

Abstract
Experimental cerebral malaria (ECM) resulting from Plasmodium berghei ANKA infection involves T lymphocytes. However, the mechanisms of T cell-mediated pathogenesis remain unknown. We found that, in contrast to ECM-susceptible C57BL6 mice, perforin-deficient (PFP-KO) mice were resistant to ECM in the absence of brain lesions, whereas cytoadherence of parasitized erythrocytes and massive accumulation of activated/effector CD8 lymphocytes were observed in both groups of mice. ECM is induced in PFP-KO mice after adoptive transfer of cytotoxic CD8+ cells from infected C57BL6 mice, which were directed to the brain of PFP-KO mice. This specific recruitment might involve chemokine/chemokine receptors, since their expression was up-regulated on activated CD8 cells, and susceptibility to ECM was delayed in CCR5-KO mice. Thus, lymphocyte cytotoxicity and cell trafficking are key players in ECM pathogenesis.
Cerebral malaria (CM)4 is one of the most severe complications of Plasmodium falciparum infection and the main cause of death in humans (1). The murine model of Plasmodium berghei ANKA (PbA) reproduces relevant clinical and pathological features of the human disease (2). Although many aspects of the pathogenesis of CM in experimental models have been described, the precise mechanisms leading to this complication are not fully understood. The initial observation of athymic nude mice not developing this neurological syndrome upon infection with PbA demonstrated the requirement for T lymphocytes in experimental cerebral malaria (ECM) pathogenesis (3). The respective roles of T cell subsets in the pathogenesis of ECM have been analyzed by depletion of CD4 or CD8 T cells in C57BL/6 mice that did not develop ECM (4). CD8 T lymphocytes are highly involved in ECM pathogenesis, since mice homozygous for the null allele of the β2-microglobulin gene that lack functional CD8 T cells (4) as well as CD8 knockout (KO) mice (5) were resistant to ECM. In addition, a selective increase in Vβ8+ CD8 T cells has been observed during ECM, suggesting a critical role for the T cell-mediated immune response after PbA infection and its neurological consequence (5). However, little is known about the precise mechanisms of CD8 T cell induction of ECM pathogenesis. We investigated the role of cytolytic effector mechanisms mediated by CD8 cells in the development of ECM and the involvement of T lymphocytes in brain cell trafficking. Indeed, in the past years, trafficking of immunocompetent cells has been highly implicated in the establishment of the immune response and disease progression (6). Based on chemokine receptor-deficient mice models, chemokine/chemokine receptors have been recently implicated in various pathologies, such as multiple sclerosis in human or murine experimental autoimmune encephalomyelitis or after transient focal cerebral ischemia (7, 8, 9, 10). Indeed, migration of leukocytes to the CNS has been described as a critical factor during experimental autoimmune encephalomyelitis (11, 12, 13). In our study we used a variety of bioengineered deficiencies ideal 1) for probing the role of immune elements in cerebral malaria pathogenesis, 2) to investigate the mechanisms by which T cells mediate cerebral malaria pathology, and 3) to analyze the importance of lymphocyte trafficking to the CNS during ECM pathogenesis.
Materials and Methods
Mice
Six- to 8-wk-old female C57BL/6 wild-type (WT) mice were purchased from Charles River Breeding Laboratories (Saint-Aubin les Elbeufs, France). Age-matched perforin-deficient mice (PFP-KO), lpr (Fas-natural mutation-inducing lymphoproliferative disease), and gld (Fas-ligand natural mutation-inducing generalized lymphadenopathy) mice on the C57BL6 background were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred in our animal facilities (Nouvelle Animalerie Commune, Pitié-Salpétrière, Paris, France). C57BL/6-TgH (recombinase-activating gene 2 (RAG-2) KO) mice were obtained from Centre de Développement des Techniques Avancées pour l’expérimentation animale (Orléans, France). All these mice are of the normally susceptible H-2b phenotype. CCR-5 KO mice from The Jackson Laboratory were backcrossed on the C57BL/6 background in our animal facility.
Parasites
Clone 1.49L of the PbA strain was a gift from Dr. D. Walliker (Institut of Genetics, Edinburgh, U.K.). This parasite was maintained in C57BL/6 mice and induces ECM, characterized by hemi- or paraplegia, ataxia, deviation of the head, and convulsions between 6–8 days postinfection (14). The erythrocytic stages of the parasite were stocked in liquid nitrogen as parasitized red blood stage (pRBC) per milliliter in Alsever’s solution containing 10% glycerol. The parasite infection was induced by i.p. injection of 106 pRBCs. Parasitemia was determined daily on Giemsa-stained blood smears and was expressed as the percent parasitemia. We choose to infect the mice with a single dose of 106 pRBCs because lower doses (102) of parasites were not able to induce ECM in C57BL6 susceptible mice.
Histology
Brains were recovered after perfusion with 1× PBS, formalin fixed, (AFA, fixing liquid; Carlo Erba, Val de Reuil, France) then paraffin-embedded for preparation of histological sections. Five-micrometer-thick sections were cut, stained with H&E, and examined by light microscopy.
Preparation of brain cell suspensions
The brains were obtained from WT susceptible mice at the coma stage of ECM and at the same time from KO animals (day 6). Briefly, brains were removed and homogenized in RPMI 1640 medium (Life Technologies, Cergy Pontoise, France) by passing through sterile meshes to obtain a single-cell suspension. Percoll (Pharmacia Biotech, Uppsala, Sweden) was added at a final concentration of 35%, and cells were centrifuged at 400 × g for 20 min at 20°C. The cell pellets were washed twice and subjected to flow cytometric analyses or T lymphocyte enrichment.
