
INTRODUCTION
The coronavirus disease 2019 (COVID-19) pandemic has burdened countries worldwide since early 2020. By mid-2022, COVID-19 cases had reached 564,266,029 and caused more than 6 million deaths (Worldometers, 2022). It is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), attacking some organs, particularly the lungs (Morse et al., 2020).
SARS-CoV-2 belongs to the subfamily Orthocoronavirinae in the family Coronaviridae and order Nidovirales (Cui et al., 2019). It is an enveloped positive-stranded RNA (+ssRNA) virus with a genome size of 26–32 kb coding structural and non-structural proteins (nsp1-16) (Gil et al., 2020; Ji et al., 2020; Mody et al., 2021). Among these proteins, 3C-like protease (3CLpro) has been reported to be essential for controlling the multiplication cycle of SARS-CoV-2 (Mody et al., 2021; Petrou et al., 2022; Zhu et al., 2022). It cleaves polyproteins 1ab at 11 sites into mature functional proteins (nsp5-16) (Lee et al., 2020). Based on cleavage sequence variation, 3CLpro presents in all genera of coronaviruses, such as Alpha-, Beta-, Gamma-, and Deltacoronavirus (Kiemer et al., 2004; Prescott, 2022). Therefore, inhibition on 3CLpro may halt the spread of coronavirus infections, especially SARS-CoV-2.
Several studies have suggested that natural products can inhibit 3CLpro (Liu et al., 2021; Su et al., 2021). Myricetin exhibits IC50 of 0.30 μM to 3CLpro by interacting with the catalytic site residues (Cys145 and His41) (Su et al., 2021). Meanwhile, baicalein shows a potent inhibitory activity to 3CLpro with IC50 of 0.39 μM (Liu et al., 2021). Both flavonoids have high inhibition activities since their IC50 values are below 20 μM (Liu et al., 2008) and shed light on the potency of other flavonoids as 3CLpro inhibitors.
Another source of flavonoids is the genus Erythrina, which is grown in tropical and subtropical regions, including Indonesia (Fahmy et al., 2018; Herlina et al., 2011). Some 378 flavonoids have been isolated from Erythrina plants. They are flavones, flavonols, flavanones, chalcones, isoflavans, isoflav-3-enes, isoflavanones, isoflavones, pterocarpans, 6α-hydroxypterocarpans, pterocarpenes, coumestans, 3-arylcoumarins, coumaronochromones, 2-arylbenzofurans, 3-aryl-2,3-dihydrobenzofurans, and biflavonoids. Lee et al. (2009) demonstrated that alpinumisoflavone from the genus Erythrina potentially inhibits the growth of human immunodeficiency virus type 1. Furthermore, vitexin isolated from the genus Erythrina potentially inhibits the expression of DNA in herpes simplex virus type 1 (Fahmy et al., 2020). Nonetheless, despite their potencies, flavonoids from Erythrina have not been investigated for their inhibition activities to SARS-CoV-2, especially targeting 3CLpro. Hence, the study on flavonoids from Erythrina as 3CLpro inhibitors is vital in the development of anti-SARS-CoV-2 agents.
Nowadays, in silico approaches are the most commonly used methods to accelerate drug discovery from natural products (Ramírez and Caballero, 2018). One of them is molecular docking, which has been utilized in various studies (Hardianto et al., 2021; Petrou et al., 2022; Raschi et al., 2009; Ul Qamar et al., 2017). Molecular docking has assisted Yamamoto et al. (2022) in virtually screening 3CLpro inhibitors from the Enamine library before in vitro assays. It was also used by Ahmad et al. (2021) as one of the methods to elucidate the molecular mechanism of PF-07321332, an oral drug for COVID-19 treatment, in inhibiting 3CLpro. Therefore, we used molecular docking to screen 378 flavonoids from Erythrina as 3CLpro potential inhibitors. Meanwhile, to evaluate the drug-likeness of the screened flavonoids as orally active drugs, we employed Lipinski’s rule of five (Ro5) (Lipinski et al., 2012). We also performed molecular dynamics (MD) simulations to top hits for more realistic energy prediction (Forli et al., 2016).
Another vital aspect of drug development is the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of compounds in the body. Absorption, distribution, metabolism, and excretion (ADME) data provide information to assess drug molecule candidates in crossing the small intestine, which eventually are distributed to target cells, metabolized, and cleared from the human body. Metabolism is an important process in activating some drug molecules. However, it may also convert drug molecules into toxic forms (Daina et al., 2017; Gil et al., 2020; Kavaliauskas et al., 2020). Hence, we evaluate the ADMET properties of flavonoids from Erythrina using an in silico approach via the pkCSM web server (Pires et al., 2015). Our study suggests that flavonoids from Erythrina have potential as 3CLpro inhibitors, which help guide further in vitro and in vivo experiments in COVID-19 drug development.
MATERIALS AND METHODS
Flavonoid structures preparation
Fahmy et al. (2018) reported that 378 flavonoids have been isolated from the genus Erythrina. The two-dimensional (2D) structures of the flavonoids were sketched in Chemaxon MarvinSketch (Chemaxon, 2020). The protonated states of the flavonoids at physiological pH (7.4) were predicted using the same program, Chemaxon MarvinSketch. Their three-dimensional (3D) structures with the protonated states at physiological pH were saved in .pdb format. MGLTools 1.5.6 (Forli et al., 2016) was utilized to prepare the 3D structures for molecular docking.
Molecular docking validation
The crystal structure of 3CLpro was retrieved from the Protein Data Bank (PDB) website (https://www.rcsb.org) with PDB ID 6LU7 (Jin et al., 2020). The 3D structures of 3CLpro and its inhibitor (N3) (Saur et al., 2021; Yang et al., 2005) were separated in BIOVIA Discovery Studio (DS) 2020 Client and saved as separated pdb files, while the water and ion molecules were discarded. Subsequently, both structures were subjected to AutoDockTools 1.5.6 for the preparation of molecular docking validation. Nonpolar hydrogen atoms were removed, whereas the polar ones were retained. The Gasteiger and Kollman atomic charges (Forli et al., 2016) were added to the N3 and 3CLpro structures, respectively. The active torsions of the N3 structure were set as suggested by AutoDockTools 1.5.6. Both 3CLpro and N3 structures were saved as a pdbqt format. The dimension of the grid box was set to cover the whole structure of N3. Grid maps at the binding site of 3CLpro were computed using the AutoGrid4.2 program. A Lamarckian genetic algorithm (GA) was selected in molecular docking. The number of GA runs and population size were set to 100 and 300, respectively. Meanwhile, the maximum number of evaluations was 2.500.000 times (medium). The rest of the search parameters were set to defaults, and so were docking parameters. Molecular docking validation was performed by redocking the N3 structure to the binding site of 3CLpro. The validation step was considered a success if the root mean square deviation (RMSD) of the docked N3 was below 2.00 Å (Gohlke et al., 2000; Kissler et al., 2020).
