Chloroplast Genomes and Comparative Analyses among Thirteen Taxa within Myrsinaceae s.str. Clade (Myrsinoideae, Primulaceae)


Abstract
:1. Introduction
2. Results and Discussion
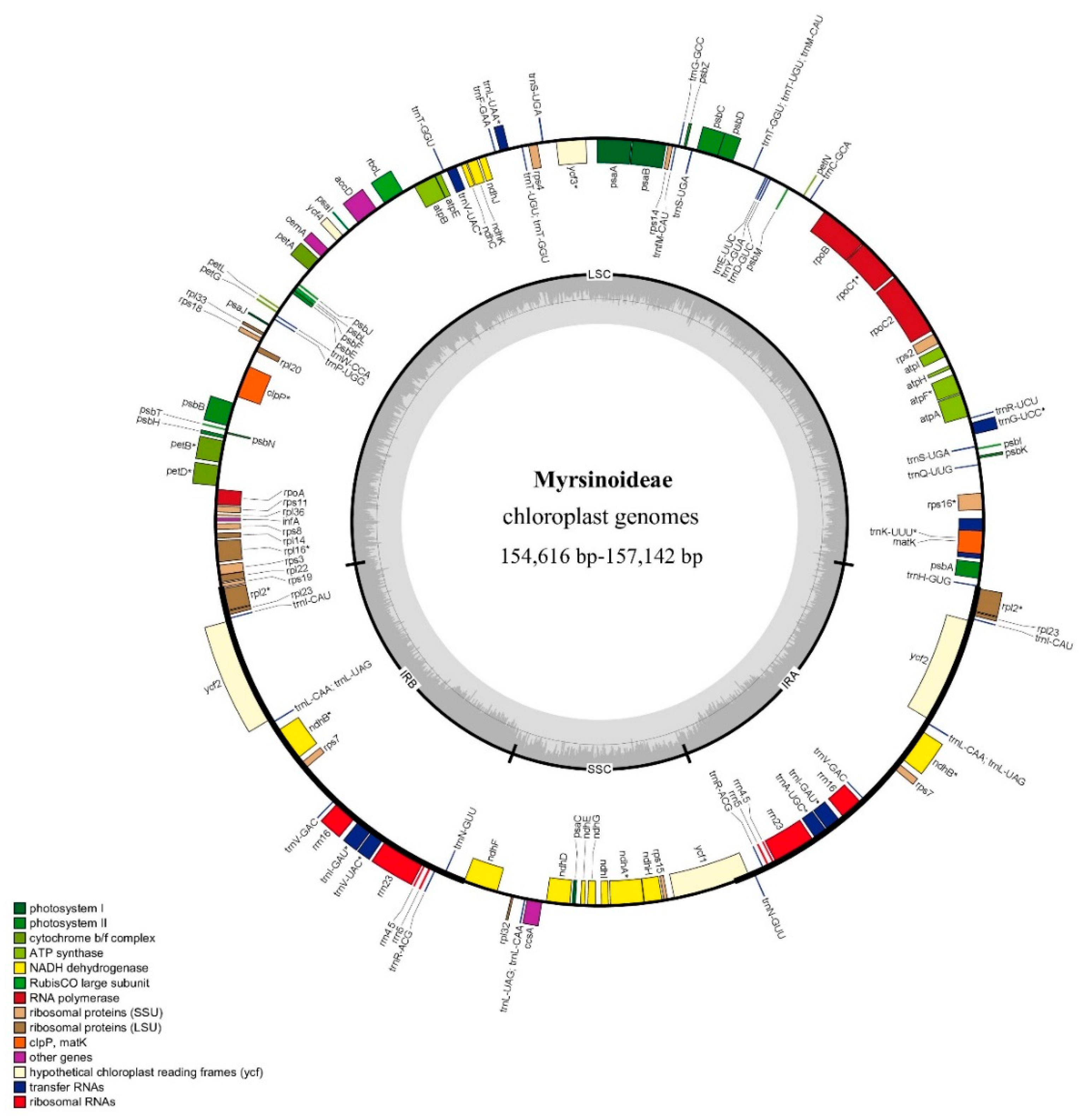
2.1. Chloroplast Genomes Features
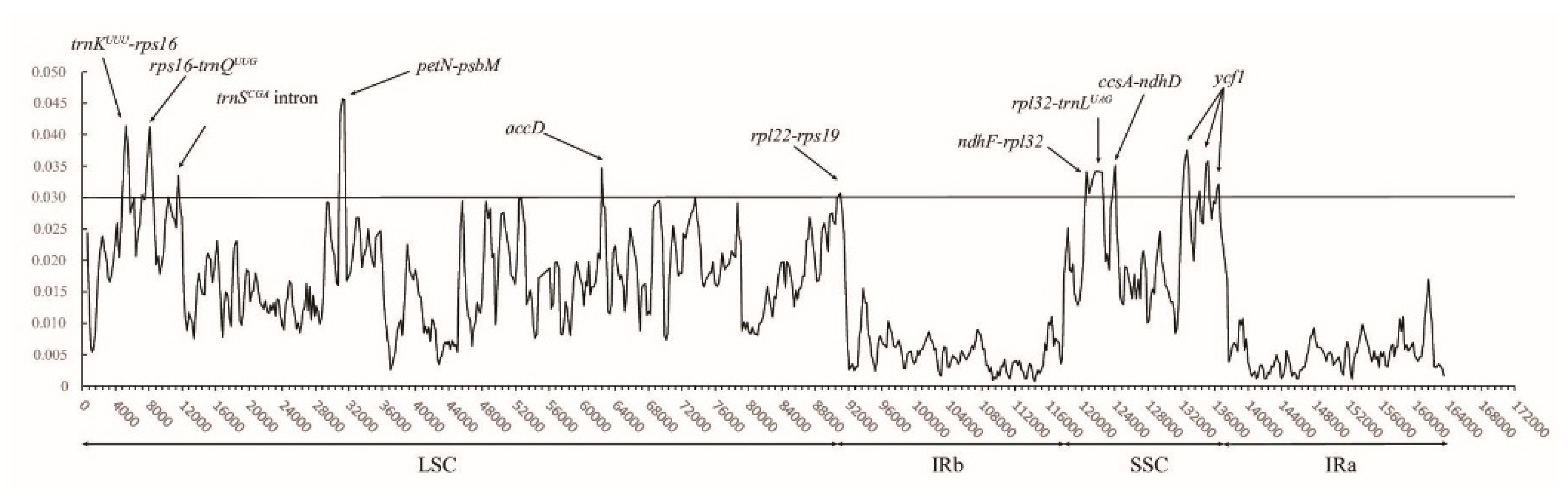
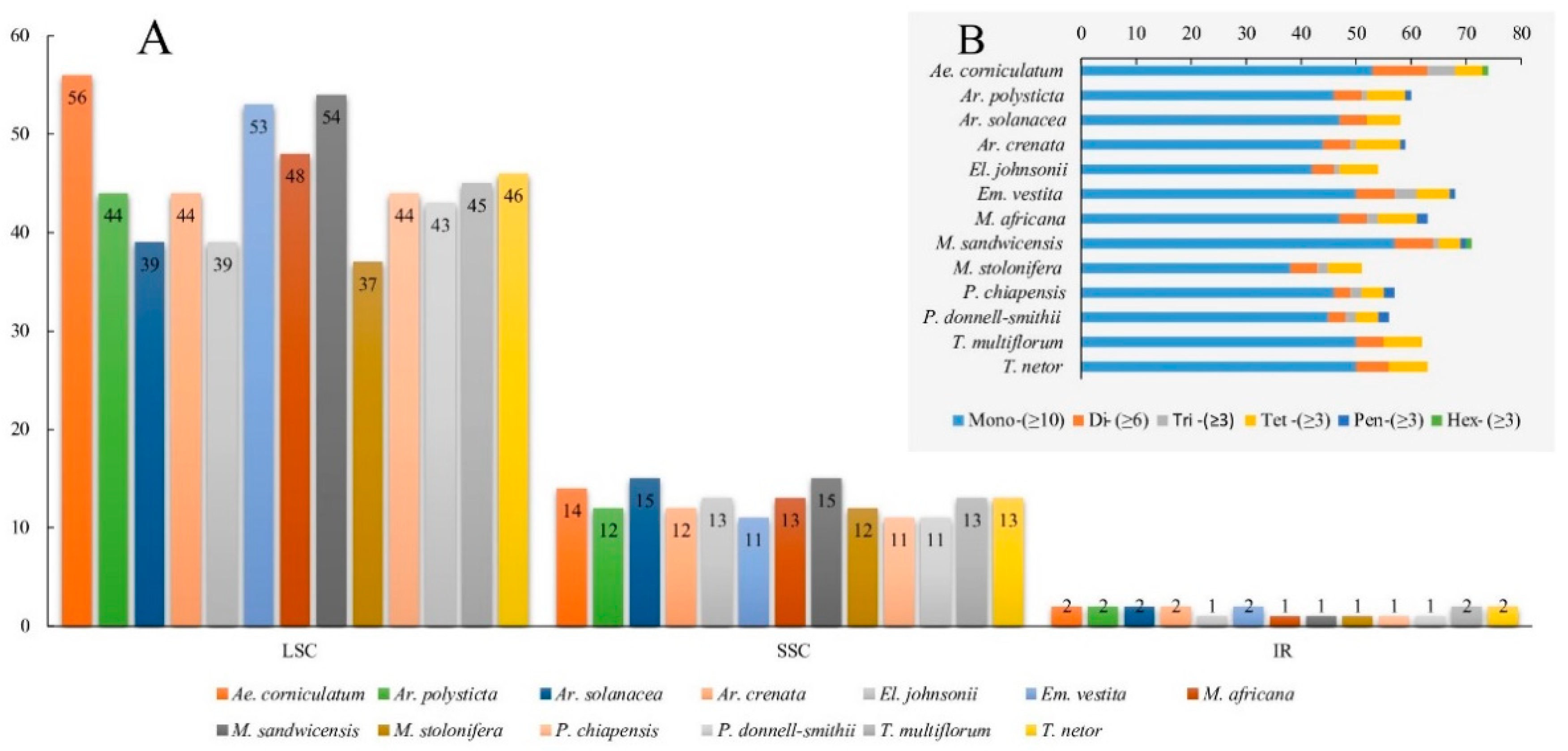
2.2. Hypervariable Regions and CpSSRs
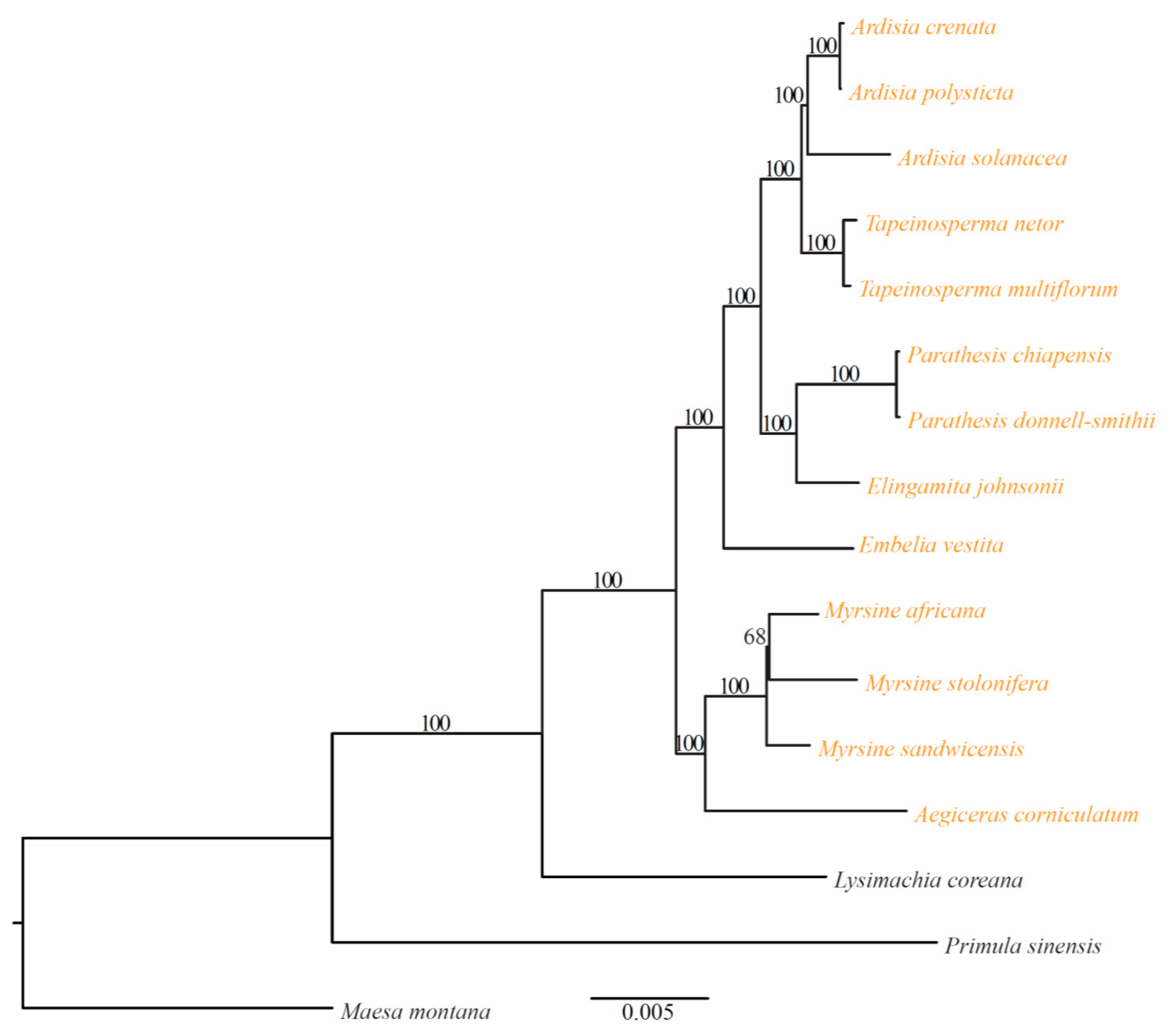
2.3. Phylogenetic Relationships
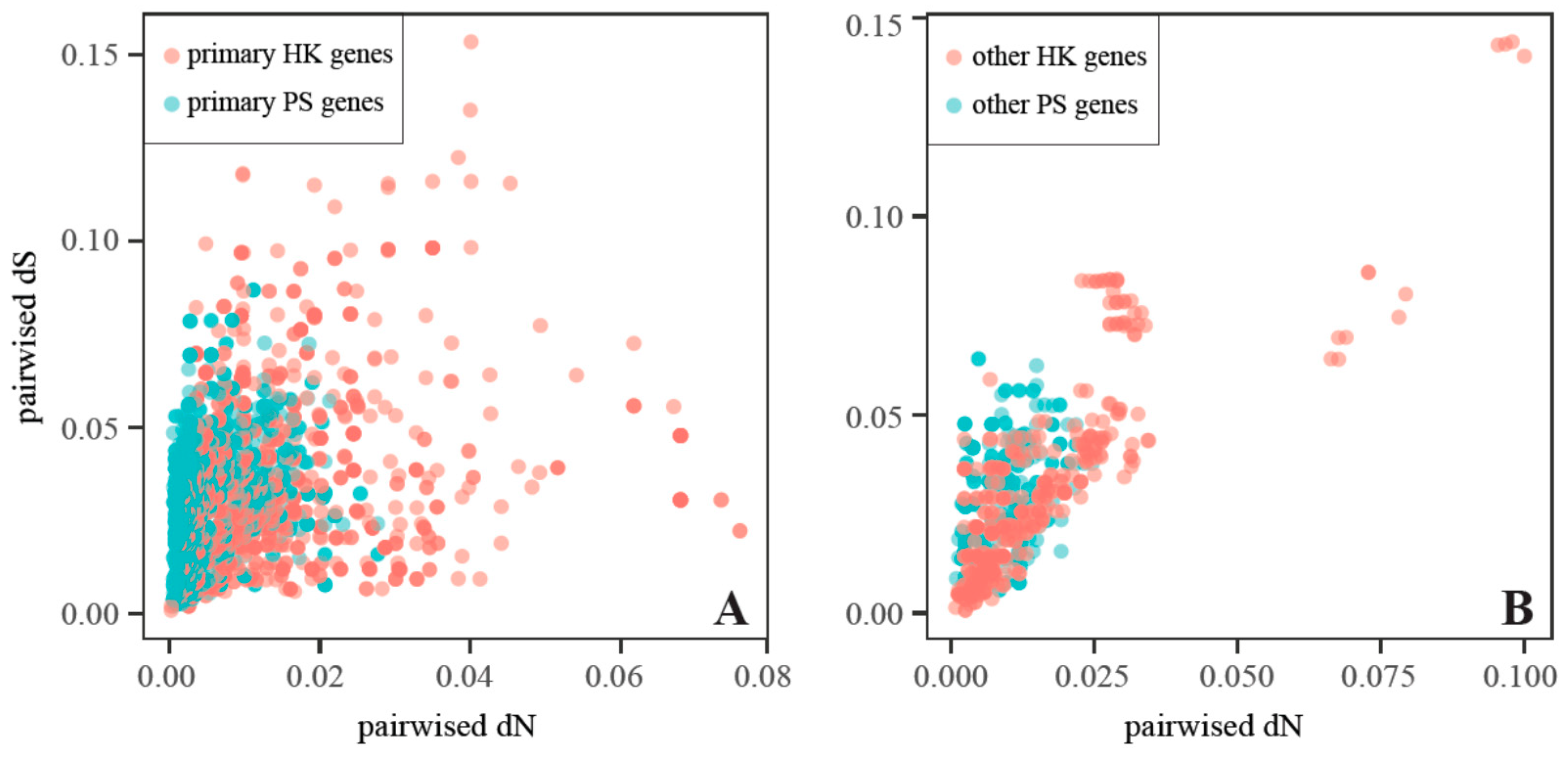
2.4. Substitution Rates and Their Variations among Genes
3. Materials and Methods
3.1. Sampling
3.2. DNA Extraction and Sequencing
3.3. Chloroplast Genome Assembly and Annotation
3.4. Identification of CpSSRs and Hypervariable Regions
3.5. Phylogenetic Analyses
3.6. Substitution Rate Estimation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [Green Version]
- Ståhl, B.; Anderberg, A.A. Myrsinaceae. In Flowering Plants. Dicotyledons: Celastrales, Oxalidales, Rosales, Cornales, Ericales; Kubitzki, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Kallersjo, M.; Bergqvist, G.; Anderberg, A.A. Generic realignment in primuloid families of the Ericales s.l.: A phylogenetic analysis based on DNA sequences from three chloroplast genes and morphology. Am. J. Bot. 2000, 87, 1325–1341. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Yuan, Y.M.; Hu, C.M.; Ge, X.J.; Zhao, N.X. Molecular phylogeny of Lysimachia (Myrsinaceae) based on chloroplast trnL-F and nuclear ribosomal ITS sequences. Mol. Phylogenet. Evol. 2004, 31, 323–339. [Google Scholar] [CrossRef]