Flow cytometric analyses
Brain cells were stained for FACS analysis according to standard protocols in cold 1× PBS containing 2% FCS and 0.01% sodium azide (FACS buffer) with the following Abs: CyChrome-labeled anti-CD4, FITC-labeled anti-CD3 (145-2C11, hamster IgG1), PE-labeled anti-CD4 (H129.19, rat IgG2a), PerCP-labeled anti-CD8α (53-6.7, rat IgG2a), PE-conjugated anti-CD44 (Pgp-1), PE-labeled anti-CD69 (Mel-14), and biotin-labeled CD62L (H1.2F3) mAbs, and streptavidin-allophycocyanin was used at the secondary step (BD PharMingen, France). The whole blood was stained before proceeding to erythrocyte lysis. RBC were eliminated using cell lysis buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA), and cells were washed in FACS buffer. Cells were washed twice before flow cytometric analyses. A total of 10,000–20,000 living events were analyzed using a four-color FACSCalibur flow cytometer with ProCellQuest software (BD Biosciences, Mountain View, CA).
In vivo study of CFSE-labeled cell migration
Splenocytes were prepared from naive and infected mice on day 6 postinfection. Cell suspensions were prepared by passing through sterile meshes. Erythrocytes were lysed using RBC lysis buffer as described above. Cells were washed twice in RPMI 1640 medium (Life Technologies), counted by trypan blue exclusion staining, and adjusted to 107 cells/ml. Staining of spleen cells with CFSE (Molecular Probes, Eugene, OR) was performed as described by Lyons and Parish (15). Briefly, cells were resuspended at 1 × 107 cells/ml in 1× PBS. A 5-mM stock solution of CFSE in DMSO was added to a final concentration of 10 μM, and cells were incubated for 30 min at 37°C in a water bath. Cells were washed twice with ice-cold 10% FCS/RPMI 1640 and once with 1× PBS, then resuspended in 1× PBS. Cells were administered i.v. to the mice (5 × 106), and some of these mice were infected 1 h later with 106 parasites by the i.p administration route. Single-cell suspensions of brains and spleens of the recipients were analyzed between days 6–8 following adoptive cell transfer.
Adoptive transfer of CD8+ cells
Cell suspensions from the spleen of WT and PFP-KO mice were prepared by the methods described above and pooled, and CD8+ cells were purified with MACS beads according to the manufacturer’s instructions (Miltenyi Biotech, Paris, France). Briefly, cells were suspended in MACS buffer and incubated for 15 min at 4°C with magnetic Abs specific for CD8 T cells. Cells were washed, and the suspension was applied to a MACS-MS+ separation column (Miltenyi Biotech) in an appropriate MACS separator, washed three times, and eluted in 1 ml of MACS buffer. Cells were stained with PerCP-conjugated anti-CD8 Ab, and the purity was evaluated by FACS analysis. After two or three rounds of column separation, we obtained >98% CD8+ T lymphocytes enrichments.
Recipient mice (WT, PFP-KO, or RAG2-KO) were administered i.v. with either total spleen cells or purified CD8+ or CD8− cells (5 × 106). After 1 h mice were infected i.p. with 106 pRBC. Controls included mice that received spleen cells without pRBC.
RT-multiplex PCR (RT-MPCR)
Total RNA was isolated from purified CD8+ T lymphocytes. RT-PCR was performed to evaluate cytokine and chemokine receptor gene expressions. Total RNA was extracted using the QIAamp RNA Blood Mini kit (Qiagen, Courtaboeuf, France). cDNA was generated using the Omniscript Reverse Transcriptase kit (Qiagen). RT-MPCR analysis was performed using the Quantitative PCR CytoXpress Detection kit for mouse cytokines (Mouse Th1/Th2 Set 2 Signaling CytoXpress Multiplex-PCR kit; BioSource, Europe, Montrouge, Belgium), which were designed to detect the expression of IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12p40, IL-13, and GAPDH genes. The quantitative PCR CytoXpress Detection kit for mouse CXCR and CCR receptors (Mouse CXCR Chemokine Receptors and Mouse CCR Receptor Set 1; BioSource Europe) was designed to detect the expression of mouse CXCR1 and -2, CXCR3, CXCR4, and CXCR5 for the CXCR genes; CCR1, CCR2, CCR3, CCR4, and CCR5 for the CCR genes; and GAPDH genes. cDNA amplification was performed on a automated PerkinElmer DNA thermocycler (model 480; Palo Alto, CA) using the time-temperature profile according to manufacturer’s instructions. Following PCR, the amplicons were analyzed in a 2% agarose gel electrophoresis containing ethidium bromide.
Chromium release assays
Standard chromium release assays were performed as previously described (16). Target cells (P815) were labeled with 60 μCi/106 cells of Na251CrO4 (Comissariat d’Energie Atomique, Saclay, France) for 2 h at 37°C and washed twice with RPMI 1640 medium. In the CD3-redirected assays, anti-CD3 mAb (UCHT1) was added to target cells (3 × 103) at 5 μg/ml in triplicate to round-bottom 96-well microtiter plates (PolyLabo, Strasbourg, France) before addition of effector cells at various E:T cell ratios. After a 4-h incubation period at 37°C, the supernatants were collected, and chromium release was measured on a gamma counter. Spontaneous 51Cr release values were 15–25% of total incorporated radioactivity. The percentage of specific release 51Cr was calculated as: percentage = 1 − (experimental − spontaneous release/total radioactivity − spontaneous release) × 100.
Statistical analysis
Data handling, analysis, and graphic representation were performed using PRISM 2.01 (GraphPad, San Diego, CA). Statistical analysis was performed by paired two-sample t test for means and by the nonparametric Mann-Whitney U test. Statistical significance was accepted at p < 0.05.
