
Complete plastid genome sequence of Primula sinensis (Primulaceae): structure comparison, sequence variation and evidence for accD transfer to nucleus
- Published
- Accepted
- Received
- Academic Editor
- Vsevolod Makeev
- Subject Areas
- Genomics, Plant Science
- Keywords
- Primula sinensis, Chloroplast genome, Illumina sequencing, Gene transfer
- Copyright
- © 2016 Liu et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Complete plastid genome sequence of Primula sinensis (Primulaceae): structure comparison, sequence variation and evidence for accD transfer to nucleus. PeerJ 4:e2101 https://doi.org/10.7717/peerj.2101
Abstract
Species-rich genus Primula L. is a typical plant group with which to understand genetic variance between species in different levels of relationships. Chloroplast genome sequences are used to be the information resource for quantifying this difference and reconstructing evolutionary history. In this study, we reported the complete chloroplast genome sequence of Primula sinensis and compared it with other related species. This genome of chloroplast showed a typical circular quadripartite structure with 150,859 bp in sequence length consisting of 37.2% GC base. Two inverted repeated regions (25,535 bp) were separated by a large single-copy region (82,064 bp) and a small single-copy region (17,725 bp). The genome consists of 112 genes, including 78 protein-coding genes, 30 tRNA genes and four rRNA genes. Among them, seven coding genes, seven tRNA genes and four rRNA genes have two copies due to their locations in the IR regions. The accD and infA genes lacking intact open reading frames (ORF) were identified as pseudogenes. SSR and sequence variation analyses were also performed on the plastome of Primula sinensis, comparing with another available plastome of P. poissonii. The four most variable regions, rpl36–rps8, rps16–trnQ, trnH–psbA and ndhC–trnV, were identified. Phylogenetic relationship estimates using three sub-datasets extracted from a matrix of 57 protein-coding gene sequences showed the identical result that was consistent with previous studies. A transcript found from P. sinensis transcriptome showed a high similarity to plastid accD functional region and was identified as a putative plastid transit peptide at the N-terminal region. The result strongly suggested that plastid accD has been functionally transferred to the nucleus in P. sinensis.
Introduction
Chloroplast is one of the most important organelles in green plant cells, and plays a central role in plant photosynthesis. Sequence data from chloroplast genomes (or plastomes) has been widely used in phylogenetic studies, because of its recombination-free and maternal inheritance (Graham & Olmstead, 2000). More importantly, they are structurally highly conserved, which facilitates PCR primer design and sequencing (Shaw et al., 2005; Shaw et al., 2014). However, obtaining accurate phylogenies using a small standardized set of chloroplast genes is challenging in some rapidly evolving plant groups, such as Gaertnera (Malcomber, 2002). Recently, chloroplast phylogenomics has been successfully used in several plant groups since the advent of 454 and/or Illumina technologies (Moore et al., 2006; Cronn et al., 2008; Ma et al., 2014; Ruhfel et al., 2014; Parks, Cronn & Liston, 2009), which offer an increasingly easy-to-access source of characters to resolve ambiguous phylogenetic relationships in some rapidly evolved plant groups (Folk, Mandel & Freudenstein, 2015; Wysocki et al., 2015).
The structure or functional change for the chloroplast genome is interesting as well. The chloroplast is considered to be a descendant of cyanobacterium-like progenitors (Raven & Allen, 2003). Since its endosymbiotic origin, the size of the chloroplast genome has been greatly reduced (Timmis et al., 2004). This shrunken genome is the consequence of the loss or transfer of genes to the nucleus (Martin et al., 2002). Loss of genes has been found in many lineages of angiosperms (Blazier, Guisinger & Jansen, 2011; Li et al., 2013). Meanwhile, only four genes transferred to nucleus have been confirmed by several studies, including infA, accD, rpl22 and rpl32 (Millen et al., 2001; Ueda et al., 2007; Magee et al., 2010; Jansen et al., 2011; Park, Jansen & Park, 2015). To better understand this transfer, it is necessary to explore more data from both the plastid and nucleus at a wide range of angiosperms.
The genus Primula L. is one of the largest genera in the family Primulaceae, and it was characterized by a rapid speciation at East Himalaya-Hengduan Mts. Region (Hu, 1994; Richards, 2003; Yan et al., 2015). Understanding the chloroplast genome of this genus will benefit us in constructing a solid phylogeny of the genus in the future. However, the complete chloroplast sequences of species in this genus still have been poorly understood except for a recently released chloroplast genome of P. poissonii Franch. without additional analyses (Yang, Li & Li, 2014).
In this study, we released a complete chloroplast genome of an endemic Primula species in China, P, sinensis Sabine ex Lindley, by using high-throughput sequencing technology. To start with this plastome sequence, firstly, we characterized gene content, sequence variation, and compared with other related species, which will facilitate further phylogenetic studies of the genus. Secondly, we verified the accD gene lacking intact open reading frames (ORF) from P. sinensis plastid and search clue in its transcriptome to get lines of evidences for functional transfer to nucleus.
Materials and Methods
Library preparation and Illumina sequencing
Fresh leaves of P. sinensis were collected from the South China Botanical Garden, Chinese Academy of Sciences (CAS). Modified CTAB method was used to isolate whole-genome DNA (Porebski, Bailey & Baum, 1997). RNAs in the initial extracts was digested by RNase A to acquire pure genomic DNA. Eight primer pairs involved in this study were designed according to Yang, Li & Li (2014), to amplify the whole chloroplast genome sequence. Primers were designed to cover inverted repeat region only once. A region of approximately 16 kb was amplified by each primer pair. Long-range PCR was performed with 25 μl reaction system by using Primestar GXL DNA polymerase (TaKaRa Bio., Dalian, China). Reactions were initially denatured for 1 min at 95 °C, followed by 35 cycles of 10 sec at 94 °C, 1 min at 60 °C, and 15 min at 68 °C. Eventually the additional extension step performed on 5 min at 68 °C.