- Anderberg, A.A.; Manns, U.; Källersjö, M. Phylogeny and floral evolution of the Lysimachieae (Ericales, Myrsinaceae): Evidence from ndhF sequence data. Willdenowia 2007, 37, 407–421. [Google Scholar] [CrossRef]
- Oh, I.C.; Schönenberger, J.; Motley, T.J.; Myrenås, M.; Anderberg, A.A. Phylogenetic relationships among endemic Hawaiian Lysimachia (Ericales: Primulaceae): Insights from nuclear and chloroplast DNA sequence data. Pac. Sci. 2013, 67, 237–251. [Google Scholar] [CrossRef]
- Yan, H.F.; Zhang, C.Y.; Anderberg, A.A.; Hao, G.; Ge, X.J.; Wiens, J.J. What explains high plant richness in East Asia? Time and diversification in the tribe Lysimachieae (Primulaceae). New Phytol. 2018, 219, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Yesson, C.; Toomey, N.H.; Culham, A. Cyclamen: Time, sea and speciation biogeography using a temporally calibrated phylogeny. J. Biogeogr. 2009, 36, 1234–1252. [Google Scholar] [CrossRef]
- Strijk, J.S.; Bone, R.E.; Thebaud, C.; Buerki, S.; Fritsch, P.W.; Hodkinson, T.R.; Strasberg, D. Timing and tempo of evolutionary diversification in a biodiversity hotspot: Primulaceae on Indian Ocean islands. J. Biogeogr. 2014, 41, 810–822. [Google Scholar] [CrossRef]
- Rose, J.P.; Kleist, T.J.; Lofstrand, S.D.; Drew, B.T.; Schonenberger, J.; Sytsma, K.J. Phylogeny, historical biogeography, and diversification of angiosperm order Ericales suggest ancient Neotropical and East Asian connections. Mol. Phylogenet. Evol. 2018, 122, 59–79. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S.; Yi, T.S.; Li, D.Z.; Soltis, D.E. Plastome phylogenetics: 30 years of inferences into plant evolution. Adv. Bot. Res. 2018, 85, 293–313. [Google Scholar]