Results
Development of ECM pathogenesis after PbA infection is dependent on perforin gene expression
Pioneering studies have shown the requirement for CD8 T cells in the development of ECM pathogenesis (4). However, the mechanisms by which these cells exert their action remain unclear. Fas/Fas ligand and perforin pathways have been clearly implicated in CD8-mediated cytotoxic effector functions against pathogens (17, 18). To determine whether and which cytotoxic functions were required for the development of ECM, C57BL/6 (WT), lpr, gld, and PFP-KO mice were infected with PbA (Fig. 1,a). Infected WT, lpr, and gld mice developed ECM between days 6 and 10 postinfection, whereas none of the PFP-KO mice showed any sign of ECM (Fig. 1 a), but died from anemia with increased parasitemia between days 17 and 28. The development of ECM pathogenesis after PbA infection is therefore dependent on perforin gene expression.
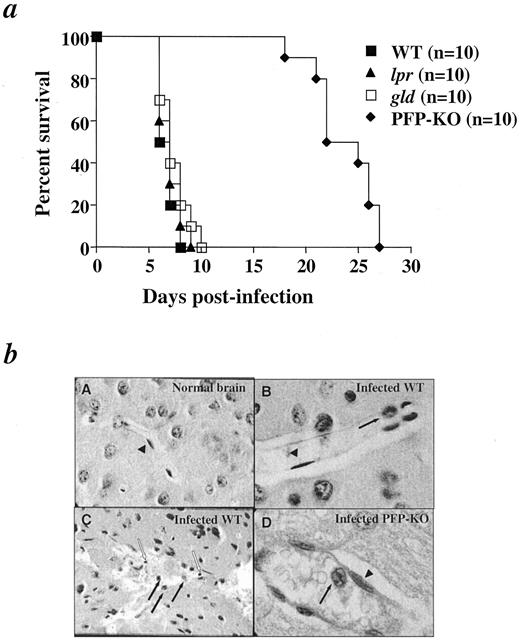
The development of ECM pathogenesis after PbA infection is dependent on perforin gene expression. a, Percent survival of C57BL/6 WT, lpr, gld, and perforin-deficient (PFP-KO) mice following i.p. PbA infection with 106 pRBC. The growth rate of parasites was comparable in all strains of mice on day 6 (parasitemia: C57BL6, 5.1 ± 1.5%; lpr, 5.6 ± 2.4%; gld, 6.2 ± 2.4%; PFP-KO, 4.8 ± 0.8%). Experiments were performed three times with 10 mice in each group. b, Histologic analyses of brain sections of ECM-susceptible WT and ECM-resistant PFP-KO mice at the coma stage in WT susceptible mice and at the same time in PFP-KO resistant mice. Brains were removed after perfusion with 1× PBS and were fixed for histological examinations. A, Section of a normal mouse brain showing a healthy uninterrupted blood vessel with normal architecture and integrity (magnification, ×250). B, Sagittal section of a WT mouse brain at the coma stage showing sequestration and cytoadherence of pRBC (filled arrows). Endothelial cells (▴; magnification, ×750). C, Severe lesions in the cortex of infected WT mice showing a disrupted vessel, with destruction of endothelial cells, extravascular pRBC (filled arrows), and mononuclear cell infiltration (empty arrows; magnification, ×500). D, Sagittal section of infected PFP-KO mouse brain on day 6 postinfection showing cytoadherence to a vessel wall (filled arrow; magnification, ×1000). ▴, Endothelial cells.
The development of ECM pathogenesis after PbA infection is dependent on perforin gene expression. a, Percent survival of C57BL/6 WT, lpr, gld, and perforin-deficient (PFP-KO) mice following i.p. PbA infection with 106 pRBC. The growth rate of parasites was comparable in all strains of mice on day 6 (parasitemia: C57BL6, 5.1 ± 1.5%; lpr, 5.6 ± 2.4%; gld, 6.2 ± 2.4%; PFP-KO, 4.8 ± 0.8%). Experiments were performed three times with 10 mice in each group. b, Histologic analyses of brain sections of ECM-susceptible WT and ECM-resistant PFP-KO mice at the coma stage in WT susceptible mice and at the same time in PFP-KO resistant mice. Brains were removed after perfusion with 1× PBS and were fixed for histological examinations. A, Section of a normal mouse brain showing a healthy uninterrupted blood vessel with normal architecture and integrity (magnification, ×250). B, Sagittal section of a WT mouse brain at the coma stage showing sequestration and cytoadherence of pRBC (filled arrows). Endothelial cells (▴; magnification, ×750). C, Severe lesions in the cortex of infected WT mice showing a disrupted vessel, with destruction of endothelial cells, extravascular pRBC (filled arrows), and mononuclear cell infiltration (empty arrows; magnification, ×500). D, Sagittal section of infected PFP-KO mouse brain on day 6 postinfection showing cytoadherence to a vessel wall (filled arrow; magnification, ×1000). ▴, Endothelial cells.
To determine histopathologic alterations during ECM pathogenesis, we examined the brains of ECM-susceptible WT mice at the coma stage and those of ECM-resistant PFP-KO resistant mice at the same time (day 6; Fig. 1). Our studies first focused on pRBC that adhere to the endothelium and microvascular destruction, since these events were shown to be a critical step in ECM pathogenesis as described in previous studies (19). In all infected mice, blood vessels were distended, and spaces around them wereenlarged in infected WT or PFP-KO mice (Fig. 1,b, B and D) compared with normal brains of uninfected-WT mice (Fig. 1,bA), suggesting severe edema. Most endothelial cells of the microvasculature showed morphologic changes consisting of amoeboid appearance and suggesting activation in infected WT mice as well as in PFP-KO (Fig. 1,b, B and D, ▴). The lumens of the unruptured vessels were congested with sequestrated pRBC adhering to the vessel walls in ECM-susceptible WT mice as well as in ECM-resistant PFP-KO mice (Fig. 1,b, B and D, filled arrows). By contrast, differences concerning cell destruction were found between PFP-KO and WT mice. Indeed, widespread microvasculature lesions with death of endothelial cells (Fig. 1,bC) were only observed in infected-WT mice at the coma stage, but were never observed in PFP-KO mice. Similarly, parasites scattered throughout extravascular spaces were observed only in the brains of infected WT mice, but not in PFP-KO (Fig. 1,bC, full arrows). Correlated to the cell wall destruction, mononuclear cell infiltrations were observed in these severe brain lesions of infected WT mice (Fig. 1 bB, empty arrows). Together, these data indicated that the cytoadherence of pRBC observed in both strains of mice was not sufficient to predict brain damage. In this regard, the ECM pathogenesis would be correlated to both mononuclear cell infiltration and endothelial cell destruction. These observations prompted us to investigate the characterization and functions of infiltrating cells.