Virtual screening campaigns
The preparation of flavonoid structures in AutoDockTools 1.5.6 followed the steps in the molecular docking validation above. We performed virtual screening campaigns using Raccoon v1.0b on an Ubuntu 20.04 operating system. The grid map and docking parameters used in molecular docking validation were employed as templates. The virtual screening results were analyzed in AutoDockTools 1.5.6.
Lipinski’s Ro5 calculation
Hits from virtual screening campaigns were subjected to Lipinski’s Ro5 (Cheng et al., 2012; Lipinski et al., 2012) in the SwissADME (http://www.swissadme.ch/) web server. This step was to assess the potency of hits as orally active drug candidates. Lipinski’s Ro5 evaluates hits based on their molecular weights (MWs), logP values, and hydrogen bond donors (HBDs) and acceptors (HBAs).
ADMET prediction
ADMET prediction was conducted through the pkCSM web server (http://biosig.unimelb.edu.au/pkcsm). The absorption criteria consist of water solubility, Caco2 permeability, human intestinal absorption, P-glycoprotein substrate, and P-glycoprotein I and II inhibitors. Hit distribution prediction involves the steady-state volume of distribution (VDss), fraction unbound, blood–brain barrier (BBB), and central nervous system (CNS) permeabilities. The in silico metabolism study includes the prediction of hits as inhibitors of CYP1A2, CYP2C19, CYP2C9, and CYP2D6. In addition, pkCSM also predicts hits as CYP2D6 and CYP3A4 substrates. Total clearance and renal organic cation transporter 2 (OCT2) are two criteria to evaluate the excretion of hits. In evaluating the safety profile of hits, we used pkCSM to predict AMES toxicity and hepatoxicity. Moreover, we employed the web server to predict the potency of hits as hERG I and II inhibitors. We utilized a tidyverse package (Wickham et al., 2019) in an R environment to assist with ADMET analysis.
MD simulations
MD simulations were performed to evaluate the binding energy of the selected compounds compared with the N3 and GC376 inhibitors. The ligand was deployed in the AMBER force field with GAFF (Wang et al., 2004) and the 3CLpro with ff14SB (Maier et al., 2015). Each protein–ligand was solvated in a TIP3P water model as an explicit solvent with a minimum boundary distance of 10 Å. Also, the numbers of Na+ and Cl− ions were added to each protein–ligand complex to acquire the neutralization and physiological state. All systems were minimized, heated gradually to 310 K, and equilibrated, which eventually produced 100 ns trajectories. Afterward, binding energy values were computed from the last frame MD trajectory using molecular mechanics with a generalized born and surface area solvation (MMGBSA) method.
Visualization
Molecular interactions between the best hits, which have good ADMET profiles, and the binding site of 3CLpro were visualized using BIOVIA DS 2020 Client. The generated images were processed further using GNU Image Manipulation Program 2.10.
RESULTS AND DISCUSSION
Molecular docking validation
As the validation step in the virtual screening campaign, we conducted a redocking procedure on the crystal structure of SARS-CoV-2 3CLpro (hereafter mentioned as 3CLpro) and its ligand (N3), which is deposited in the PDB as 6LU7. Such a procedure ensures that the stochastic search method reaches a global minimum (Forli et al., 2016). We achieved a RMSD of less than 2 Å in the redocking procedure by applying grid box dimensions of 26 × 32 × 32 with 1 Å spacing. The best pose of N3 has a binding energy score of −10.14 kcal/mol in binding to 3CLpro.
Virtual screening campaigns
In the next step, we performed a virtual screening campaign of 378 Erythrina flavonoids from Erythrina against 3CLpro (Table S1). These flavonoids include 18 flavones, 3 flavonols, 91 flavanones, 14 chalcones, 11 isoflavans, 5 isoflav-3-enes, 43 isoflavanones, 99 isoflavones, 42 pterocarpans, 14 6α-hydroxypterocarpans, 7 pterocarpenes, 5 coumestans, 3 3-arylcoumarins, 1 coumaronochromone, 15 2-arylbenzofurans, 2 3-aryl-2,3-dihydrobenzofurans, and 1 bioflavonoid. Every compound was subjected to a 50-run molecular docking, and the results are summarized in Table S1.
Of 378 Erythrina flavonoids, 108 possess binding energy scores stronger than that of N3 (−10.14 kcal/mol) (Table 1). Most of these flavonoids, having high binding affinities to 3CLpro, belong to flavanones and isoflavones. Several 40 hits are flavanones with erycaffra F (57) as the strongest 3CLpro binder within the group (−11.93 kcal/mol), while 34 others are isoflavones. The best hit from the isoflavone group is 6,8-diprenylorobol (211) with a binding energy score of −12.18 kcal/mol. Isoflavanones are the third largest group of flavonoids from Erythrina with binding energy scores stronger than that of N3. This flavonoid group shares 15 hits, where the most negative binding energy (−12.04 kcal/mol) is owned by orientanol E (171). The rest of the flavonoids with tight binding to 3CLpro consist of five flavones, four isoflavans, two chalcones, two pterocarpans, two 6α-hydroxypterocarpans, one flavonol, one pterocarpene, and one biflavonoid. Meanwhile, none of the isoflav-3-enes, coumestans, 3-arylcoumarins, coumaronochromones, 2-arylbenzofurans, coumaronochromones, 2-arylbenzofurans, and 3-aryl-2,3-dihydrobenzofurans is stronger than N3 in binding to 3CLpro.
The top hit among the flavonoid groups is kaempferol-3-O-(2??-O-β-?-glucopyranosyl-6??-O-α-L-rhamnopyranosyl-β-?-glucopyranoside) (20), a flavonol, with a binding energy score of −13.23 kcal/mol. The second top hit is a flavone, vicenin-2 (16), followed by 6,8-diprenylorobol (211) and vogelin H (277) which are isoflavones. The fifth top hit is orientanol E (171), belonging to isoflavanones.
The conducted virtual screening campaign suggests that many flavonoids from Erythrina are potential 3CLpro inhibitors. Nonetheless, these compounds should fulfill drug-likeness criteria to be active as oral drugs. Therefore, in the next step, we assessed the resulting hits using Lipinski’s Ro5 through the SwissADME web server.
Lipinski’s Ro5
Several of the 108 hits were further assessed for their drug-likeness using Lipinski’s Ro5 through the SwissADME web server. According to Ro5, drugs should have a MW less than 500 Da, HBD and HBA less than 5 and 10, respectively, and logP less than 5. Compounds should fulfill three rules for the minimum requirement as drug-like molecules (Lipinski et al., 2012; Raj, 2021).