- Soltis, D.; Soltis, P.; Endress, P.; Chase, M.W.; Manchester, S.; Judd, W.; Majure, L.; Mavrodiev, E. Phylogeny and Evolution of the Angiosperms: Revised and Updated Edition; University of Chicago Press: Chicago, IL, USA, 2018. [Google Scholar]
- Qiu, Y.X.; Fu, C.X.; Comes, H.P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 2011, 59, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Morris, A.B.; McLachlan, J.S.; Manos, P.S.; Soltis, P.S. Comparative phylogeography of unglaciated eastern North America. Mol. Ecol. 2006, 15, 4261–4293. [Google Scholar] [CrossRef] [PubMed]
- CBOL Plant Working Group; Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S.; van der Bank, M.; Chase, M.W.; Cowan, R.S.; Erickson, D.L.; et al. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [Green Version]
- Hollingsworth, P.M.; Li, D.Z.; van der Bank, M.; Twyford, A.D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. 2016, 371, 20150338. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, M.M.; Fér, T.; Schmickl, R.; Leong-Škorničková, J.; Newman, M.; Kress, W.J. Resolving the rapid plant radiation of early diverging lineages in the tropical Zingiberales: Pushing the limits of genomic data. Mol. Phylogenet. Evol. 2018, 128, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, N.; Lewis, P.O.; Lemmon, E.M.; Lemmon, A.R.; Holsinger, K.E. Anchored phylogenomics improves the resolution of evolutionary relationships in the rapid radiation of Protea L. Am. J. Bot. 2017, 104, 102–115. [Google Scholar] [CrossRef]