CD8 and CD4 T lymphocytes infiltrate the brains of WT-susceptible and PFP-KO resistant mice during PbA infection
Consistent with the above observations, trafficking of immunocompetent cells has been described in the establishment of the immune response and various disease progressions (6). To quantify and characterize brain-infiltrating mononuclear cells after PbA infection, we performed flow cytometric analyses of brain cell suspensions from WT mice at the coma stage (day 6) and of PFP-KO mice on the same day. Representative flow cytometric analyses indicated that the brains from infected WT and PFP-KO mice contained similar percentages of CD3+ cells (8.6 and 7.6%, respectively; Fig. 2 a). To better enumerate the CD3 cell population in the brain, the absolute number of CD3+ cells was calculated for each mouse. Uninfected control mice contain <2000 CD3+ cells/brain, whereas 48,030 ± 18,450 CD3+ cells/brain were recovered in infected WT mice. These data clearly indicated that lymphocyte accumulation in the brain is a key event during ECM pathogenesis. However, PFP-KO mice did not develop ECM pathogenesis, although 26,800 ± 14,500 CD3+ cells/brain were recovered in these mice. In addition, in PFP-KO mice the absolute number of CD3+ cells tended to increase at later time points (data not shown). The number of accumulating CD3 cells in the brain was not significantly different in WT and PFP-KO mice (n = 4; p = 0, 11). These data demonstrated for the first time that CD3+ T lymphocytes infiltrated the brains of both WT-susceptible and PFP-KO resistant mice after PbA infection. Furthermore, these results suggest that 1) lymphocyte infiltration is an active phenomenon that does not result from nonspecific leakage of blood cells to the brain after alteration of blood-brain barrier; and 2) although required, the presence of T cell is not sufficient to induce brain damage.

CD8 and CD4 T lymphocytes infiltrate the brains of WT susceptible and PFP-KO resistant mice during PbA infection. Flow cytometric analyses of CNS-infiltrating lymphocytes in C57BL/6 WT susceptible and PFP-KO resistant mice infected with PbA. a, CD3 staining in the brain of WT at the coma stage and in PFP-KO mice on day 6 postinfection. b, CD4 and CD8 T cells in brains of WT and PFP-KO mice after infection with PbA. The percentage positive is indicated.
CD8 and CD4 T lymphocytes infiltrate the brains of WT susceptible and PFP-KO resistant mice during PbA infection. Flow cytometric analyses of CNS-infiltrating lymphocytes in C57BL/6 WT susceptible and PFP-KO resistant mice infected with PbA. a, CD3 staining in the brain of WT at the coma stage and in PFP-KO mice on day 6 postinfection. b, CD4 and CD8 T cells in brains of WT and PFP-KO mice after infection with PbA. The percentage positive is indicated.
To further investigate potential differences between various subsets of T lymphocytes in infected PFP-KO resistant vs WT susceptible mice, we evaluated absolute numbers of CD4+ and CD8+ cells (Fig. 2). Representative flow cytometric analyses of CD4 and CD8 cells showed 5.3 and 4.5% of CD8+ cells and 2.8 and 3% of CD4+ cells in infected-WT and PFP-KO, respectively. Both WT susceptible and PFP-KO resistant mice had similar percentages and absolute numbers of CD4+ T cells present within the brain cell suspension (3.7 ± 1.9% (14,080 ± 8,570 CD4+ cells/brain) and 3.4 ± 1% (11,040 ± 7,350 CD4+ cells/brain), respectively; n = 4; p = 0.9). The percentages and absolute numbers of CD8 T cells were also similar in WT mice with CM (8.6 ± 4.8% (33,320 ± 23,230 CD8+ cells/brain) and in PFP mice without ECM on day 6 postinfection (7.4 ± 2.3% (20,390 ± 9,172 CD8+ cells/brain); n = 4; p = 0.4). The absolute numbers of brain-infiltrating CD8+ cells were, on the average, 2-fold higher than those of CD4 cells in both groups of mice. These results were consistent with the total number of CD3+ cells infiltrating the brain. In conclusion, despite differences between WT and PFP-KO mice in their susceptibility to develop ECM, no significant differences were found in the accumulation of CD8 and CD4 T lymphocytes in the brains of these two strains of mice after PbA infection.
Brain-infiltrating CD8 and CD4 T lymphocytes in WT-susceptible and PFP-KO-resistant mice are differentiated memory T lymphocytes
We then further evaluated the differentiation and activation status of brain-infiltrating CD8 and CD4 lymphocytes of PbA-infected WT mice at the coma stage and PFP-KO mice on day 6 postinfection. As shown in a representative flow cytometric analysis (Fig. 3,a), brain-infiltrating CD8 were mainly effector/memory T cells, CD44+CD62L− (60 and 50%, in WT and PFP-KO mice, respectively). Averages of 56.8 ± 6.6 and 52.7 ± 6% of CD8+CD44+CD62L− cells were detected in WT and PFP-KO mice, respectively (Fig. 3,b). Similar results were observed for CD4 cells, with averages of 56.8 ± 18.2 and 56.3 ± 5.8% of CD4+CD44+CD62L− cells in WT and PFP-KO mice, respectively (Fig. 3,b). No naive CD44−CD62L+ cells were detected in the brains of these mice (Fig. 3 b). In addition, the majority of CD4 (65–70%) and CD8 (58–70%) cells also expressed the CD69 activation marker (data not shown). No significant differences were observed between these two strains of mice regarding the differentiation and activation status of T lymphocytes. These results together showed that brain-infiltrating CD8 and CD4 cells were mainly differentiated memory cells in both WT susceptible and PFP-KO resistant mice after PbA infection.

