef] [PubMed]
- Khoury, D.S.; Cromer, D.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Subbarao, K.; Kent, S.J.; Triccas, J.A.; Davenport, M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 1205–1211. [Google Scholar] [CrossRef]
- Erickson, A.K.; Pfeiffer, J.K. Spectrum of disease outcomes in mice infected with YFV-17D. J. Gen. Virol. 2015, 96, 1328–1339. [Google Scholar] [CrossRef] [Green Version]
- Bassi, M.R.; Kongsgaard, M.; Steffensen, M.A.; Fenger, C.; Rasmussen, M.; Skjodt, K.; Finsen, B.; Stryhn, A.; Buus, S.; Christensen, J.P.; et al. CD8+ T cells complement antibodies in protecting against yellow fever virus. J. Immunol. 2015, 194, 1141–1153. [Google Scholar] [CrossRef] [Green Version]
- van der Most, R.G.; Harrington, L.E.; Giuggio, V.; Mahar, P.L.; Ahmed, R. Yellow fever virus 17D envelope and NS3 proteins are major targets of the antiviral T cell response in mice. Virology 2002, 296, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Oreshkova, N.; Cornelissen, L.A.; de Haan, C.A.; Moormann, R.J.; Kortekaas, J. Evaluation of nonspreading Rift Valley fever virus as a vaccine vector using influenza virus hemagglutinin as a model antigen. Vaccine 2014, 32, 5323–5329. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.B.; Suh, Y.S.; Ryu, J.I.; Jang, H.; Oh, H.; Koo, B.S.; Seo, S.H.; Hong, J.J.; Song, M.; Kim, S.J.; et al. Soluble Spike DNA Vaccine Provides Long-Term Protective Immunity against SARS-CoV-2 in Mice and Nonhuman Primates. Vaccines 2021, 9, 307. [Google Scholar] [CrossRef]
- Freyn, A.W.; Pine, M.; Rosado, V.C.; Benz, M.; Muramatsu, H.; Beattie, M.; Tam, Y.K.; Krammer, F.; Palese, P.; Nachbagauer, R.; et al. Antigen modifications improve nucleoside-modified mRNA-based influenza virus vaccines in mice. Mol. Ther. Methods Clin. Dev. 2021, 22, 84–95. [Google Scholar] [CrossRef]
- Tu, Y.; Hu, Y.; Fan, G.; Chen, Z.; Liu, L.; Man, D.; Liu, S.; Tang, C.; Zhang, Y.; Dai, W. Protective effects of membrane-anchored and secreted DNA vaccines encoding fatty acid-binding protein and glutathione S-transferase against Schistosoma japonicum. PLoS ONE 2014, 9, e86575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelissen, L.A.; de Leeuw, O.S.; Tacken, M.G.; Klos, H.C.; de Vries, R.P.; de Boer-Luijtze, E.A.; van Zoelen-Bos, D.J.; Rigter, A.; Rottier, P.J.; Moormann, R.J.; et al. Protective efficacy of Newcastle disease virus expressing soluble trimeric hemagglutinin against highly pathogenic H5N1 influenza in chickens and mice. PLoS ONE 2012, 7, e44447. [Google Scholar] [CrossRef]
- Roberts, A.; Deming, D.; Paddock, C.D.; Cheng, A.; Yount, B.; Vogel, L.; Herman, B.D.; Sheahan, T.; Heise, M.; Genrich, G.L.; et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Heeney, J.L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 2010, 8, 62–73. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Foreign Insert (Domain *) | Virus | Strain | Amino Acids Used for the Inserts ** |
|---|---|---|---|
| RBD | MERS-CoV | EMC2012 | 364–606 |
| S1 | 18–750 | ||
| Full length spike | 18–1353 | ||
| RBD | SARS-CoV | MA15 | 306–527 |
| S1 | 14–665 | ||
| RBD | MHV | A59 | 15–296 |
| S1 | 15–712 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oreshkova, N.; Myeni, S.K.; Mishra, N.; Albulescu, I.C.; Dalebout, T.J.; Snijder, E.J.; Bredenbeek, P.J.; Dallmeier, K.; Kikkert, M. A Yellow Fever 17D Virus Replicon-Based Vaccine Platform for Emerging Coronaviruses. Vaccines 2021, 9, 1492. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9121492
Oreshkova N, Myeni SK, Mishra N, Albulescu IC, Dalebout TJ, Snijder EJ, Bredenbeek PJ, Dallmeier K, Kikkert M. A Yellow Fever 17D Virus Replicon-Based Vaccine Platform for Emerging Coronaviruses. Vaccines. 2021; 9(12):1492. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9121492
Chicago/Turabian StyleOreshkova, Nadia, Sebenzile K. Myeni, Niraj Mishra, Irina C. Albulescu, Tim J. Dalebout, Eric J. Snijder, Peter J. Bredenbeek, Kai Dallmeier, and Marjolein Kikkert. 2021. "A Yellow Fever 17D Virus Replicon-Based Vaccine Platform for Emerging Coronaviruses" Vaccines 9, no. 12: 1492. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9121492