- Folk, R.A.; Mandel, J.R.; Freudenstein, J.V. A protocol for targeted enrichment of intron-containing sequence markers for recent radiations: A phylogenomic example from Heuchera (Saxifragaceae). Appl. Plant Sci. 2015, 3, 1500039. [Google Scholar] [CrossRef] [PubMed]
- Li, H.T.; Yi, T.S.; Gao, L.M.; Ma, P.F.; Zhang, T.; Yang, J.B.; Gitzendanner, M.A.; Fritsch, P.W.; Cai, J.; Luo, Y.; et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 2019, 5, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Ruhfel, B.R.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J.G. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 2014, 14, 23. [Google Scholar] [CrossRef]
- Mower, J.P.; Vickrey, T.L. Structural diversity among plastid genomes of land plants. In Advances in Botanical Research; Chaw, S.M., Jansen, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 58, pp. 263–292. [Google Scholar]
- McKain, M.R.; Johnson, M.G.; Uribe-Convers, S.; Eaton, D.; Yang, Y. Practical considerations for plant phylogenomics. Appl. Plant Sci. 2018, 6, e1038. [Google Scholar] [CrossRef] [PubMed]
- Son, O.; Park, S.J. Complete chloroplast genome sequence of Lysimachia coreana (Primulaceae). Mitochondrial DNA 2016, 27, 2263–2265. [Google Scholar] [PubMed]