The result of the Ro5 assessment suggested that only five hits failed to satisfy these three rules (Table S2). Three of them belong to flavones, namely, apigenin-7-O-rhamnosyl-6-C-glucoside (14), vicenin-1 (15), and vicenin-2 (16). The others are kaempferol-3-O-(2??-O-β-?-glucopyranosyl-6??-O-α-L-rhamnopyranosyl-β-?-glucopyranoside) (20) and auriculatin 4’-O-glucoside (189), which are a flavonol and an isoflavanone, respectively. Compounds (20) and (16) are the top first and second hits in the conducted virtual screening campaign, but they do not fulfill the minimum requirement for potential orally active drugs. This issue can be addressed by implementing synthetic carriers to improve the solubility, bioavailability, and stability of (20) and (16). For example, Uekama (1999) used cyclodextrin as the synthetic carrier, whereas Choudhury (2020), Du and Chen (2019), and Vyas et al. (2014) used a liposome. Various strategies can improve drug bioavailability, such as lipid nanoparticles or sugar moieties. A study demonstrated that the lipid group increased passive diffusion across epithelial barriers. Also, the polyhydroxylated nature of sugars provides increasing water solubility and facilitates transport across the BBB. Hence, some combinations may enhance the solubility and permeability of drugs (Blanchfield and Toth, 2004).
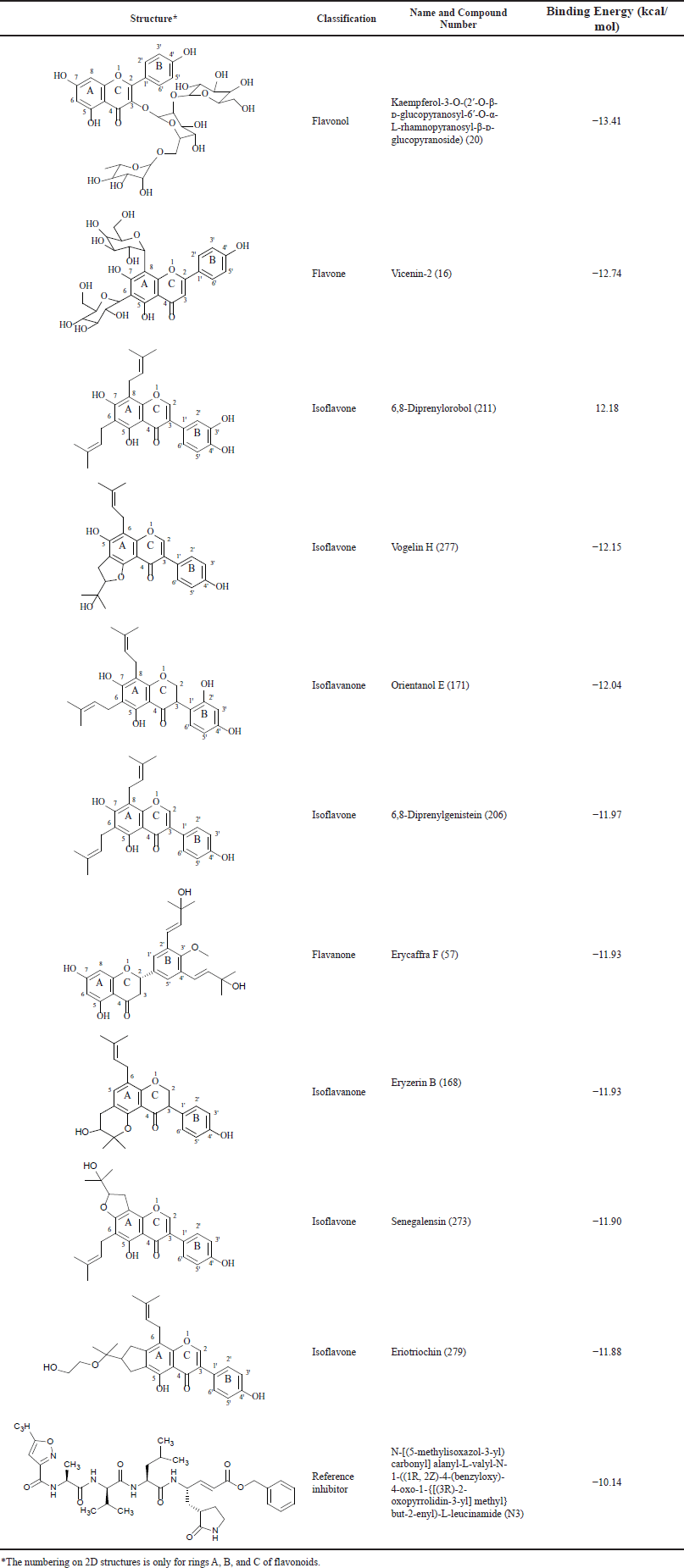 | Table 1. Top 10 hits from virtual screening campaigns possessing binding energy values stronger than that of the N3 inhibitor. [Click here to view] |
Most of the hits, 103 flavonoids, fulfill the minimum requirement of Ro5. Some 101 satisfy all the rules of Ro5, whereas vitexin (11) and bis-sigmodiol (378) violate the rule of HBA number (Table S2). These results suggest that those 103 flavonoids are drug-like molecules and potential orally active drug candidates.
In silico ADMET
Oral is the most popular route for drug administration due to its convenience for patients (Alqahtani et al., 2021). Through oral administration, drug molecules follow a journey including ADME. Therefore, 103 flavonoids satisfying Ro5 also should be further assessed according to their ADME criteria inside the body. Moreover, their toxicities should be evaluated to describe their safety profile. In this work, we utilized the pkCSM web server (http://biosig.unimelb.edu.au/pkcsm/), which offers more comprehensive feature predictions than SwissADME, to study the in silico absorption, metabolism, excretion, and toxicity.
In silico predictions to assess absorption criteria of 103 flavonoids include water solubility, human intestinal absorption, Caco2 permeability, P-glycoprotein substrate, and P-glycoprotein I and II inhibitors. Prediction results suggest that the 103 flavonoids have good water solubility and human intestinal absorption (Table S3), which are expected since they fulfill Ro5, as predicted by SwissADME. Nonetheless, only 69 flavonoids have good Caco2 permeabilities, with predicted values greater than 0.900. As one of the top hits, compound (171) has a predicted value of 0.771, which fails to satisfy Caco2 permeability. It is worth noting that the Caco2 permeability assay uses a cell line originating from a human colorectal carcinoma which represents the discrimination of drug absorption in the large intestine (Artursson and Karlsson, 1991). In terms of P-glycoprotein-related absorption, the pkCSM web server predicts the 103 flavonoids as P-glycoprotein substrates, implying that they are readily pumped back to the lumen, lowering their absorption. However, 85 of them are P-glycoprotein I inhibitors, 78 are P-glycoprotein II inhibitors, and 64 are inhibitors for both proteins. Compounds (171), (211), and (277), which are on the top hit list, are inhibitors for both P-glycoproteins I and II. Inhibition of P-glycoproteins I and II can increase the bioavailability of molecules that are P-glycoprotein substrates (Finch and Pillans, 2014).
