Cytokines in Inflammatory Disease


1
Experimental Radiology, Department of Radiology and Nuclear Medicine, Otto von Guericke University Magdeburg, 39120 Magdeburg, Germany
2
Department of Cardiology with Emphasis on Electrophysiology, University Heart Centre, University Hospital Hamburg-Eppendorf, 20251 Hamburg, Germany
3
Department of Trauma, Hand and Reconstructive Surgery, Goethe University, 60590 Frankfurt, Germany
4
Experimental Radiology, Department of Radiology and Nuclear Medicine, Otto von Guericke University Magdeburg, 39120 Magdeburg, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2019, 20(23), 6008; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236008
Submission received: 4 October 2019
/
Revised: 24 November 2019
/
Accepted: 25 November 2019
/
Published: 28 November 2019
(This article belongs to the Special Issue Metabolic Bone Diseases: Pathophysiology and Molecular Biology)
Abstract
:This review aims to briefly discuss a short list of a broad variety of inflammatory cytokines. Numerous studies have implicated that inflammatory cytokines exert important effects with regard to various inflammatory diseases, yet the reports on their specific roles are not always consistent. They can be used as biomarkers to indicate or monitor disease or its progress, and also may serve as clinically applicable parameters for therapies. Yet, their precise role is not always clearly defined. Thus, in this review, we focus on the existing literature dealing with the biology of cytokines interleukin (IL)-6, IL-1, IL-33, tumor necrosis factor-alpha (TNF-α), IL-10, and IL-8. We will briefly focus on the correlations and role of these inflammatory mediators in the genesis of inflammatory impacts (e.g., shock, trauma, immune dysregulation, osteoporosis, and/or critical illness).
1. Introduction
The first line of defense against molecules that are either pathogen-derived or endogenous danger signals (or quite often both) has evolved over millions of years. It is composed of players and mediators that are common to most vertebrates and invertebrates, as well as even plants [1]. In general, immunity does not only differentiate between self and not-self but also between dangerous and not dangerous [2]. Inflammation-inducing pathogen-associated molecular patterns (PAMPs) include highly conserved structures [3] such as cytosine-phosphate-guanin motifs [4], heat shock proteins (HSP), peptidoglycans (PGN), and lipopolysaccharides (LPS) [5], while danger-associated molecular patterns (DAMPs) are originally intracellular proteins or nucleic acids normally not found outside the cell, such as chromatin-associated protein high-mobility group box 1 (HMGB1), adenosine triphosphate (ATP), uric acid, deoxyribonucleic acid (DNA), and degraded extracellular matrix-like heparan sulphate and hyaluronan [6,7]. Both PAMPs and DAMPs are recognized via pattern recognition receptors (PRRs) [8,9,10]. Toll-like receptors (TLR), nucleotide-binding oligomerization domain (NOD)-like receptors (NLR) and mannose binding lectin (MBL) are major PRRs implicated in inflammatory pathways [5,11]. Upon their activation, PRR transduces signals intracellularly [12], e.g., via mitogen-activated protein (MAP) kinase signaling pathways to nuclei where diverse transcription factors, such as nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-κB) get activated and induce a cellular response [13]. This response often involves the induction of adhesion molecules accelerating inflammation and diapedesis of effector cells of the innate immunity [14]. Furthermore, it leads to the induction, production and the release of pro-inflammatory mediators, including cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1, IL-6 and IL-10, or type 1 interferons (IFNs) [15]. Additionally, those inflammatory mediators themselves can induce, for example, DAMPs to potentiate inflammation [16]. Summarized, the messenger molecules such as cytokines are highly important in the orchestration of the inflammatory response to self- or not-self danger molecules. Meanwhile, the role of the immune system in various inflammatory diseases, traumas, and bone pathologies, such as osteoporosis, osteoarthritis, and rheumatoid arthritis is well described. In this review, we will briefly describe IL-1, IL-6, IL-8, IL-10, and TNF-α cytokines.
2. Cytokines
Cytokines are small secreted proteins (<40 kDa), which are produced by nearly every cell to regulate and influence immune response [5]. The release of pro-inflammatory cytokines will lead to activation of immune cells and production as well as the release of further cytokines [17]. Therefore, in the past when the term “cytokine storm” arose, it explained inflammation as a sudden release of cytokines to upregulate an inflammatory process [18]. However, recent research indicates that a simultaneous release of pro- and anti-inflammatory cytokines are mandatory in any immune response [19].