- Ren, T.; Yang, Y.C.; Zhou, T.; Liu, Z.L. Comparative plastid genomes of Primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 2018, 19, 1050. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Wolf, P.G.; Karol, K.G. Plastomes of Bryophytes, Lycophytes and Ferns. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 89–102. [Google Scholar]
- Liu, T.J.; Zhang, C.Y.; Yan, H.F.; Zhang, L.; Ge, X.J.; Hao, G. Complete plastid genome sequence of Primula sinensis (Primulaceae): Structure comparison, sequence variation and evidence for accD transfer to nucleus. PeerJ 2016, 4, e2101. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.N.; Li, H.T.; Milne, R.; Zhang, T.; Ma, P.F.; Yang, J.; Li, D.Z.; Gao, L.M. Comparative analyses of plastid genomes from fourteen Cornales species: Inferences for phylogenetic relationships and genome evolution. BMC Genom. 2017, 18, 965. [Google Scholar] [CrossRef]
- Guo, X.Y.; Liu, J.Q.; Hao, G.Q.; Zhang, L.; Mao, K.S.; Wang, X.J.; Zhang, D.; Ma, T.; Hu, Q.J.; Al-Shehbaz, I.A.; et al. Plastome phylogeny and early diversification of Brassicaceae. BMC Genom. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Wang, Y.W.; He, P.Z.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C.X. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef]
- Shi, C.; Liu, Y.; Huang, H.; Xia, E.H.; Zhang, H.B.; Gao, L.Z. Contradiction between plastid gene transcription and function due to complex posttranscriptional splicing: An exemplary study of ycf15 function and evolution in angiosperms. PLoS ONE 2013, 8, e59620. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef]