PCR products were mixed to build pair-end library using Nextera XT DNA Library Prep Kit (Illumina Inc., San Diego, CA, USA). PCR products mixture was fragmented into ∼300 bp size by the Nextera XT transposome. Library Sequencing acquired 2× 250 bp paired reads using Illumina Miseq Desktop Sequencer at Kunming Institute of Botany, CAS. All reads data were deposited to NCBI SRA database with an accession number SRP068226.
Plastome assembly and annotation
Reads were assembled using CLC Genomics Workbench v7.5.1 (CLC Bio., Aarhus, Denmark) after removing adaptors and trimmed low quality reads. Assembly was conducted twice separately with two different k-mer value 60 and 64. Contigs generated by assembling were subjected to BLAST searches against the complete chloroplast sequence of P. poissonii (NC_024543). Then, relative position and direction of each hitting contig were determined. Subsequently, hitting contigs were assembled manually to acquire complete chloroplast sequence in Geneious R7 (Biomatters, Auckland, New Zealand). The resulting plastome sequence was used as the reference, which was subsequently verified by remapping initial reads. Regions of four SC-IR junctions were identified by Sanger sequencing using four pair primers. Primer sequences used in this study can be found (Table S1).
Plastome annotation was performed using DOGMA (http://dogma.ccbb.utexas.edu/) (Wyman, Jansen & Boore, 2004) and compared with other Primulaceae species in alignment matrix. Annotations of each gene was adjusted to appropriate start and stop codons in accordance with the genetic codon for plant plastid. Incomplete genes identified from P. sinensis were verified by Sanger sequencing. The names of a few genes were updated according to the latest study, including ycf3 to pafI and ycf4 to pafII (Wicke et al., 2011). The annotated chloroplast genome sequence was submitted to GenBank (accession number: KU321892). Finally, a circular genome map was illustrated using OGDRAW (http://ogdraw.mpimp-golm.mpg.de/) (Lohse et al., 2013).
Sequence variance and SSR analysis
To further identify highly variable regions in chloroplast genome of Primula, sequences of P. sinensis and P. poissonii were compared according to variability (Pi). Alignment was performed with MAFFT version 7 (Katoh & Standley, 2013). The genetic diversity (Pi) were calculated in each split regions (400 bp) of alignment using DnaSP version 5 (Librado & Rozas, 2009). The adjacent split regions were overlapped each other with 300 bp. Gaps in the alignment were excluded from analysis. With the variance of recommended barcode rbcL + matK + ITS as reference (Yan et al., 2015), the top 40 regions with most variable sites, whose aligned lengths are longer than 200 bp were extracted for the next informative character (PICs) analysis. We followed the method of Shaw et al. (2005) to count manually the numbers of nucleotide substitutions and indels for each regions and plot them in Fig. 3.
To detect and locate simple sequence repeats (SSRs), GMATo v1.2 (Wang, Lu & Luo, 2013) was used to screen the chloroplast sequence of two Primula species. The parameter settings of mononucleotide and dinucleotide to hexanucleotide were at least eight repeat units and four repeat units, respectively.
RNAseq and accD gene characterizing
Total RNA was extracted using a modified CTAB method. Quantified total RNA (concentration ≥ 100 ng/μL; rRNA ratio ≥ 1.5) was delivered to Majorbio (Shanghai, China), where cDNA sequencing was performed with Illumina Hiseq4000 platform. Raw data were filtered and deposited in the Sequence Reads Archive (SRA) database under accession number SRX1665905. The cleaned reads were assembled de novo using Trinity with the default parameters to obtain 48,887 unisequences. Eight different software, namely TargetP (Emanuelsson et al., 2000), Protein Prowler (Hawkins & Bodén, 2006), BacelLo (Pierleoni et al., 2006), CELLO2GO (Yu et al., 2014), Euk-mPLoc2 (Chou & Shen, 2010), EuLoc (Chang et al., 2013), HybridGO-Loc (Wan, Mak & Kung, 2014), and Predotar (Small et al., 2004), were used to identify the subcellular location signals from N-terminal sequence of putative nuclear-encoded protein.
Phylogenetic analysis
Plastome of P. sinensis together with eight other plastomes published previously from different genera in Ericales was involved in this phylogenetic analysis, and Agrostemma githago from Caryophyllales was used as outgroup. All plastomes used in this study are available in GenBank (Table S2). A total of 57 plastid protein-coding genes were concatenated to generate three data sets according to the different strategies, such as all CDS, codon1+2 and condon3, respectively. On the other hand, the fourth datasets were generated including 30 pt-accD genes from six families in Ericales and putative n-accD gene in P. sinensis. All sequences from four datasets were aligned using the default option implemented in MAFFT version 7 (Katoh & Standley, 2013). Maximum likelihood (ML) trees was constructed with RAxML (RAxML-VI-HPC, http://www.trex.uqam.ca/) using GTR + Γ nucleotide substitution model (Stamatakis, 2006). Branch supports were assessed with 500 bp replicates.
Results and Discussion
Chloroplast genome assembly
The P. sinensis chloroplast genome was sequenced using Illumina Miseq sequencer, producing total number of 2× 250 bp pair-end reads 2,640,572. The mean coverage depth is about 3838.8×, ranging from 40× minimum to 25814× maximum. Two assemblies with different k-mer values successfully generated identically complete sequence with no gaps. Six contigs from assembly with k-mer value 60 were matched to reference plastome sequence, which was used to determine relative position and direction respectively. A new draft chloroplast genome was generated by identifying overlap regions manually. The draft genome was then checked and corrected according to quality and coverage of each base position by reads remapping (Fig. S1). The annotated genome was deposited into GenBank under the accession number KU321892.