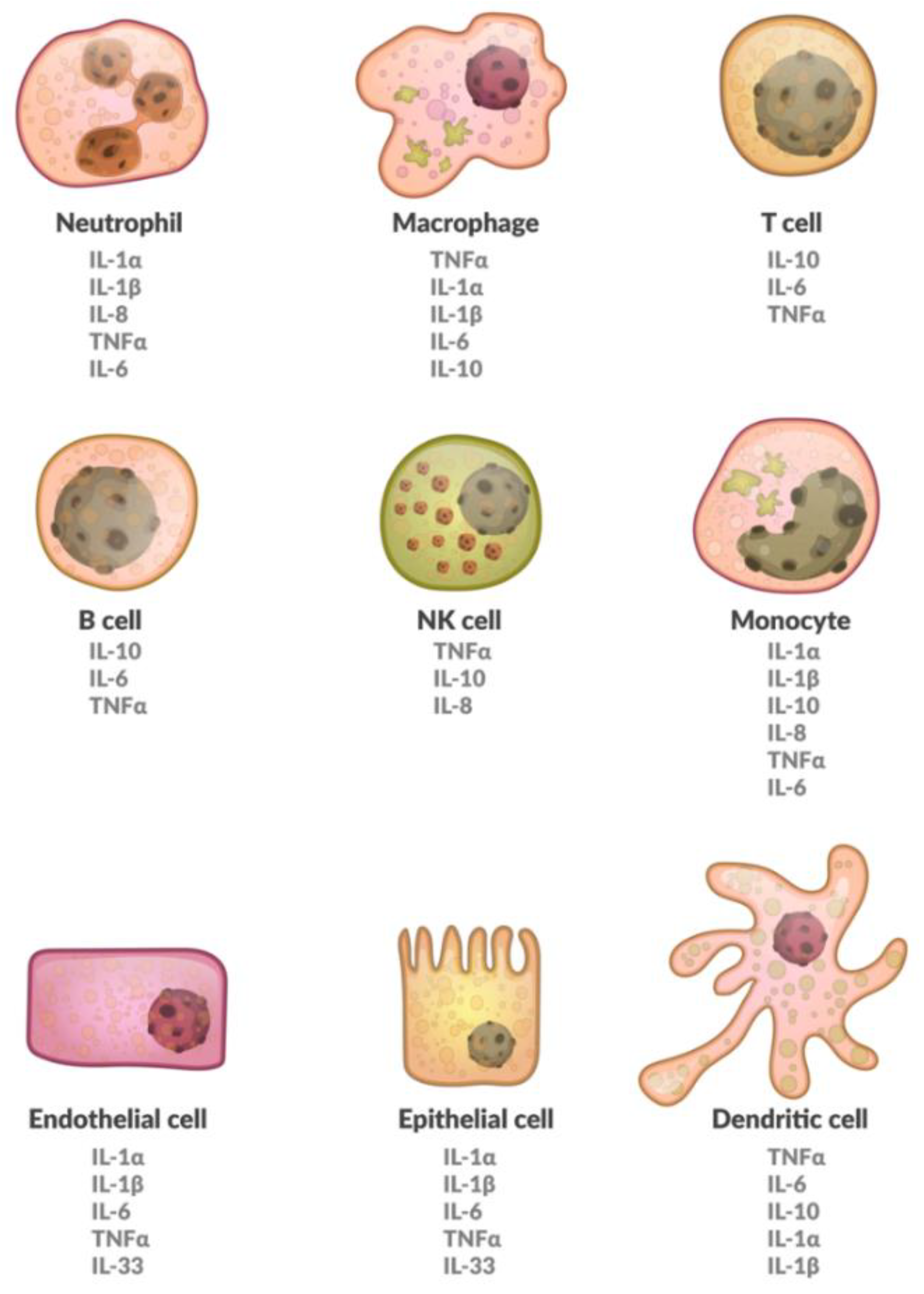
Cytokines suffer from a somewhat inconsistent nomenclature; they are referred to as interleukins, chemokines, or growth factors among many other names [20]. Cytokines are made up of so-called superfamilies, not necessarily describing common genes, but rather similar structures [21]. Furthermore, different cell populations can produce the same cytokine. The effects of cytokines depend on the targeted cell, making them pleiotropic [20]. Also, different cytokines may have the same effect and are therefore redundant. They may, however, also have a synergistic effect. Finally, they potentially trigger signaling cascades, giving the smallest amounts of protein the chance to be devastating in consequence [22]. A brief overview on various cells expressing different cytokines has been provided in Figure 1. Furthermore, a brief overview of cytokines that are involved in osteoporosis is shown in Table 1.