- Haigh, J. The accumulation of deleterious genes in a population—Muller’s Ratchet. Theor. Popul. Biol. 1978, 14, 251–267. [Google Scholar] [CrossRef]
- Martin, W.; Stoebe, B.; Goremykin, V.; Hansmann, S.; Hasegawa, M.; Kowallik, K.V. Gene transfer to the nucleus and the evolution of chloroplasts. Nature 1998, 393, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant. Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Weising, K.; Nybom, H.; Pfenninger, M.; Wolff, K.; Kahl, G. DNA Fingerprinting in Plants: Principles, Methods, and Applications, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Wolf, P.G. Plastid Genome Diversity. In Plant Genome Diversity Volume 1: Plant Genomes, Their Residents, and Their Evolutionary Dynamics; Wendel, J.F., Greilhuber, J., Dolezel, J., Leitch, I.J., Eds.; Springer: Vienna, Austria, 2012. [Google Scholar]
- Wicke, S.; Schneeweiss, G. Next-generation organellar genomics: Potentials and pitfalls of high-throughput technologies for molecular evolutionary studies and plant systematics. In Next Generation Sequencing in Plant Systematics; Hörandl, E., Appelhans, M., Eds.; Koeltz Scientific Books: Königstein, Germany, 2015. [Google Scholar]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Wang, T.J.; Wu, C.W.; Wang, Y.N.; Chaw, S.M. Plastome evolution in the sole hemiparasitic Genus Laurel Dodder (Cassytha) and insights into the plastid phylogenomics of Lauraceae. Genome Biol. Evol. 2017, 9, 2604–2614. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.M.K. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 1987, 19, 11–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: Enabling high-impact science for phylogenetics researchers with limited resources. In Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the Campus and Beyond, Chicago, IL, USA, 16–20 July 2012; pp. 1–8. [Google Scholar]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ruhlman, T.A.; Weng, M.L.; Hajrah, N.H.; Sabir, J.S.M.; Jansen, R.K. Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol. Evol. 2017, 9, 1766–1780. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Muller, K.F.; dePamphilis, C.W.; Quandt, D.; Bellot, S.; Schneeweiss, G.M. Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc. Natl. Acad. Sci. USA 2016, 113, 9045–9050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Voucher No./Herbarium Code | GenBank Accession | Size (bp) | GC Content | Gene No. | Protein Coding Gene | tRNA | rRNA |
|---|---|---|---|---|---|---|---|---|
| Aegiceras comiculatum | Liu150016/IBSC | MN167882 | 157,241 | 36.9% | 114 | 80 | 30 | 4 |
| Ardisia solanacea | 17988*/K | MN094783 | 156,518 | 37.1% | 114 | 80 | 30 | 4 |
| Ardisia polysticta | No data | KC465962 | 156,506 | 37.1% | 113 | 80 | 29 | 4 |
| Aridisa crenata | Yxk160038/IBSC | KM719568 | 156,876 | 37.1% | 113 | 80 | 30 | 4 |
| Elingamita johnsonii | 958*/K | MN094784 | 156,180 | 37.0% | 113 | 79 | 30 | 4 |
| Embelia vestita | Liu150050/IBSC | MN167884 | 157,238 | 36.9% | 114 | 80 | 30 | 4 |
| Myrsine africana | 30087*/K | MN165129 | 156,433 | 37.0% | 114 | 80 | 30 | 4 |