Features of the P. sinensis chloroplast genome
The chloroplast genome of P. sinensis has a total length of 150,859 bp. It is divided into three parts: 82,064 bp of large single-copy (LSC) region, 17,725 bp of small single-copy (SSC) region, and two inverted repeat (IR) regions with 25,535 bp of one copy in length. The nucleotide composition of this genome has a GC content of 37.2%. Comparative analysis revealed that the genome structure of P. sinensis shared a high similarity structure to other Primulaceae species (P. poissonii NC_024543, Lysimachia coreana NC_026197, Ardisia polysticta KC465962) (Ku, Hu & Kuo, 2013; Son & Park, 2014) (Table 1).
| Primula sinensis (KU321892) | Primula poissonii (NC_024543) | Lysimachia coreana (NC_026197) | Ardisia polystica (KC465962) | |
|---|---|---|---|---|
| Plastome size | ||||
| Total size | 150,859 | 151,664 | 155,386 | 156,506 |
| LSC size | 82,064 | 83,444 | 85,229 | 86,078 |
| SSC size | 17,725 | 17,822 | 17,951 | 18,328 |
| IR size | 25,535 | 25,199 | 26,103 | 26,050 |
| Base content (%) | ||||
| Total A content | 31.1 | 31.1 | 31.1 | 31.2 |
| Total T content | 31.7 | 31.8 | 31.8 | 31.8 |
| Total C content | 19 | 18.8 | 18.9 | 18.9 |
| Total G content | 18.2 | 18.2 | 18.2 | 18.2 |
| LSC GC content | 35.2 | 34.9 | 35 | 34.9 |
| SSC GC content | 30.5 | 30.1 | 30.5 | 30.2 |
| IR GC content | 42.8 | 42.9 | 42.9 | 43 |
| Total GC content | 37.2 | 37 | 37.1 | 37.1 |
| Number of genes | ||||
| Total | 112 | 113 | 113 | 114 |
| Protein encoding | 78 | 79 | 79 | 80 |
| tRNA | 30 | 30 | 30 | 30 |
| rRNA | 4 | 4 | 4 | 4 |
A total of 112 genes were detected in this chloroplast genome, which could be classified into 78 protein-coding genes, 30 tRNA genes and four rRNA genes. Among them, seven coding genes, seven tRNA genes and four rRNA genes have two copies due to their locations in the IR regions. As we expected, split genes also exist in this plastome. Among them, 15 genes contain an intron and two genes have two introns (Table 2; Fig. 1).
| Category | Class | Gene |
|---|---|---|
| Genetic apparatus | DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1*, rpoC2 |
| Maturase | matK | |
| Large ribosomal subunits | rpl2*(x2), rpl14, rpl16*, rpl20, rpl22, rpl23(x2), rpl32, rpl33, rpl36 | |
| Small ribosomal subunits | rps2, rps3, rps4, rps7(x2), rps8, rps11, rps12, rps14, rps15, rps16*, rps18, rps19 | |
| Protease | clpP** | |
| Ribosomal RNAs | rrn4.5(x2), rrn5(x2), rrn16(x2), rrn23(x2) | |