After absorption via the small intestine, drug molecules are distributed to their targets and tissues through the blood and lymphatic circulatory systems. Therefore, distribution is a vital pharmacokinetic parameter for monitoring the number of drug molecules reaching target cells (Twardowski and Ksiazek, 1983; Zhivkova et al., 2015). For the distribution aspect, pkCSM predicts the VDss, fraction unbound in humans, BBB permeability, and CNS permeability.
VDss represents the propensity of drug molecules to reside in the plasma or distribute in tissues. Drugs with higher VDss values are distributed more in tissue than in plasma, thus requiring greater doses, and vice versa. Only four flavonoids are predicted to have high values of distribution volumes (log VDss > 0.45). Some 24 flavonoids have low log VDss values below −0.15, including compound (211) as one of the top hits. The values of log VDss for the other 75 flavonoids are between −0.15 and 0.45 (Table S4). Therefore, the majority of the flavonoids complying with Ro5 may be more distributed in plasma. However, all these flavonoids have high fractions of bound forms to proteins in the blood plasma, indicated by low values of predicted unbound states. These predictions may reflect the low bioavailability of the flavonoids and their inefficiency in diffusing the membrane cells. A study conducted by Vandyck and Deval (2021) suggests that the low bioavailability of 3CLpro inhibitors can be addressed by injection or inhalation administration. Moreover, cyclodextrin delivery systems such as 2-hydroxypropyl-β-cyclodextrin (Glisoni et al., 2012; Jeulin et al., 2009; Nguyen and Van Den Mooter, 2014), randomly methylated β-cyclodextrin (Pathak et al., 2010), and α-cyclodextrin (αCD) are other solutions for the low bioavailability issue.
Another distribution criterion is BBB permeability. The BBB plays a key role in limiting the distribution of foreign molecules, which may be toxic, to the brain (Faria et al., 2012). BBB permeability can be expressed as logBB, which reflects the ability of drug molecules to enter the brain. None of the 103 hits are readily distributed to the brain (logBB > 0.3) and 45 of them poorly crossed the BBB (logBB < −1) (Table S4). However, the blood–barrier permeability surface product (logPS) prediction, which is derived from more direct measurement, suggests that 38 hits can easily enter the CNS, while 18 others are not permeable, such as compounds (16) and (20). A study conducted by Zanin et al. (2020) revealed that SARS-CoV-2 infects the brain. Therefore, those 38 hits potentially inhibit the virus’s growth in the brain. Nevertheless, those hits should be free from mutagenesis and carcinogenesis properties (Gyebi et al., 2021).
The next step after drug distribution is metabolism or biotransformation. Metabolism may occur before or after drug molecules reach target cells. It is an essential process in activating drug molecules and inactivating target cells and the excretion system, respectively (Wilde et al., 2019). Cytochrome P450 or the CYP superfamily are groups of enzymes that highly participate in drug metabolism, with CYP2C, CYP2D, and CYP3A subfamilies as the most active (Tushar et al., 2007). The pkCSM web server provides a series of predictions for drug molecules or secondary metabolites likely to be substrates or inhibitors for such proteins. It predicts that none of the hits is a CYP2D6 substrate and 14 hits are CYP3A4 substrates. These 14 hits are potentially metabolized by CYP3A4, increasing their elimination rates and decreasing their bioavailabilities in the body. However, further metabolism prediction suggests that 10 of them are CYP3A4 inhibitors, along with the other 30 hits. Other cytochromes, CYP1A2 and CYP2C19, are potentially inhibited by 29 and 89 hits, respectively. Meanwhile, CYP2C9 may be inhibited by 90 hits (Table S5). Of all hits, compound (259) may inhibit five cytochromes, including CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. Inhibition of one or more cytochromes can increase the bioavailability of other hits from Erythrina plants.
The last step in pharmacokinetics is excretion, which reduces the concentration of drug molecules at their active sites. Slow total clearance of drug molecules via renal or hepatic excretion assists in the maintenance of therapeutic effects. Total clearance affects the bioavailability and half-life of drug molecules, which determine their dose sizes and regimens. The pkCSM web server provides a total clearance prediction, in log(CLtot), representing a combination of renal and hepatic clearance. Some 10 hits are classified as high total clearance with log(CLtot) more than 0.7. Those hits could be quickly cleared from the blood by the liver. Some 56 hits are intermediate total clearance, with log(CLtot) values between 0.3 and 0.7, whereas some 37 hits are low total clearance, with log(CLtot) less than 0.3 (Table S6). The other excretion parameter is the renal OCT2 substrate. OCT2 has a key role in the distribution and renal elimination of endogenous and drug molecules. Its substrates may have adverse interactions with OCT2 inhibitors. Therefore, predicting all hits as OCT2 substrates is beneficial for their potential contraindication and clearance (Pires et al., 2015). Interestingly, pkCSM only predicts erysubin D (329) as an OCT2 substrate. This prediction indicates the safety profile of the majority of the flavonoids from Erythrina plants in terms of contraindication due to OCT2.
Drug candidates often fail in the late phases of development due to their toxicities. Therefore, a preliminary toxicity study through pkCSM would be beneficial. All 103 hits are not hERG I inhibitors, but 37 of them inhibit hERG II (Table S7). Inhibition of hERG causes heart rhythm abnormality, which may lead to fatal arrhythmia (Raschi et al., 2009). Another toxicity parameter provided by pkCSM is AMES toxicity. The pkCSM web server predicts 96 hits are AMES-toxicity-free, while the other 7 have a positive result prediction (Table S7), suggesting their carcinogenicity potentials. For hepatoxicity prediction, 15 hits potentially damage the liver (Table S7). In total, pkCSM predicts 33 hits free from hepatoxicity and mutagenicity. Additionally, they are non-hERG I and II inhibitors. Two of them, orientanol E (171) and erycaffra F (57), are the fifth and seventh on the top hit list.
Non-bonded interactions analysis
Among 33 flavonoids that are considered hepatoxicity- and mutagenicity-free according to in silico ADMET above, orientanol E (171) and erycaffra F (57) are the fifth and seventh on the top hit list of virtual screening (Tables S1 and S7). Therefore, they are potential 3CLpro inhibitor candidates, and their detailed interaction analysis with the enzyme is interesting to explore.