Brain-infiltrating CD8 and CD4 T lymphocytes in WT susceptible and PFP-KO resistant mice are differentiated memory T lymphocytes. a, Representative flow cytometric analyses of CNS-infiltrating lymphocytes and PBL in WT-susceptible (coma stage) and PFP-KO resistant mice infected with PbA on day 6. The analysis of CD44 and CD62L expression was gated on CD8+ cells: upper panels, brain; lower panels, PBL. B, Frequencies of CD62L−CD44high memory CD4 and CD8 cells were measured in the brain (three pools of three brains) and PBL (n = 5) of WT (coma stage) and PFP-KO mice on day 6. The p values are indicated.
Brain-infiltrating CD8 and CD4 T lymphocytes in WT susceptible and PFP-KO resistant mice are differentiated memory T lymphocytes. a, Representative flow cytometric analyses of CNS-infiltrating lymphocytes and PBL in WT-susceptible (coma stage) and PFP-KO resistant mice infected with PbA on day 6. The analysis of CD44 and CD62L expression was gated on CD8+ cells: upper panels, brain; lower panels, PBL. B, Frequencies of CD62L−CD44high memory CD4 and CD8 cells were measured in the brain (three pools of three brains) and PBL (n = 5) of WT (coma stage) and PFP-KO mice on day 6. The p values are indicated.
We then compared these data to the distribution of differentiated memory subsets in the periphery of infected WT and PFP-KO: peripheral blood (Fig. 3,b), spleen, and lymph nodes (data not shown). Both naive (CD44−CD62L+) and memory (CD44+CD62L−) cells were detected in the peripheral blood of infected mice (Fig. 3,a), whereas no naive cells were found in the brains of infected mice. A major increase in CD44+CD62L− cells (Fig. 3,b) and CD69 (data not shown) was observed in infected mice on day 6 postinfection compared with uninfected mice for both CD8 and CD4 subsets. However, a lower frequency of CD8+ or CD4+ CD44+CD62L− PBL was detected in PFP-KO mice than in WT mice after PbA (n = 5; p < 0.005; Fig. 3 b). Similar results were found in the spleen and lymph nodes of WT susceptible and PFP-KO resistant mice (data not shown). Thus, activated-differentiated CD4 and CD8 T cells similarly infiltrated the brains of both WT-susceptible and PFP-KO mice after PbA infection. It can be hypothesized from these findings that T lymphocytes first became activated and differentiate in the periphery and then migrate to the brains of PbA-infected mice.
CFSE-labeled immune splenocytes from WT mice infiltrate the CNS of PFP-KO mice and induce ECM pathology
Following the above hypothesis, we tested whether 1) activated and differentiated cells can migrate to the brain; 2) such migration is dependent on the presence of parasites; 3) a similar phenomenon occurs in WT and PFP-KO mice; and 4) T lymphocyte migration to the brain is involved in the onset of ECM pathology. We then performed a series of adoptive transfer experiments with CFSE-labeled splenocytes from WT mice infected with PbA on day 6 postinfection. Recipient mice were either WT susceptible or PFP-KO resistant mice that were infected with parasites 1 h after cell transfer. CFSE-labeled cells were tracked 6 days later in brains and spleens (Fig. 4,a) of the recipient mice at the coma stage on day 6 postinfection. The results showed an infiltration of CFSE+ lymphocytes in the spleens and brains of both infected WT and PFP-KO mice. Approximately 1–2% of CFSE+ cells were recovered from the spleens, while 0.1–0.2% CFSE+ cells were recovered from the brains, of WT and PFP-KO recipients (Fig. 4,a). When CFSE-labeled splenocytes from infected WT mice were transferred into uninfected WT recipients, no CFSE+ cells were found in the brains of these mice (Fig. 4,a), and the mice did not develop ECM. These results indicate that the presence of parasite in the recipient mice was necessary for brain infiltration and disease induction. In addition, naive CFSE-labeled splenocytes (from uninfected WT mice) migrated to the spleen, but not to the brain, of infected WT mice (Fig. 4 a). Furthermore, 70% of CFSE-labeled cells isolated from infected WT mice were activated CD62L-CD44+. These results were consistent with the absence of the naive cell phenotype in the brain.

CFSE-labeled immune splenocytes from WT mice infiltrate the CNS of PFP-KO mice and induce ECM pathology. a, CFSE-labeled Splenocytes (5 × 106 cells/mice) were prepared from PbA-infected or uninfected WT mice on day 6 postinfection and adoptively transferred into infected or uninfected WT or PFP-KO recipient mice as indicated. Recipient mice were then infected with 106 pRBC. Flow cytometric analyses of CFSE+ cells gated on live lymphocytes infiltrating the brain (left panel) and the spleen (right panel) of WT and PFP-KO recipient mice on day 6 postinfection. Percentages of cells recovered from the brain and spleen are indicated. b, Percent survival of PFP-KO or recipient WT or PFP-KO mice (n = 5) after adoptive transfer of CFSE-labeled WT splenocytes (i.v. injection of 5 × 106 cells/mice). Recipient mice WT or PFP-KO mice died from ECM, and PFP-KO mice died after day 21 from anemia. The p values are indicated.