| Transfer RNAs | trnH-GUG, trnK-UUU*, trnQ-UUG, trnS-GCU, trnG-UCC*, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-GCC, trnfM-CAU, trnS-GGA, trnT-UGU, trnL-UAA*, trnF-GAA, trnV-UAC*, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU(x2), trnL-CAA(x2), trnV-GAC(x2), trnI-GAU*(x2), trnA-UGC*(x2), trnR-ACG(x2), trnN-GUU(x2), trnL-UAG | |
| Light-dependent photosynthesis | Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| NAD(P)H dehydrogenase complex | ndhA*, ndhB*(x2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| F-type ATP synthase | atpA, atpB, atpE, atpF*, atpH, atpI | |
| PS I assembly factor | pafI**, pafII | |
| Cytochrome b6/f complex | petA, petB*, petD*, petG, petL, petN | |
| Light-independent photosynthesis | Inner membrane protein | cemA |
| Cytochrome c biogenesis protein | ccsA | |
| Large subunit of Rubisco | rbcL | |
| Other | Function unknown | ycf1, ycf2(x2), ycf15(x2) |
| Pseudogene | Subunit of acetyl-CoA-carboxylase | accD |
| Translation initiation factor | infA |
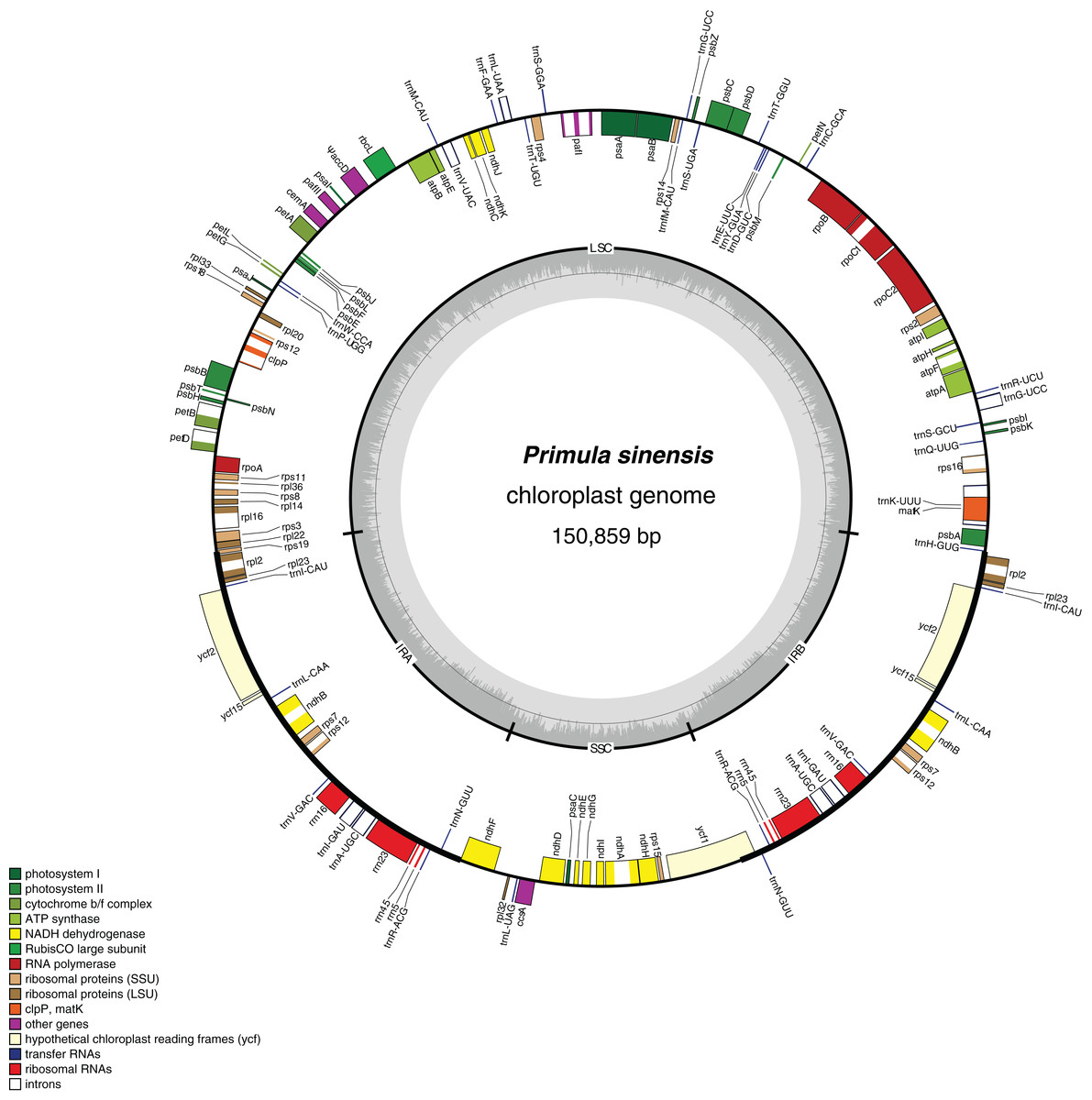
Figure 1: Chloroplast genome map of Primula sinensis (GenBank accession number KU321892).
Genes on the outside of the outer circle indicate the clockwise direction of transcription; those on the inside indicate the counterclockwise direction. The bar graphs on the inner circle reveal GC content in dark grey with the 50% threshold line.{kind=link}
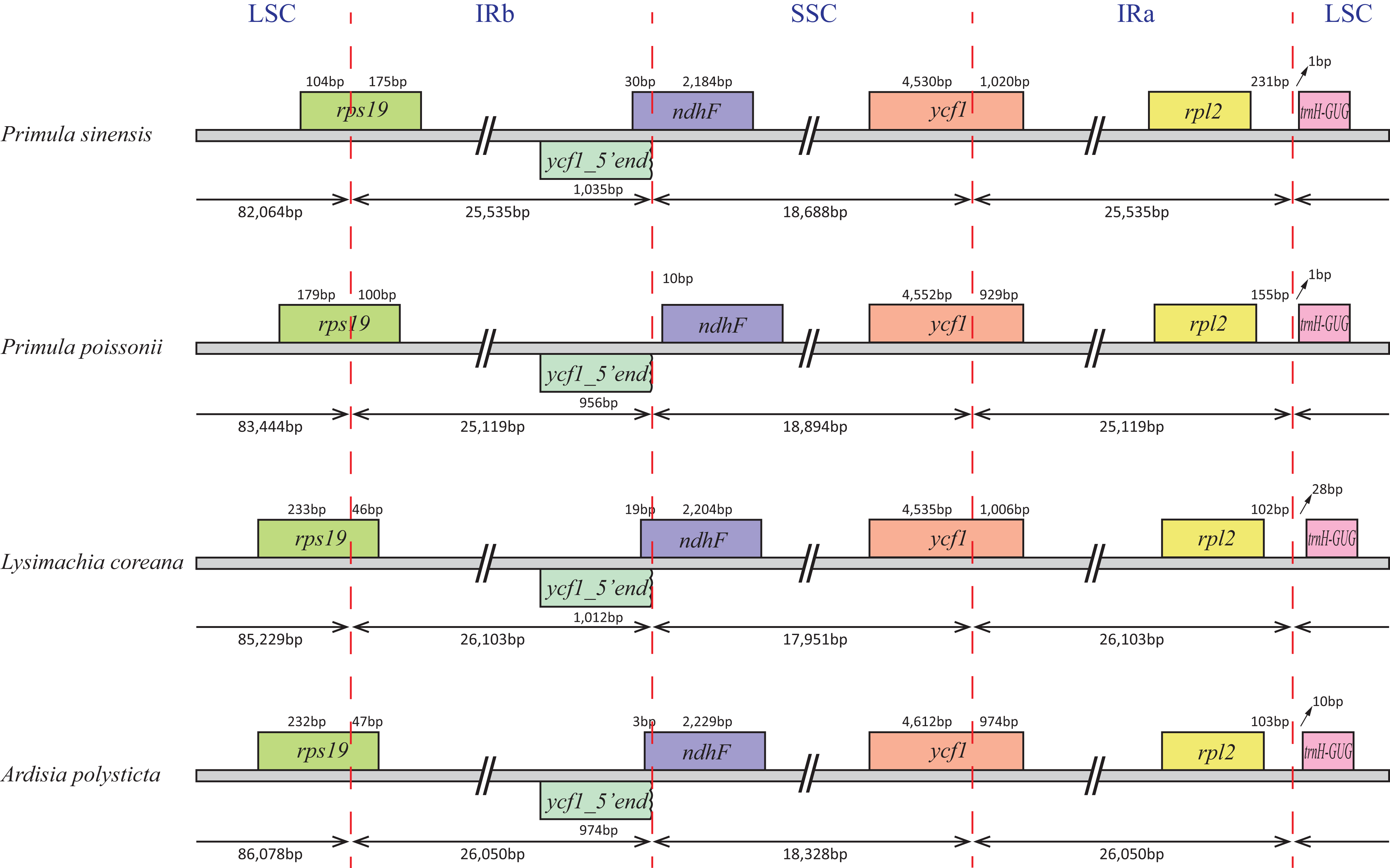
In particular, the rps12 gene is interrupted into three pieces which resulted in trans-splicing because of its first exon located in LSC, while its second and third exons located in IRs (Hildebrand et al., 1988). Notably, the phenomena that two coding regions sharing overlapped sequence with different reading frames was found in psbD and psbC, atpE and atpB, and rps3 and rpl22. In addition, three genes, rps19, ndhF and ycf1, cross the LSC-IRb, IRb-SSC, SSC-IRa boundary, respectively. Furthermore, the ndhF 3′-terminal sequence shares the region in the IRb with the rest of ycf1 5′-terminal sequence, while the IRb-SSC boundary of P. poissonii was separated from the start codon of ndhF with 10 bp length (Fig. 2). Significantly, the accD in P. sinensis were identified as pseudogene with an extremely reduced ORF. Meanwhile, the infA lacked intact ORF in P. sinensis, P. poissonii and L. coreana, which strongly indicated that pseudogenization remains occurred in certain angiosperm groups. Further studies are necessary to focus on the mechanism of occurrence of these pseudogenes and applications of orthologous genes for phylogenetic analysis.