We performed interaction analysis for all poses of orientanol E (171) and erycaffra F (57) in their best clusters (Tables S8–S10). The results (Tables S8–S11) showed that both flavonoids interact with His41 and Cys145, dyad catalytic residues, which are important to 3CLpro inhibition activity (Konwar and Sarma, 2021; Tahir Ul Qamar et al., 2020). Orientanol E (171) forms a salt bridge with His41 through its C7-OH, which is predicted in a deprotonated state (Fig. 1). Such an interaction presents in all poses docked to the binding site of 3CLpro. Furthermore, it also interacts with His41 via π-π T-shape stacking, where its occurrence is 80.5% in all poses in the best cluster. Meanwhile, erycaffra F (57) interacts with His41 solely through a π-alkyl with an occurrence of 92.1% in all poses (Tables S10 and S11). For Cys145, both flavonoids form a hydrophobic alkyl interaction which emerges in 90.2% and 81.6% among all poses of orientanol E (171) and erycaffra F (57), respectively.
Separate studies conducted by Cannalire et al. (2022) and Zhang et al. (2020) suggest that the interaction with His163 is essential for the inhibition of 3CLpro, as demonstrated on baicalein. Orientanol E (171) and erycaffra F (57) also form interactions with His163 (Figs. 1 and 2). For erycaffra F (57), the interaction with this residue is through an H-bond, which appears in all poses (Tables S10 and S11). In the case of orientanol E (171), His163 makes a hydrophobic π-alkyl with almost all poses of the flavanone (Tables S8 and S9). Additionally, orientanol E (171) forms a hydrophobic alkyl interaction with Met49. It is another vital residue for the inhibition of 3CLpro, situated on the floor of the binding pocket. Xiang et al. (2022) suggested that the interaction with this residue hinders the native substrate from docking at the active site of 3CLpro.
Orientanol E (171) also interacts with Met165 and Pro168 with an occurrence of more than 80% within poses in the best molecular docking hit cluster (Tables S8 and S9). Moreover, it builds interactions with some residues, including His41, Ala191, Arg188, Gln189, Leu167, Cys145, Gln192, Glu166, Met165, Thr190, Cys44, Pro52, His164, His172, and Tyr54. These interactions occur in 80% poses of the best hit cluster. Nonetheless, it has unfavorable interactions. For example, the best pose of orientanol E (171) exhibits such an unfavorable interaction with Glu166. Meanwhile, erycaffra F (57) interacts with Met165, Pro168, Arg188, Gln189, and Gln192, which almost exist in all poses (80% occurrence). Additionally, it interacts with the binding site of 3CLpro without any unfavorable interaction. It also forms another interaction with Leu167, Pro168, Ala191, His172, Glu166, Arg188, His41, Ser144, Tyr54, Cys145, Gln192, Gln189, Gln192, Met49, Asp187, Thr190, Leu141, and Met49. However, the interactions do not occur in the majority of poses, with occurrence in less than 80%. Orientanol E (171) has seven types of nonbonded interactions, including attractive charge, conventional hydrogen bond, alkyl, π-alkyl, π-π T-shaped, π-sigma, and π-sulfur, whereas erycaffra F (57) forms six interaction categories (carbon-hydrogen bond, conventional hydrogen bond, alkyl, amide π-stacked, π-alkyl, and dan π-sigma). Overall, the number of interactions built by erycaffra F (57) is higher than that by orientanol E (171) (Table 2, Table S1). These nonbonded interactions may explain the more negative intermolecular energy of erycaffra F (57) than orientanol E (171), although the best pose of orientanol E (171) has more binding energy than that of erycaffra F (57).
Another binding energy component in molecular docking is torsional energy. Isa et al. (2022) and Kumar et al. (2020) showed that torsional bonds maintain ligand flexibility in the rigid binding site of 3CLpro. According to molecular docking analysis, orientanol E (171) has lower torsional energy than erycaffra F (57) (Table 4), which is in agreement with their binding energy values. The torsional energy values suggest that orientanol E (171) may have a higher potency than erycaffra F (57) in binding to the active site of 3CLpro due to its flexibility.
MD simulations
Molecular docking is widely used in structural-based virtual screening due to its rapid evaluation, which was implemented in this study to screen flavonoids in the genus Erythrina against 3CLpro. However, it suffers several limitations, such as employing a rigid receptor and simplifying a scoring function (Forli et al., 2016). Forli et al. (2016) recommended MD simulation method to refine the top hits obtained from molecular docking; MD also mimics a physiological condition which helps to estimate the strength of binding energy between the ligand and the target protein as an experimental study (Kumar et al., 2020). Therefore, we subjected 3CLpro-bound orientanol E (171) and erycaffra F (57) to MD simulations, yielding a 100 ns trajectory for each complex.
Additionally, we conducted MD simulations on the 3CLpro-bound N3 and GC376 as the references. N3 is a potent SARS-CoV-2 3CLpro inhibitor, which can also inhibit multiple coronaviruses, like MERS-CoV and SARS-CoV (Jin et al., 2020). Meanwhile, GC376 has been widely investigated as a potential inhibitor targeting 3CLpro in Vero cells against FCoV, SARS-CoV, MERS-CoV, and SARS-CoV-2 (Fu et al., 2020; Sharun et al., 2021). It is also employed as a standard inhibitor in a commercial 3CLpro assay kit (BPS Bioscience, 2022). Hence, the utilization of N3 and GC376 as the references assists in predicting the inhibition potential of orientanol E (171) and erycaffra F (57) to 3CLpro.