2.1. Interleukin-6
Interleukin-6 has been shown to play important roles in autoimmune diseases, bacterial infections and metabolic side effects have been observed also [23]. It is composed of four α-helices, comparable to other members of the IL-6 family [23]. It is translated as a 184 amino acid long protein that undergoes glycosylation and is secreted by T-cells, monocytes, endothelial cells, and fibroblasts [17]. Interestingly, IL-6 was first described for its effects on adaptive immunity, like promoting cluster of differentiation (CD)4+ T-cells via IL-21 production, and promoting T-cell differentiation towards T-helper2 cells (Th2) and Th17 cells [24]. The very first reference was as a B-cell stimulatory factor by the Kishimoto group in 1986 [25]. It has pro- and anti-inflammatory properties, which are described further below. Signaling is achieved via two different mechanisms; one of which is IL-6 binding to its membrane-bound IL-6 receptor (mbIL6R) [26]. This complex subsequently recruits two molecules of membrane-bound glycoprotein (gp) 130, a process that leads to downstream signaling via Janus kinases/signal transducer and activator of transcription (STAT) kinases, phosphoinositide 3-kinase (PI3K), and MAP kinases like p38 [27,28]. A major limitation for a sustainable reaction to IL-6 is the availability of the mbIL6R, which is only expressed on certain cell types, while gp130 is found in almost every cell [29]. This implies that the systemic influence of IL-6 via classic signaling is rather limited [17]. The second mechanism of IL-6 recognition is dependent on the soluble IL-6 receptor (sIL6R), which is expressed via mRNA splicing or proteolysis by a disintegrin and metalloproteinase (ADAM) proteases [17]. Interestingly, ADAM proteases cannot be activated only by other cytokines, such as IL-1β or TNF-α [30], they can be induced by bacterial toxins as well [31]. In the case of sIL6R expression, IL-6 binds to the sIL6R and builds an IL6/sIL6R complex, which in turn activates gp130 on mbIL6R-less cells [32]. This process is termed trans-signaling and is responsible for most of IL-6 inflammation-inducing capabilities [23]. Currently, similar to the C-reactive protein (CRP), IL-6 is used to “monitor” inflammation levels in patients with cancer, infection, or autoimmune diseases [33,34]. The reason for using IL-6 as a biomarker is its central role in activating and maintaining the inflammatory response. For instance, the clinical quantification of IL-6 is a strong predictor for mortality in pancreatic and cardiovascular disease [35,36]. However, unfortunately, its anti-inflammatory properties, further described below, are so far neglected in clinical practice. While early inflammation is dominated by neutrophils, later states of inflammation are dominated by monocytes. IL-6 is essential in this so-called leukocyte switch [37]. Subsequently, it reduces neutrophil recruitment via suppression of chemokines attracting polymorphonuclear leukocytes (PMNL), like the chemokine (C-X-C motif) ligand (CXCL)1 and CXCL8 (IL-8), while upregulating monocyte attracting chemokines CC-chemokine ligand (CCL)2/monocyte chemotactic protein (MCP)-1 and CCL8/MCP-2 in vitro and in vivo [37]. Furthermore, cell adhesion molecules like vascular cell adhesion molecule (VCAM) 1, intercellular cell adhesion molecules (ICAM) and E-selectin are upregulated by IL-6 in a fever range mice model [38]. In models with gp130 knockout mice, the ability of IL-6 to enhance macrophage-colony stimulating factor (M-CSF) receptor expression, thereby accelerating monocyte differentiation to macrophages, was linked to its gp130 MAP kinase pathway [39]. In a Staphylococcus epidermidis induced peritoneal inflammation mice model, IL-6 was mandatory for the recruitment of T-cells [40]. Interestingly, classic signaling of IL-6 is needed for regenerative and protective processes in the body. For instance, in inflammatory disease mice models and diverse knockout mice models, IL-6 was essential to liver regeneration, gut barrier repair, and suppression of inflammation in the kidney and pancreas [41,42,43].