| Myrsine sandwicensis | 38322*/HAW | MN177700 | 156,284 | 37.0% | 114 | 80 | 30 | 4 |
| Myrsine stolonifera | Liu150044/IBSC | MN167883 | 156,953 | 36.9% | 114 | 80 | 30 | 4 |
| Parathesis chiapensis | Alush Nendes 6574/ARIZ | MN177699 | 156,666 | 37.0% | 113 | 79 | 30 | 4 |
| Parathesis donnell-smithii | Alvaro Campos 3924/ARIZ | MN177698 | 156,344 | 37.0% | 113 | 79 | 30 | 4 |
| Tapeinosperma multiflorum | 9361/MO | MN177701 | 155,168 | 37.0% | 113 | 79 | 30 | 4 |
| Tapeinosperma netor | 33984*/K | MN177702 | 154,616 | 37.1% | 113 | 79 | 30 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, X.; Liu, T.; Yuan, X.; Xu, Y.; Yan, H.; Hao, G. Chloroplast Genomes and Comparative Analyses among Thirteen Taxa within Myrsinaceae s.str. Clade (Myrsinoideae, Primulaceae). Int. J. Mol. Sci. 2019, 20, 4534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184534
Yan X, Liu T, Yuan X, Xu Y, Yan H, Hao G. Chloroplast Genomes and Comparative Analyses among Thirteen Taxa within Myrsinaceae s.str. Clade (Myrsinoideae, Primulaceae). International Journal of Molecular Sciences. 2019; 20(18):4534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184534
Chicago/Turabian StyleYan, Xiaokai, Tongjian Liu, Xun Yuan, Yuan Xu, Haifei Yan, and Gang Hao. 2019. "Chloroplast Genomes and Comparative Analyses among Thirteen Taxa within Myrsinaceae s.str. Clade (Myrsinoideae, Primulaceae)" International Journal of Molecular Sciences 20, no. 18: 4534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184534