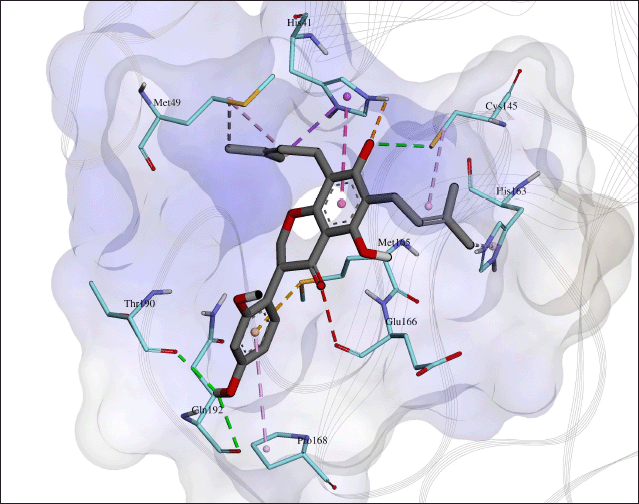 | Figure 1. The visualization of interactions between orientanol E (171) and 3CLpro. The carbon atoms for the residues on the binding site of 3CLpro are in a light-blue color, whereas those for orientanol E (171) are in grey. The orange dash line depicts the interaction of pi anion and attractive charge, the green for hydrogen bond, the purple, and pink for hydrophobic interactions including alkyl, amide-π stacked, π-alkyl, π-π T-shaped, and π-sigma. Meanwhile, the yellow dash line denotes the interaction of π-sulfur, whereas the red for unfavorable interaction. [Click here to view] |
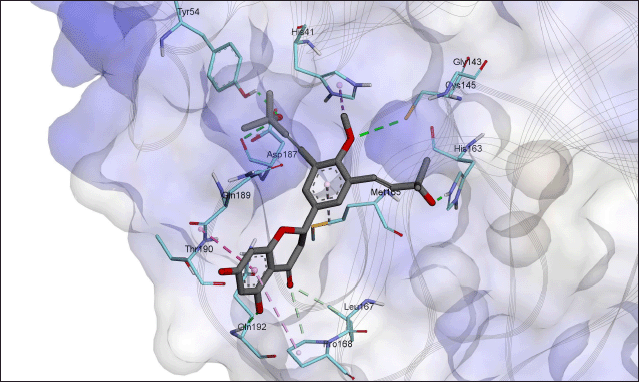 | Figure 2. Visualization of interactions between erycaffra F (57) and 3CLpro. The carbon atoms for the residues on the binding site of 3CLpro are in a light-blue color, whereas those for orientanol E (171) are in grey. The orange dash line depicts the interaction of π-anion, the green for hydrogen bond, the light green for carbon hydrogen interaction, the purple and pink for hydrophobic interactions including alkyl, amide-π stacked, π-alkyl, π-π T-shaped, and π-sigma. Meanwhile, the yellow dash line denotes the interaction of π-sulfur, whereas the red for unfavorable interaction. [Click here to view] |
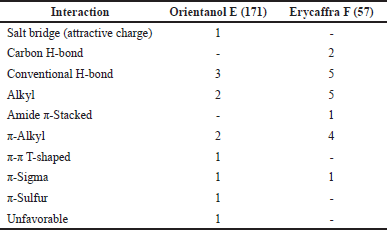 | Table 2. Non-bonded interaction types between the binding site of 3CLpro and two flavonoids, orientanol E (171), and eryvsffra F (57). These flavonoids have good ADMET profiles and are in the top 10 hits. [Click here to view] |
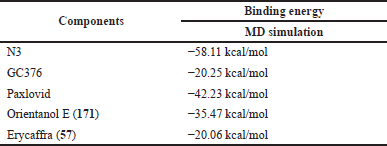 | Table 3. Binding energy values between 3CLpro and ligands obtained from MD simulation. The ligands are orientanol E (171), erycaffra F (57), N3, GC376, and paxlovid. Binding energy values from MD simulations were computed using a MMGBSA method. [Click here to view] |
Binding energy evaluations from MD simulations are tabulated in Table 3. The binding energy values change from molecular docking. These changes may be due to MD simulations accommodating complete flexibility in both ligands and 3CLpro, allowing intermolecular interaction adjustment (Hardianto et al., 2017, 2019) between them (Table S12). Based on MD simulation, N3 and Paxlovid bind 3CLpro stronger than orientanol E (171) and erycaffra F (57). Nevertheless, orientanol E (171) still has a more negative binding energy value (−35.47 kcal/mol) than GC376 (−20.25 kcal/mol), which is comparable to that of erycaffra F (57) (−20.06 kcal/mol). These results suggest that both orientanol E (171) and erycaffra F (57) are potential 3CLpro inhibitors.
CONCLUSION
In this study, we have performed structure-based virtual screening campaigns on 378 flavonoids from the genus Erythrina against SARS-CoV-2 3CLpro. These virtual screening campaigns filtered 108 hits with binding energy scores stronger than the N3 inhibitor. Some 103 of the hits satisfy Lipinski’s Ro5, suggesting their potency as oral drug candidates. Additionally, we conducted an in silico ADMET study using the pkCSM web server on the 108 hits since ADME and safety profiles of molecules are essential in drug discovery and development. The web server suggests that 33 flavonoids are hepatotoxicity- and mutagenicity-free. In addition, they are not hERG I and II inhibitors. Two of them, orientanol E (171) and erycaffra F (57), have binding energy values in the top 10 hits. Binding energy evaluation from MD simulation also supports both orientanol E (171) and erycaffra F (57) as potential 3CLpro inhibitors. Both flavonoids satisfy Ro5 and have good absorption profiles, but their distribution properties are poor according to pkCSM prediction. These issues can be anticipated using delivery systems such as liposome and cyclodextrin. The pkCSM web server predicts both flavonoids as nonsubstrates for CYP2D6 and CYP3A4, suggesting their high bioavailability in the body. According to the web server, they may be cleared from the body through feces. In summary, this study suggests that flavonoids from Erythrina may have the potency to be developed as herbal medicines to treat COVID-19 through inhibition of 3CLpro. Further in vitro and in vivo experiments, however, are required to confirm this notion.
ACKNOWLEDGMENTS
The authors are grateful to the Ministry of Research and Technology/National Research and Innovation Agency for funding through the Basic Research Grants for Higher Education (PDUPT) Scheme (No. 094/PG.02.00PT/2022) and the Universitas Padjadjaran for ALG Tati Herlina (No. 2064/UN6.3.1/PT.00/2022).
CONFLICTS OF INTEREST
All authors declare that they have no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Ahmad B, Batool M, Ain QU, Kim MS, Choi S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int J Mol Sci, 2021; 22:17. CrossRef
Alqahtani MS, Kazi M, Alsenaidy MA, Ahmad MZ. Advances in oral drug delivery. J Front Pharmacol, 2021; 12:618411. CrossRef
Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. J Biochem Biophys Res Commun, 1991; 175(3):880–5. CrossRef
Blanchfield J, Toth I. Lipid, sugar, and liposaccharide based delivery systems 2. Curr Med Chem, 2004; 11:2375–82. CrossRef
BPS Bioscience. 3CL protease, untagged (SARS-CoV-2) assay kit. BPS Bioscience, Inc., 2022. [Online] Available via https://bpsbioscience.com/3cl-protease-untagged-sars-cov-2-assay-kit-78042 (Accessed 23 March 2022).
Cannalire R, Cerchia C, Beccari AR, Di Leva FS, Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J Med Chem, 2022; 65(4):2716–46. CrossRef
ChemAxon. Marvin sketch 20.11.0 software. 2020. [Online] Available via http://www.chemaxon.com (Accessed 11 February 2020).