CFSE-labeled immune splenocytes from WT mice infiltrate the CNS of PFP-KO mice and induce ECM pathology. a, CFSE-labeled Splenocytes (5 × 106 cells/mice) were prepared from PbA-infected or uninfected WT mice on day 6 postinfection and adoptively transferred into infected or uninfected WT or PFP-KO recipient mice as indicated. Recipient mice were then infected with 106 pRBC. Flow cytometric analyses of CFSE+ cells gated on live lymphocytes infiltrating the brain (left panel) and the spleen (right panel) of WT and PFP-KO recipient mice on day 6 postinfection. Percentages of cells recovered from the brain and spleen are indicated. b, Percent survival of PFP-KO or recipient WT or PFP-KO mice (n = 5) after adoptive transfer of CFSE-labeled WT splenocytes (i.v. injection of 5 × 106 cells/mice). Recipient mice WT or PFP-KO mice died from ECM, and PFP-KO mice died after day 21 from anemia. The p values are indicated.
A striking consequence of these adoptive transfer experiments was that PFP-KO mice developed ECM pathogenesis and died at the same rate as WT mice (Fig. 4 b). Thus, ECM was induced in PFP-KO mice after adoptive transfer of cells from infected WT mice, which were directed to the brain of PFP-KO mice. These results further highlight the importance of lymphocyte trafficking to the CNS after PbA infection in the development of ECM and suggest that only activated lymphocytes expressing perforin from infected mice can induce ECM pathology. These data prompted us to further characterize the cytotoxic/effector capacities of CD8 lymphocytes from infected WT and PFP-KO mice.
Cytotoxic CD8 lymphocytes are required for the development of ECM pathogenesis
The initial observation of the resistance of PFP-KO mice to ECM together with the major infiltration of CD8 and CD4 cells in the brain suggest that mostly cytotoxic mechanisms were involved in the development of ECM. We further tested the capacity of CD8+ and CD8− subsets to induce ECM and characterize their cytolytic functions and cytokine mRNA expression. We chose RAG2-KO mice because these mice lack peripheral T and B lymphocytes and did not develop ECM (Fig. 5,a). After infection with PbA, RAG2-KO mice died from anemia with increased parasitemia between days 20 and 28 postinfection (data not shown). Splenic CD8+ (purity, ∼99% CD8 cells, as assessed by flow cytometric analysis) and CD8 cells from WT mice on day 6 postinfection were magnetically sorted and adoptively transferred into ECM-resistant, RAG2-KO recipient mice (Fig. 5,a). RAG2-KO recipient mice were then infected with parasites. RAG2-KO became moribund, with all signs of ECM pathogenesis, after adoptive transfer of either total splenocytes or CD8+ cells, but not after transfer of CD8− cells from infected WT donors (Fig. 5,a). In addition, RAG2-KO mice did not develop the ECM pathology after adoptive transfer of CD8+ cells isolated from PFP-KO donor mice (Fig. 5 a). These results demonstrated that only perforin gene-expressing CD8 cells were able to induce ECM.

Perforin-associated cytolytic mechanisms are required for the development of ECM pathogenesis. a, Percent survival of recipient RAG2-KO mice after adoptive transfer of total, CD8−, or CD8+ splenocytes (i.v. injection of 5 × 106 cells/mice) isolated from infected WT or PFP-KO mice on day 6 postinfection. Recipient mice (n = 3) were infected 1 h after cell transfer with 106 pRBC. Purified CD8+ fractions contained 98–99% CD8+ lymphocytes, as assessed by flow cytometric analyses. b, Anti-CD3-redirected chromium release assay of purified CD8+ effector cells from WT and PFP-KO infected mice (E, effector cells). Target cells (T) were from the P815 cell line. c, IFN-γ, IL-2, IL-4, IL-5, IL-10, IL12-p40, IL-13 cytokine, and GAPDH gene expression determined by RT-MPCR with CD8+ T cells from WT and PFP-KO infected mice.
Perforin-associated cytolytic mechanisms are required for the development of ECM pathogenesis. a, Percent survival of recipient RAG2-KO mice after adoptive transfer of total, CD8−, or CD8+ splenocytes (i.v. injection of 5 × 106 cells/mice) isolated from infected WT or PFP-KO mice on day 6 postinfection. Recipient mice (n = 3) were infected 1 h after cell transfer with 106 pRBC. Purified CD8+ fractions contained 98–99% CD8+ lymphocytes, as assessed by flow cytometric analyses. b, Anti-CD3-redirected chromium release assay of purified CD8+ effector cells from WT and PFP-KO infected mice (E, effector cells). Target cells (T) were from the P815 cell line. c, IFN-γ, IL-2, IL-4, IL-5, IL-10, IL12-p40, IL-13 cytokine, and GAPDH gene expression determined by RT-MPCR with CD8+ T cells from WT and PFP-KO infected mice.
To further explore functional differences between CD8 cells from PFP-KO and WT mice, we tested their effector functions, such as cytolytic capacity (Fig. 5,b) and IFN-γ expression (Fig. 5,c). Because the Ag specificity of the CD8 T cells in PbA infection was unknown, we used a CD3-redirected assay to analyze their cytotoxic capacity in a chromium release assay. We showed that in vivo stimulated CD8 T cells from PbA-infected WT mice exhibited significantly higher cytotoxicity toward P815 targets than CD8 T cells from PFP-KO mice (80 vs 16% cytotoxicity, respectively; Fig. 5,a). Naive CD8 T cells from uninfected mice did not mediate any cytotoxic functions (<5% at 90/1 E:T cell ratio). The low level of cytotoxic activities observed with CD8 T cells from PFP-KO mice could be due to Fas/Fas-ligand-induced cytotoxic pathways (17). The cytokine expression was also analyzed by RT-MPCR (Fig. 5,c). We showed that IFN-γ mRNA was expressed in CD8 cells from both infected WT and PFP-KO, but not in naive CD8 from uninfected mice (Fig. 5). These results demonstrated that in vivo stimulated CD8 T cells from WT mice did not differ from those of PFP-KO mice in their cytokine expression, but differed in their cytolytic capacity.