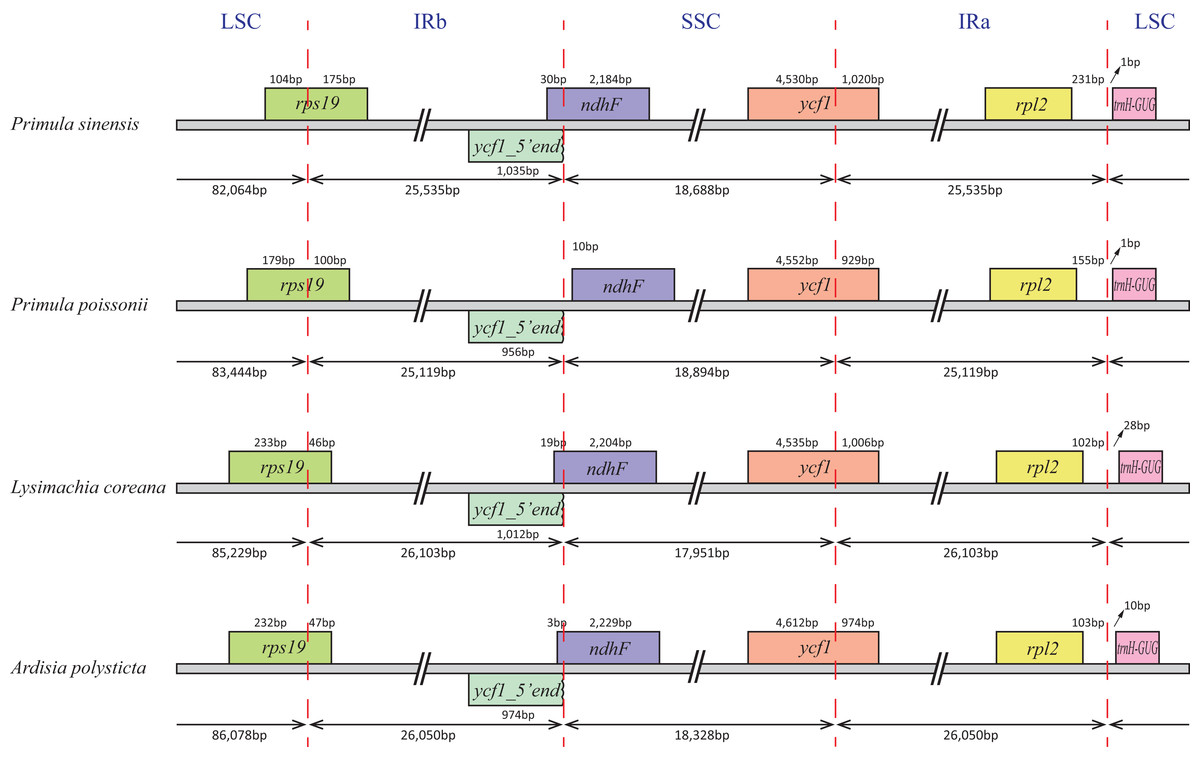
Figure 2: Comparison of the SC-IR boundary of four plastomes from Primulaceae.
{kind=link}
SSR analysis of Primula sinensis
Perfect SSRs were screened in P. sinensis and P. poissonii conducted by GMATo v1.2. Mono-, di- and tri-nucleotide repeats were found in both species (Table 3). The total number of SSRs in P. sinensis chloroplast genome is 193, of which 148 homopolymers, 44 dipolymers and 1 tripolymers are respectively found in this genome. Among them, 85.49% SSRs are only composed of A/T bases. Similar quantity level and base proportion of SSRs were also found in P. poissonii (Tables S3–S5).
| Taxon | Length (bp) | GC% | Number of SSRs | |||
|---|---|---|---|---|---|---|
| Homo (> 8 units) | Di (> 4 units) | Tri (> 4 units) | Total | |||
| Primula sinensis | 150,859 | 37.20% | 148 | 44 | 1 | 193 |
| Primula poissonii | 151,664 | 37% | 129 | 46 | 2 | 177 |
Sequence variation in two Primula plastomes
Although the chloroplast genome of P. sinensis has a similar genome structure in gene contents and orders in comparison with P. poissonii, there are considerable differences in noncoding regions, especially in intergenic sequence (IGS) regions. Highly divergence regions are potential molecular genetic markers for population genetics studies. We therefore compared the regional divergence of chloroplast genome sequences of these two Primula species. The Pi value generated by DnaSP version 5 was used to indicate the level of divergence between P. sinensis and P. poissonii (Fig. S2). Then, the top 40 regions with most variable sites, with aligned length longer than 200 bp, were extracted from the alignment for further analysis (PIC calculation).