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model, 2012; 52(11):3099–105. CrossRef
Choudhury A. Liposome: a carrier for effective drug delivery. J App Pharm Res, 2020; 8(1):22–8. CrossRef
Cui J, Li F, Shi ZL. Origin and evolution of pathogenic Coronaviruses. J Nat Rev Microbiol, 2019; 17(3):181–92. CrossRef
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. J Sci Rep, 2017; 7(1):42717. CrossRef
Du Y, Chen B. Combination of drugs and carriers in drug delivery technology and its development. J Drug Des Devel Ther, 2019; 13:1401–8. CrossRef
Fahmy NM, Al-Sayed E, El-Shazly M, Singab AN. Comprehensive review on flavonoids biological activities of Erythrina plant species. J Ind Crops Prod, 2018; 123:500–38. CrossRef
Fahmy NM, Al-Sayed E, Moghannem S, Azam F, El-Shazly M, Singab AN. Breaking down the barriers to a natural antiviral agent: antiviral activity and molecular docking of Erythrina speciosa extract, fractions, and the major compound. J Chem Biodivers, 2020; 17(2):e1900511. CrossRef
Faria A, Mateus N, Calhau C. Flavonoid transport across blood-brain barrier: implication for their direct neuroprotective actions. J Nutr Aging, 2012; 1(2):89–97. CrossRef
Finch A, Pillans P. P-glycoprotein and its role in drug-drug interactions. J Aust Prescr, 2014; 37(4):137–9. CrossRef
Forli S, Huey R, Pique ME, Sanner M, Goodsell DS, Arthur J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. J Nat Protoc, 2016; 11(5):905–19. CrossRef
Fu L, Ye F, Feng Y, Yu F, Wang Q, Wu Y, Zhao C, Sun H, Huang B, Niu P, Song H, Shi Y, Li X, Tan W, Qi J, Gao GF. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun, 2020; 11(1):1–8. CrossRef
Gil C, Ginex T, Maestro I, Nozal V, Barrado-Gil L, Cuesta-Geijo MÁ, Urquiza J, Ramírez D, Alonso C, Campillo NE, Martinez A. COVID-19: drug targets and potential treatments. J Med Chem, 2020; 63(21):12359–86. CrossRef
Glisoni RJ, Cuestas ML, Mathet VL, Oubiña JR, Moglioni AG, Sosnik A. Antiviral activity against the hepatitis C virus (HCV) of 1-indanone thiosemicarbazones and their inclusion complexes with hydroxypropyl-β- cyclodextrin. Eur J Pharm Sci, 2012; 47(3):596–603. CrossRef
Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol, 2000; 295(2):337–56. CrossRef
Gyebi GA, Ogunyemi OM, Ibrahim IM, Ogunro OB, Adegunloye AP, Afolabi SO. SARS-CoV-2 host cell entry: an in silico investigation of potential inhibitory roles of terpenoids. J Genet Eng Biotechnol, 2021; 19(1):113. CrossRef
Hardianto A, Khanna V, Liu F, Ranganathan S. Diverse dynamics features of novel protein kinase C (PKC) isozymes determine the selectivity of a fluorinated balanol analogue for PKC?. BMC Bioinform, 2019; 19(13):342. CrossRef
Hardianto A, Yusuf M, Hidayat IW, Ishmayana S, Soedjanaatmadja UMS. Exploring the potency of nigella sativa seed in inhibiting sars-cov-2 main protease using molecular docking and molecular dynamics simulations. Indones J Chem, 2021; 21(5):1252–62. CrossRef
Hardianto A, Yusuf M, Liu F, Ranganathan S. Exploration of charge states of balanol analogues acting as ATP-competitive inhibitors in kinases. BMC Bioinform, 2017; 18(16):572. CrossRef
Herlina T, Nurlelasari, Kurnia D, Supratman U, Udin Z. In vitro anticancer and antimalarial Erystagallin-A from Erythrina variegata (L.) stem bark. Med Plants Int J Phytomed Relat Ind, 2011; 3(1):9–13. CrossRef
Isa MA, Mustapha A, Qazi S, Raza K, Allamin IA, Ibrahim MM, Mohammed MM. In silico molecular docking and molecular dynamic simulation of potential inhibitors of 3C-like main proteinase (3CLpro) from severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) using selected african medicinal plants. J Adv Trad Med, 2022; 22(1):107–23. CrossRef
Jeulin H, Venard V, Carapito D, Finance C, Kedzierewicz F. Effective ribavirin concentration in mice brain using cyclodextrin as a drug carrier: evaluation in a measles encephalitis model. J Antivir Res, 2009; 81(3):261–6. CrossRef
Ji W, Wang W, Zhao X, Zai J, Li X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J Med Virol, 2020; 92(4):433–40. CrossRef
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. J Nat, 2020; 582(7811):289–93. CrossRef
Kavaliauskas P, Grybaite B, Mickevicius V, Petraitiene R, Grigaleviciute R, Planciuniene R, Gialanella P, Pockevicius A, Petraitis V. Synthesis, ADMET properties, and in vitro antimicrobial and antibiofilm activity of 5-nitro-2-thiophenecarbaldehyde N-((E)-(5-nitrothienyl)methylidene)hydrazone (KTU-286) against staphylococcus aureus with defined resistance mechanisms. J Antibiot, 2020; 9(9):1–17. CrossRef
Kiemer L, Lund O, Brunak S, Blom N. Coronavirus 3CLpro proteinase cleavage sites: possible relevance to SARS virus pathology. BMC Bioinform, 2004; 5:1–9. CrossRef
Kissler S, Tedijanto C, Goldstein E, Grad Y, Lipsitch M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. J Sci, 2020; 368(6493):860–8. CrossRef
Konwar M, Sarma D. Advances in developing small molecule SARS 3CLpro inhibitors as potential remedy for corona virus infection. J Tetrahedron, 2021; 77:131761. CrossRef
Kumar S, Sharma PP, Shankar U, Kumar D, Joshi SK, Pena L, Durvasula R, Kumar A, Kempaiah P, Poonam, Rathi B. Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: molecular docking, molecular dynamics simulation, and structure-activity relationship studies. J Chem Inf Model, 2020; 60(12):5754–70. CrossRef
Lee JS, Won KO, Jong SA, Yong HK, Mbafor JT, Wandji J, Fomum ZT. Prenylisoflavonoids from Erythrina senegalensis as novel HIV-1 protease inhibitors. J Planta Med, 2009; 75(3):268–70. CrossRef
Lee J, Worrall LJ, Vuckovic M, Rosell FI, Gentile F, Ton AT, Caveney NA, Ban F, Cherkasov A, Paetzel M, Strynadka, NCJ. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. J Nat Commun, 2020; 11(1):5877. CrossRef
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. J Adv Drug Deliv Rev, 2012; 64:4–17. CrossRef