Brain-infiltrating effector-memory cytotoxic CD8 T cells express chemokine receptor gene, which could be partially involved in the development of ECM pathogenesis
Our above data indicated that activated and differentiated cytotoxic CD8 cells migrated and accumulated in the brain during ECM pathogenesis. The role of chemokines/chemokine receptors in cell trafficking has been described during the establishment of the immune response and disease progression (6). To better understand how CD8 T cells were targeted to the CNS following PbA infection, we studied the pattern of chemokine receptor expression on purified CD8+ T cells recovered from spleens (Fig. 6,a) and brains (Fig. 6,b) of mice at the coma stage of ECM and at the same time from PFP-KO mice (data not shown). We found that activated CD8+ cells showed increased expression of mRNA coding for CCR-2 and CCR-5 as well as CXCR-3 and CXCR-4, whereas naive CD8 cells (uninfected WT mice) expressed only CXCR4. A similar pattern of chemokine receptor expression was observed in CD8+ cell fractions from both infected WT or PFP-KO mice regardless of their origins, spleen or brain (Fig. 6, a and b). We thus investigated the role of the chemokine receptor, CCR-5, in ECM pathogenesis using CCR5-deficient (CCR5−/−) mice and their littermates (CCR5+/+; Fig. 6 c). We found a modest, but consistent, delayed effect of CCR5-KO on the onset of ECM after PbA infection (n = 10; p = 0.0007). Although limited to the study of one chemokine receptor, these data indicate for the first time the role of these molecules in ECM pathogenesis and emphasize the importance of CD8 cells with regard not only to their cytotoxic capacities, but also to their specific pattern of migration after PbA infection.

Brain-infiltrating effector-memory cytotoxic CD8 T cells express the chemokine receptor gene, which could be partially involved in the development of ECM pathogenesis. Pattern of chemokine receptor expression in brain (a) and spleen (b) CD8+ T lymphocytes (98% purity) from PbA-infected WT mice at the coma stage. Similar results were observed with CD8+ cells from PFP-KO mice (data not shown). RT-MPCR for CXC (upper panel) and CC (lower panel) chemokine receptors: CXCR1, -2, -3, -4, and -5; CCR1, -2, -3, -4, and –5; and GAPDH. c, Percent survival of CCR5-deficient mice and CCR5 WT littermates after infection with PbA (n = 10; p = 0.0007). Mice died from ECM pathogenesis.
Brain-infiltrating effector-memory cytotoxic CD8 T cells express the chemokine receptor gene, which could be partially involved in the development of ECM pathogenesis. Pattern of chemokine receptor expression in brain (a) and spleen (b) CD8+ T lymphocytes (98% purity) from PbA-infected WT mice at the coma stage. Similar results were observed with CD8+ cells from PFP-KO mice (data not shown). RT-MPCR for CXC (upper panel) and CC (lower panel) chemokine receptors: CXCR1, -2, -3, -4, and -5; CCR1, -2, -3, -4, and –5; and GAPDH. c, Percent survival of CCR5-deficient mice and CCR5 WT littermates after infection with PbA (n = 10; p = 0.0007). Mice died from ECM pathogenesis.
Discussion
Our data brought striking evidence of the implication of brain-infiltrating cytotoxic effector CD8 lymphocytes in the development of ECM pathogenesis. We found that in contrast to ECM-susceptible C57BL6 mice, PFP-KO mice were resistant to ECM in absence of brain lesions, whereas cytoadherence of parasitized erythrocytes and a massive accumulation of activated/effector CD8 lymphocytes were observed in both groups of mice. ECM can be induced in PFP-KO mice when cytotoxic CD8 cells from infected C57BL6 mice infiltrated the brain of PFP-KO mice. This specific recruitment might involve chemokine/chemokine receptors, since their expression was up-regulated on activated CD8 cells, and susceptibility to ECM was delayed in CCR5-KO mice.
PFP-KO mice were totally resistant to ECM pathogenesis, whereas mice with mutations in Fas or Fas ligand genes remained susceptible. Indeed, granule exocytosis and Fas/Fas ligand activation pathways are two distinct mechanisms involved in the death of infected target cells by CD8 lymphocytes (17, 20). The Fas-mediated pathway, mainly operative with CD4 T cells, is thought to be primarily involved in immune regulation and maintenance of tolerance (21). Granule exocytosis is mediated by perforin and granzymes and plays a major role in the control of infection (22). Our results clearly demonstrated that perforin-dependent, but not Fas/Fas ligand-dependent, cytotoxic pathways were involved in ECM pathogenesis. Previous reports have observed the resistance of PFP-KO mice to ECM. However, the mechanism involved was not investigated (23). Perforin-mediated cytolysis is the primary pathway of cytotoxicity used by both CD8 T cells and NK cells (22); however, the role of NK cells in the development of ECM has been excluded by Yanez et al. (4). These results together with previous data on the requirement for CD8 cells in the development of ECM (4, 5) further suggest that mainly CD8+ T cells are critical in T cell-mediated immune responses after PbA infection and its neurological consequence.