The result shows the genetic diversity (Pi) varied from 0–0.47 (Table S6). Most of the variation occurs in the non-coding regions of the LSC and SSC, while less variable characters were found in IRs. The four most variable loci, namely rpl36–rps8, rps16–trnQ, trnH–psbA and ndhC–trnV, were identified. All of these loci have been reported previously (Shaw et al., 2007). Remarkably, rpl36–rps8 exhibits the improved degree of variation. A total of 41 regions had been extracted to calculate their PICs (Fig. 3). Comparing the result with sequence variance, PICs are considered to be affected by sequence length apparently. For example, the trnH-psbA spacer has the lower number of PICs as its aligned length is only 213 bp. In contrast, ycf1 have the high PICs with the sequence length coming up to 2,500 bp in total. However, difficulties in primer designing and PCR amplification limit them (such as ycf1) for further phylogenetic use. We therefore recommend rpl32–trnL, trnS–trnG, pafII–cemA, trnC–petN, trnT–trnL, trnK–rps16 as efficient chloroplast markers, considering their balanced PICs and length size, although additional researches of their utilities are necessary in future.
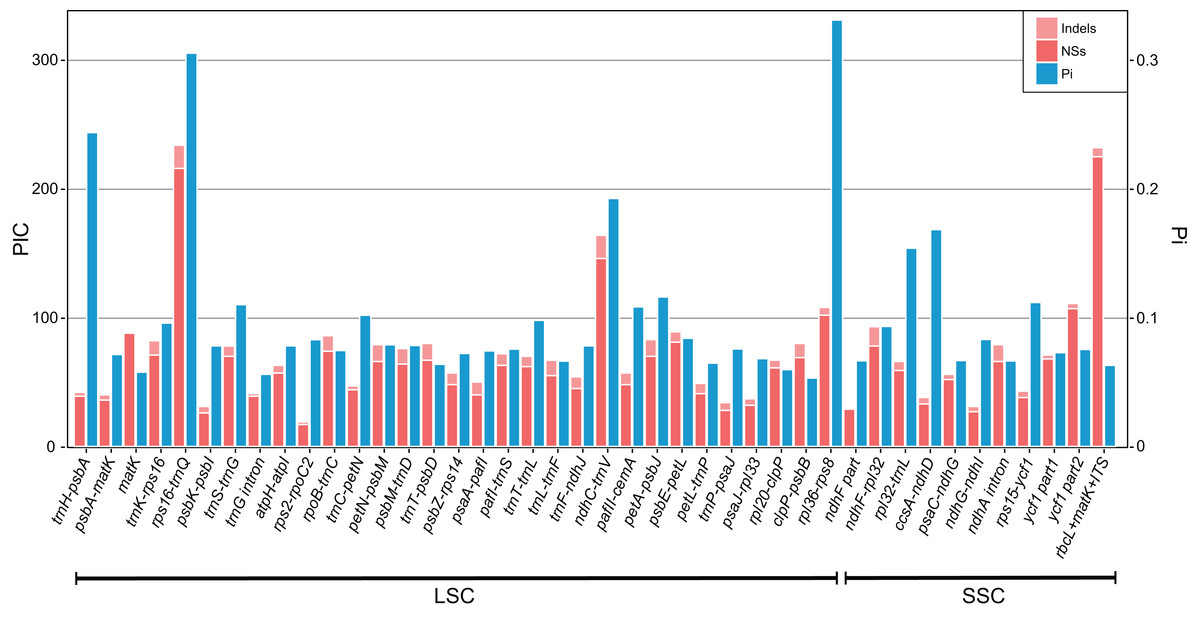
Figure 3: Bar plot that compares potential marker regions with PICs and genetic diversity (Pi).
PICs were determined as the sum of nucleotide substitutions and indels. Barcode rbcL + matK + ITS was used here as a reference as proposed previously for the barcoding analysis of genus Primula (Yan et al., 2015).{kind=link}
Pseudogenization of pt-accD and evidence for transfer to nucleus
AccD gene encodes β subunit of the Acetyl-CoA Carboxylase, which is unique related to fatty acid synthesis within the chloroplast. Gene knockout experiments in tobacco indicated that accD gene function is indispensable, suggesting it should be an essential gene (Kode et al., 2005). So far, several researches have reported that lack of a plastid accD (pt-accD) gene or pseudogenization of pt-accD are widely present in multiple distant lineages, including Acoraceae, Campanulaceae, Ericaceae, Fabaceae, Geraniaceae and Poaceae (Goremykin et al., 2005; Guisinger et al., 2008; Haberle et al., 2008; Magee et al., 2010; Fajardo et al., 2013; Harris et al., 2013; Martínez-Alberola et al., 2013), which implied the lack of pt-accD gene or events of pseudogenization occur independently. Owing to its fundamental function in plastid development, there should be some equivalent genes replaced functionally in other subcellular structures. In recent years, a few case of nuclear encoded accD genes (n-accD) originated from plastid have been found in different taxa, such as Sciadopitys verticillata (Sciadopityaceae) (Li et al., 2016), Trifolium repens (Fabaceae) (Magee et al., 2010), Trachelium caeruleum (Campanulaceae) (Rousseau-Gueutin et al., 2013). These n-accD genes share a similar 3′-terminal region with pt-accD genes which corresponds to the carboxylase domain, while the 5′-terminal regions are completely different from pt-accD. Products of these n-accD have a putative transit peptide at the 5′-terminus. The functional prediction suggests that the transit peptide guides the protein product relocation to chloroplast. Fluorescence microscopy showed that n-ACCD-GFP fusion protein was imported in plastids contained in tobacco guard cells (Rousseau-Gueutin et al., 2013), which provided strong evidence that nuclear encoded accD gene still returns to plastid to play its roles as the same as other subunits of nuclear-encoded plastid ACCase do.