Liu AL, Wang HD, Lee SM, Wang YT, Du GH. Structure–activity relationship of flavonoids as influenza virus neuraminidase inhibitors and their in vitro anti-viral activities. J Bioorg Med Chem, 2008; 16(15):7141–7. CrossRef
Liu H, Ye F, Sun Q, Liang H, Li C, Li S, Lu R, Huang B, Tan W, Lai L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J Enzyme Inhib Med Chem, 2021; 36(1):497–503. CrossRef
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput, 2015; 11(8):3696–713. CrossRef
Mody V, Ho J, Wills S, Mawri A, Lawson L, Ebert MCCJC, Fortin GM., Rayalam S. Taval S. Identification of 3-chymotrypsin like protease (3CLpro) inhibitors as potential anti-SARS-CoV-2 agents. J Commun Biol, 2021; 4(1):93. CrossRef
Morse JS, Lalonde T, Xu S, Liu WR. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. J Chem Bio Chem, 2020; 21(5):730–8. CrossRef
Nguyen DN, Van den Mooter G. The fate of ritonavir in the presence of darunavir. Int J Pharm, 2014; 475(1):214–26. CrossRef
Pathak SM, Musmade P, Dengle S, Karthik A, Bhat K, Udupa N. Enhanced oral absorption of saquinavir with Methyl-Beta-Cyclodextrin-preparation in vitro and in vivo evaluation. Eur J Pharm Sci, 2010; 41(3–4):440–51. CrossRef
Petrou A, Zagaliotis P, Theodoroula NF, Mystridis GA, Vizirianakis IS, Walsh TJ, Geronikaki A. Thiazole/thiadiazole/benzothiazole based thiazolidin-4-one derivatives as potential inhibitors of main protease of SARS-CoV-2. J Molecules, 2022; 27(7):2180. CrossRef
Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem, 2015; 58(9):4066–72. CrossRef
Prescott L. SARS-CoV-2 3CLpro whole human proteome cleavage prediction and enrichment/depletion analysis. Comput Biol Chem, 2022; 98:107671. CrossRef
Qamar MT, Ashfaq UA, Tusleem K, Mumtaz A, Tariq Q, Goheer A, Ahmed B. In silico identification and evaluation of plant flavonoids as dengue NS2B/NS3 protease inhibitors using molecular docking and simulation approach. Pak J Pharm Sci, 2017; 30(6):2119–37.
Raj R. Analysis of non-structural proteins, NSPs of SARS-CoV-2 as targets for computational drug designing. J Biochem Biophys Rep, 2021; 25:100847. CrossRef
Ramírez D, Caballero J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data. J Molecules, 2018; 23(5):1–17. CrossRef
Raschi E, Ceccarini L, De Ponti F, Recanatini M. HERG-related drug toxicity and models for predicting hERG liability and QT prolongation. J Expert Opin Drug Metab Toxicol, 2009; 5(9):1005–21. CrossRef
Saur IML, Panstruga R, Schulze-Lefert P. NOD-like receptor-mediated plant immunity: from structure to cell death. Nat Rev Immunol, 2021; 21(5):305–18. CrossRef
Sharun K, Tiwari R, Dhama K. Protease inhibitor GC376 for COVID-19: lessons learned from feline infectious peritonitis. Ann Med Surg, 2021; 61:122–5. CrossRef
Su H, Yao S, Zhao W, Zhang Y, Liu J, Shao Q, Wang Q, Li M, Xie H, Shang W, Ke C, Feng L, Jiang X, Shen J, Xiao G, Jiang H, Zhang L, Ye Y, Xu Y. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. J Nat Commun, 2021; 12(1):1–12. CrossRef
Tahir Ul Qamar M, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal, 2020; 10(4):313–9. CrossRef
Tushar T, Vinod T, Rajan S, Shashindran C, Adithan C. Effect of honey on CYP3A4, CYP2D6 and CYP2C19 enzyme activity in healthy human volunteers. J Basic Clin Pharmacol Toxicol, 2007; 100(4):269–72. CrossRef
Twardowski Z, Ksiazek A. Pharmacokinetics in renal failure. J Pol Arch Med Wewn, 1983; 65(6):491–500.
Uekama K. Cyclodextrins in drug delivery system. J Adv Drug Deliv Rev, 1999; 36(1):1–2. CrossRef
Vandyck K, Deval J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. J Virol, 2020; 49:36–40. CrossRef
Vyas A, Kumar AS, Gidwani B. Carrier-based drug delivery system for treatment of acne. Sci World J, 2014; 2014(276260):1–14. CrossRef
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem, 2004; 25(9):1157–74. CrossRef
Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen T, Miller E, Bache S, Müller K, Ooms J, Robinson D, Seidel D, Spinu, V, Yutani H. Welcome to the Tidyverse. J Open Source Softw, 2019; 4(43):1686. CrossRef
Wilde M, Pichini S, Pacifici R, Tagliabracci A, Busardò FP, Auwärter V, Solimini R. Metabolic pathways and potencies of new fentanyl analogs. J Front in Pharmacol, 2019; 10:238. CrossRef
Worldometers. Coronavirus cases. 2022. [Online] Available via https://www.worldometers.info/coronavirus/ (Accessed 14 July 2022).
Xiang R, Yu Z, Wang Y, Wang L, Huo S, Li Y, Liang R, Hao Q, Ying T, Gao Y, Yu F, Jiang S. Recent advances in developing small-molecule inhibitors against SARS-CoV-2. J Acta Pharm Sin B, 2022; 12(4):1591–623. CrossRef
Yamamoto KZ, Yasuo N, Sekijima M. Screening for inhibitors of main protease in SARS-CoV-2: in silico and in vitro approach avoiding peptidyl secondary amides. J Chem Inf Model, 2022; 62(2):350–8. CrossRef
Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Zhao Q, Zhou Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol, 2005; 3(10):e324. CrossRef
Zanin L, Saraceno G Panciani PP, Renisi G, Signorini L, Migliorati K, Fontanella MM. SARS-CoV-2 can induce brain and spine demyelinating lesions. J Acta Neurochir, 2020; 162(7):1491–4. CrossRef
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors, (PDB ID 6Y2F). J Sci, 2020; 368(6489):409–12. CrossRef
Zhivkova Z, Mandova T, Doytchinova I. Quantitative structure – pharmacokinetics relationships analysis of basic drugs: volume of distribution. J Pharm Pharm Sci, 2015; 18(3):515–27. CrossRef
Zhu J, Zhang H, Lin Q, Lyu J, Lu L, Chen H, Zhang X, Zhang Y, Chen K. Progress on SARS-CoV-2 3CLpro inhibitors: inspiration from SARS-CoV 3CLpro peptidomimetics and small-molecule anti-inflammatory compounds. J Drug Des Devel Ther, 2022; 16:1067–82. CrossRef
SUPPLEMENTARY MATERIAL
Supplementary data can be downloaded from the journal’s website