This report is the first demonstration of a massive accumulation of CD8 and CD4 T lymphocytes in the brains of both C57BL/6 and PFP-KO mice on day 6 postinfection. Although most of our studies showed results on day 6, we have also investigated cell infiltration on days 10 and 15 postinfection in perforin KO mice. We found 2-fold increased cell infiltration of both CD4+ and CD8+ T cells in the brains of PFP KO mice on days 10 and 15 postinfection compared with day 6 (data not shown). These mice did not develop cerebral symptoms, but did later with overwhelming parasitemia. Hence, the number of infiltrating T cells was indicative not of disease induction, but, rather, of perforin expression by CD8+ T cells. Our results provide striking evidence that T lymphocytes were mainly activated and differentiated specifically recruited to the brain as ascribed by CD44, CD69 up-regulation, and CD62L down-regulation. We found that T cell memory also increased in the periphery (PBL, spleen, and lymph nodes) of PbA-infected WT and PFP mice. However, the memory cell population was less expanded in PFP-KO than in WT mice. It has been demonstrated that the acquisition of perforin-dependent cytotoxic activity regulates the expansion and persistence of CD8 effector T cells in vivo (24, 25). This mechanism is independent of the role of perforin as an antimicrobial effector molecule (25). Thus, the survival and death of CD8 cells might be differently regulated in the peripheral blood of WT and PFP-KO mice after PbA infection.
Our work clearly indicated that lymphocyte accumulation in the brain is a key event during ECM pathogenesis. Infected WT mice developed ECM with severe brain damage, whereas PFP-KO mice did not develop ECM pathogenesis, while T lymphocytes also accumulated in brains. Thus, the lymphocyte infiltration is an active phenomenon that did not result from alteration of the blood-brain barrier. Adoptive transfer of CFSE-labeled splenocytes from infected WT or PFP-KO recipient mice revealed preferential migration of CFSE+ lymphocytes into the brain after the host parasite infection. In addition, ECM pathogenesis can be induced in PFP-KO mice when cytotoxic CD8+ cells from infected C57BL6 mice infiltrated the brain of PFP-KO mice. Although CD8 T lymphocytes produced IFN-γ, its secretion by these cells was not sufficient for the development of ECM. As a matter of fact, when PFP-KO CD8+ cells, which also produce IFN-γ, were transferred in vivo, mice remained resistant to ECM. It has also been shown that IFN-γ receptor KO mice (26) and IFN-γ KO mice (J. Nitcheu, unpublished observation) were resistant to the development of ECM pathogenesis. IFN-γ produced by other types of cells could play a role in ECM. Indeed, IFN-γ was shown to be required for PbA-induced endothelial ICAM-1 up-regulation (27), which might be involved in the interaction of effector CD8 lymphocytes with APC.
We have shown that several conditions are required for the onset of ECM: 1) infiltration of CD8 lymphocytes in the brain, 2) cytotoxic capacity of infiltrating CD8 lymphocytes, and 3) presence of parasites in mice. The recent definition of murine endothelial cells as semiprofessional APC (28) led us to speculate that these cells are likely to play a role in Ag presentation. Previous reports have implicated the role of ICAM-1 in the induction of ECM, suggesting that the interaction of effector to APC was a crucial step in the pathogenesis (29). The striking histological differences between the two strains of mice concerned the severe destructive lesions selectively affecting WT brain microvessels, with disrupted microvessels and widespread endothelial cell destruction. Therefore, Ag presentation by endothelial cells to inflammatory T CD8 cells was likely to occur and might lead to endothelial cell destruction. Furthermore, histopathologic examinations in WT susceptible and PFP-KO resistant mice showed similar tendencies: cytoadherence of sequestrated pRBC to the endothelium of cerebral vessels and activation of endothelial cells. In many studies sequestration of cells within the microvasculature has been invoked as a factor of pathology leading to vessel occlusion (14). In our study we showed that the cytoadherence phenomenon occurred in both resistant and susceptible mice and thus was necessary, but not sufficient, to induce ECM pathology.
The multistep process of leukocyte binding to endothelium occurred through complex interactions of adhesion molecules, chemoattractants, and their receptors, including cell rolling, adherence, diapedesis, and migration (30, 31, 32). The pattern of chemokine receptor expression on cytolytic effector cells compared with naive CD8 cells showed for the first time increased expression of CCR2, CCR5, and CXCR3 that could be involved in the preferential migration of peripheral T lymphocytes to the brain. A similar pattern of receptor expression was observed in both infected PFP and WT mice. Several ligands of these receptors might be expressed in the brain during inflammation (10, 13, 33, 34, 35, 36, 37). Although CXCR4 is relevant in the trafficking of all resting and activated lymphocytes, CXCR3, CCR2, and CCR5 expressions were likely to be involved in the recruitment of activated T cells in multiple tissues, including the brain, during neuropathologic disorders (33). CCR5, CXCR3, and their ligands, macrophage inflammatory protein-1α and IFN-γ-inducible protein 10, have been recently implicated in activated T cell migration to the CNS in demyelinating brain lesions (36). Since CCR-5 is expressed on type 1 CTLs, we chose to study available CCR5-KO mice as a first example of the functional role for these chemokine receptors. A consistent effect was found on ECM onset after PbA infection in CCR5-KO mice. Other chemokine receptors might play a synergistic role, since the onset of ECM was delayed, but not abolished, in CCR5-KO mice. Although these data should be expanded to the study of CXCR3 and CCR2 in this pathology, we think our results open new perspectives in the involvement of cell trafficking of cytotoxic/effector cells in the brain during ECM pathogenesis after PbA infection.
Acknowledgements
We thank O. Sylvie and S. Bagot for technical assistance. We also thank P. Delis for supervising the animal care facility Nouvelle Animalerie Commune de la Pitié-Salpétrière. We are particularly grateful to Profs. P. Debre, B. Autran, and J. C. Ameisen for helpful discussions.
Footnotes
This work was supported by a grant from La Fondation de Treilles (Paris, France; to J.N.) and by Grant PAL+ from Ministère de la Recherche.
Abbreviations used in this paper: CM, cerebral malaria; ECM, experimental cerebral malaria; KO, knockout; PbA, Plasmodium berghei ANKA; pRBC: parasitized RBC; RAG, recombinase-activating gene; RT-MPCR, RT-multiplex PCR; WT, wild type.