In this study, plastid accD locus in Primula sinensis shows the truncated gene with incomplete ORF. We found a portion of pt-accD gene near the start position was absent with about 400 bp in length by comparing with other plastomes from Primulaceae. This deletion has been verified by PCR amplification using a pair of primers located on both flanks. Reads mapping also showed high coverage levels on flanking regions of the deletion. Due to the presence of the deletion, pt-accD coding sequence was terminated prematurely with the introduction of the stop codon. The remaining ORF with residual sequences does not include the conserved functional region. Actually, pt-accD has become a pseudogene, with a deletion involved (Figs. S3 and S4).
Owing to its important function in plastid development, accD should probably be transferred into other subcellular structures, and still retained its catalytic activity (Rousseau-Gueutin et al., 2013). We therefore checked the transcriptome of P. sinensis using P. poissonii pt-accD as reference. The result of blast searching showed only one transcript, which was highly identical to the plastid accD functional region of P. poissonii. This transcript contained an intact ORF encoding a protein with 362 amino acids. In comparison with pt-accD sequences of other related species (such as Androsace bulleyana, Lysimachia coreana and Ardisia polystica, Fig. 4), the ORF of this transcript is different from pt-accD coding regions of other species. However, their C-terminal regions are conserved, which all contained three putative motifs, such as acetyl-CoA binding sites, carboxybiotin binding sites and carboxyltransferase catalytic sites (Lee et al., 2004). In contrast, N-terminal of the ORF shows no similarity against any plastid-encoded sequences (Fig. 4). In order to test whether the software (TargetP and Protein Prowler) affect the results of subcellular localization prediction of this putative protein-encoded sequence, we also predicted the protein sorting signals using other six software (BacelLo, CELLO2GO, Euk-mPLoc2, EuLoc, HybridGO-Loc, and Predotar). Our conclusion of the protein subcellular localization was confirmed by identical results provided by other software (data not shown). Prediction results showed a chloroplast transit peptide of 72 residues length located at the N-terminus (Fig. 4). The chloroplast transit peptides of nuclear-encoded plastid proteins (NUPTs) are necessary for targeting and import of proteins into chloroplasts. It is strongly suggested that plastid accD has been functionally transferred to the nucleus in P. sinensis. This is the fourth report in angiosperms (to our knowledge) for the transferability of accD gene with lines of solid evidences.

Figure 4: Alignment of the putative nuclear accD from Primula sinensis and the plastid accD from Primula poissonii, Androsace bulleyana, Lysimachia coreana and Ardisia polystica.
(A) indicates the putative transit peptide at the N-terminal region of nuclear accD. (B, C, D) indicates three putative motifs, acetyl-CoA binding sites, carboxybiotin binding sites and carboxyltransferase catalytic sites, respectively.{kind=link}
Phylogenetic analysis of the n-accD transcript in P. sinensis with pt-accD from other 30 species, which belongs to six families in Ericales, was performed by RAxML. This dataset contained multiple alignment of 31 sequences of the C-terminal functional regions, since the regions are relatively conserved as discussed above. The maximum likelihood tree showed n-accD from P. sinensis was located within the clade of the Primulaceae with P. poissonii as the sister group (Fig. 5). Considering the close relationship between P. sinensis and P. poissonii, this result probably indicated that accD of P. sinensis might have transferred to nucleus recently. Available data showed that complete pt-accD genes have been lost functionally from three species, Arbutus unedo (JQ067650), Vaccinium macrocarpon (JQ757046), and Chamaedaphne calyculata (KJ463365) in Ericaceae. It is likely that the missing or pseudogenization of pt-accD genes occurred accidently and independently in Ericales.
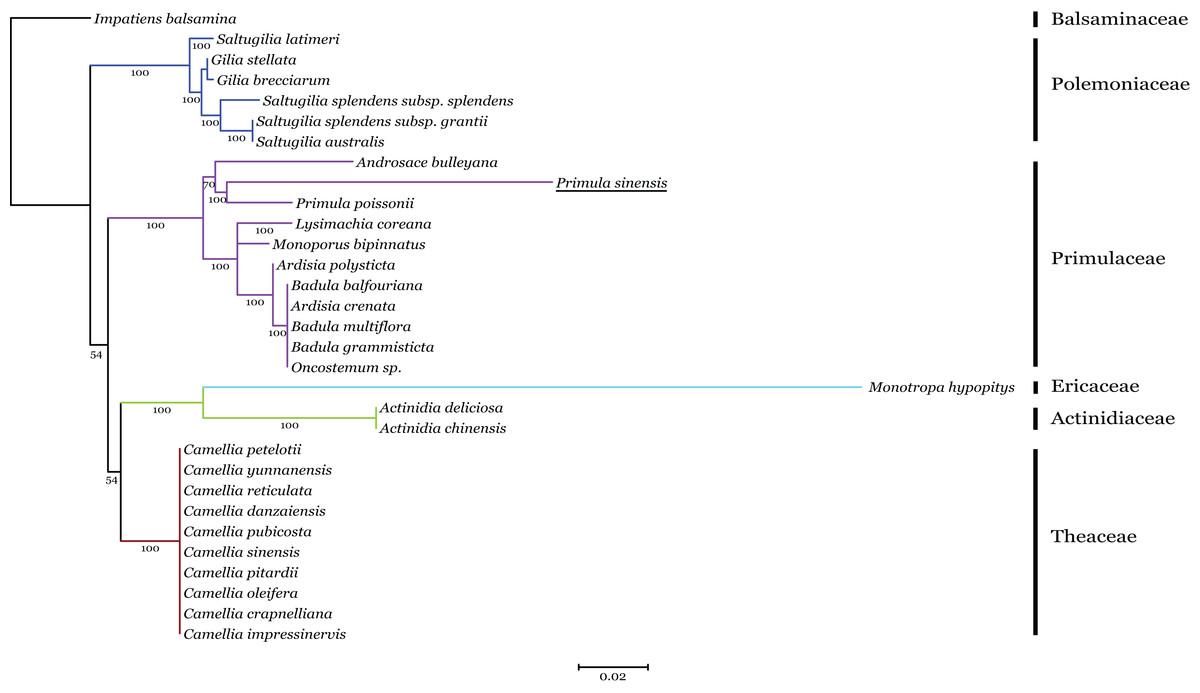
Figure 5: Phylogenetic analysis of accD sequences from six families in Ericales using the Maximum likelihood.
The underline indicates the putative n-accD gene of Primula sinensis.{kind=link}
Phylogenetic analysis based on plastome sequence
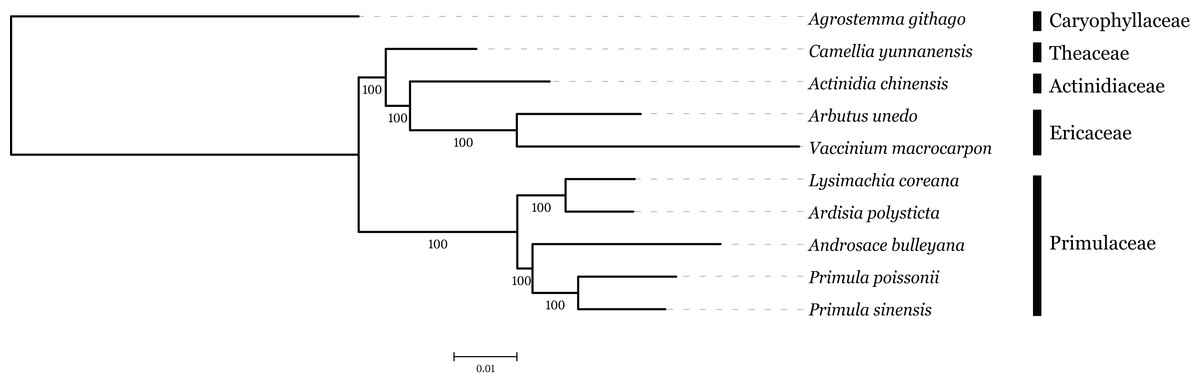
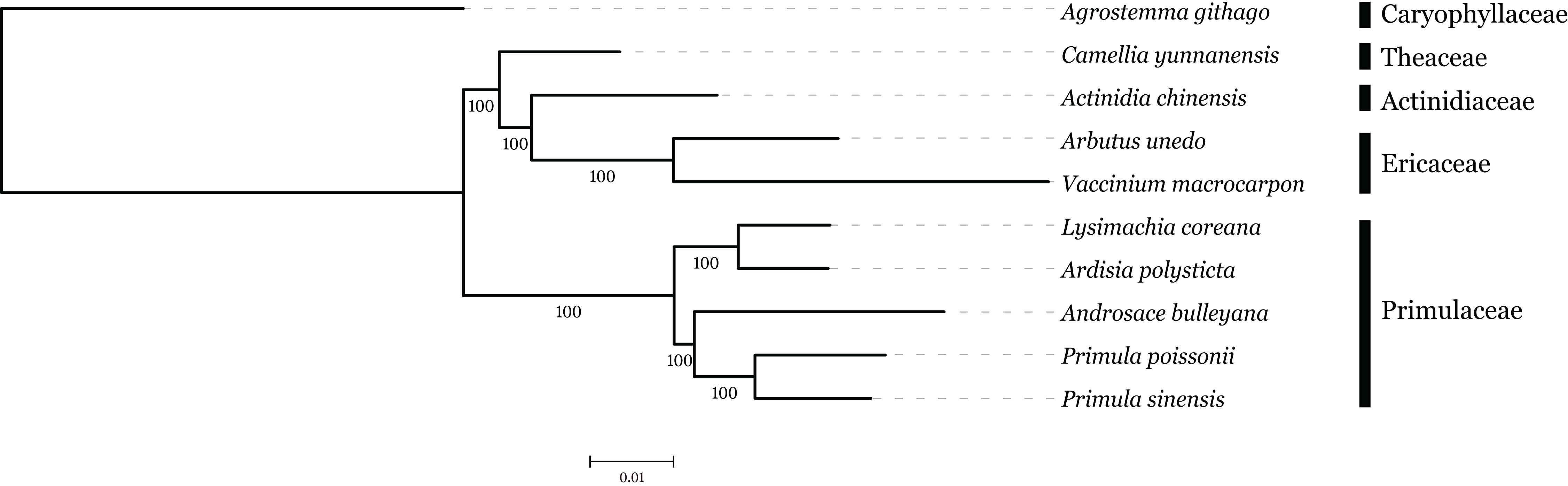
The family interrelationships within the large order Ericales, constrained by the data available, have remained unclear (Anderberg, Rydin & Källersjö, 2002; Bremer et al., 2002; Geuten et al., 2004). Recently, advances in high-throughput sequencing have provided a large amount of data, which was an improvement of the phylogenetic resolution (Wen et al., 2013; Yang et al., 2015). With the expectation, nine plastome sequences represented different genera in Ericales involved in phylogenetic analysis, with Agrostemma githago as outgroup. Phylogenetic relationships were inferred using 57 plastid protein-coding genes. As we know, the third base position of codon evolves faster than the rest of two positons with higher substitution rate. ML trees were produced by using three different datasets, all coding sequence, codon 1 + 2 and codon3, respectively. The result showed that topologies of three datasets were highly congruent with one another and all nodes were well supported (Fig. 6). Primuloideae and Myrsinoideae within Primulaceae s. l. fall in two branches separately with strong supports. Primulaceae s. l. was placed as sister to the clade that comprising Theaceae, Actinidiaceae, and Ericaceae. The phylogenetic positions of these groups are in agreement with recent studies (Stevens, 2012; Zhang et al., 2015).
{kind=link}