In clinical practice, the first association of IL-6 with cardiovascular disease and cancer was found in 1990 [44]. Enhanced levels of IL-6 were found in three patients with cardiac myxomas and removal of the tumor abolished the IL-6 levels [44]. In fact, increased pretreatment levels of IL-6 can be a predictor of survival in head and neck cancer [45]. Yet, it often remains unclear if IL-6 is only correlative to cancer or rather essential in cancer genesis. A study by Zhang et al. demonstrated that escalated levels of IL-6R in sera from nasopharyngeal carcinoma (NPC) patients are not just correlative [46]. The cytokine serves as a catalyst for the malignant transformation of Epstein–Barr infected nasopharyngeal cells to cancerous cells in vitro via STAT kinases [46].
Osteoporosis is a common disease in the aging population and studies have shown that IL-6 is potentially implicated in its pathogenesis [47]. IL-6 stimulates bone resorption. Several studies have examined the association between IL-6 gene polymorphisms and bone mineral density [47,48,49].
Another prominent use of IL-6 as a biomarker is in sepsis or after major trauma. Studies in the nineties demonstrated 1000-fold increased IL-6 levels in septic patients and correlation with the gravity of organ failure [50]. Likewise, the detection of IL-6 is correlative to invasiveness and duration of surgery [51]. Levels of IL-6 after trauma usually do not reach those of septic patients [52]. Unlike CRP, IL-6 can also help to distinguish infection from fever of unknown origin in pediatric practice [53]. Several studies confirm a predictive value of IL-6 for mortality and organ dysfunction in sepsis or after major trauma [54,55]. While IL-6 has undoubted prognostic value in early inflammation, clinical use has not seen any breakthroughs. Many physicians prefer a combination of clinical presentation, white blood count, CRP levels, and fever measurement over the expensive IL-6 determination [52].
2.2. Interleukin 1 Family
Interleukin-1α and IL-1β were the first cytokines to be discovered in 1974 by Charles A. Dinarello, and since then, they have been greatly studied [56]. In this review, we will focus on the following members of the IL-1 family: IL-1α, IL-1β, and IL-33.
Interleukin-1α and IL-1β are encoded by different genes but can be bound by the same IL-1 receptor (IL-1R) [56]. While IL-1α has a higher affinity for IL1-R1, IL-1β has a higher affinity for the soluble IL-1R2 [57]. Both are translated as 31 kDa precursor protein and cleaved into smaller 17 kDa forms, albeit with different amino acid sequences [58].
The IL-1α precursor is usually found in intracellular space, as well as constitutively in many cell types including hepatocytes, nephrotic epithelium, endothelium, and epithelial cells of the gastro-digestive tract [59].
Even in cases of severe infection, relatively low concentrations are found in extracellular space [60]. Upon stimuli such as oxidative stress or cytokine exposure, e.g., other IL-1 family cytokines, the expression of the IL-1α mRNA is inducible [61]. Nevertheless, it is not clear if post-translational modifications are needed for IL-1α to become active. In contrast to IL-1β and IL-33, the precursor form of IL-1α and recombinant human mature IL-1α have the same biological activity in inducing IL-6 and TNF-α in human peripheral blood mononuclear cells (PBMCs) and lung cancer cells [62]. Nevertheless, the secretion of IL-1α protein is well regulated. During apoptosis, cytosolic IL-1α translocates to the nucleus and binds firmly to chromatin [63], while during necrosis, it becomes released from the nucleus into the local tissue upon degradation of the cell membrane [63]. This exemplifies the properties of IL-1α as an alarmin. Whereas the release of IL-1α during the process of necrosis is explained by the loss of plasma membrane stability, the leakage of IL-1α in “healthy” cells is induced via pyroptosis [64]. This is a process of the so-called inflammation-induced apoptosis, which leads to enhanced cell membrane permeability through the formation of an inflammasome complex in an, e.g., caspase-1-dependent mechanism [64]. Caspase-1 knock-out mice displayed significantly less IL-1α protein release by monocytes upon their stimulation with LPS and ATP as compared to the wild-type mice [65]. The soluble decoy receptor IL-1R2 functions as a receptor in plasma, and limits spreading of IL-1α, thereby reducing its signaling and restraining inflammation [66]. Another unique trait among the IL-1 family is that the pro-IL-1α in its full length is implemented in the cell membrane in case of inflammation, and can operate as a fully active membrane-bound cytokine [57].
The primary sources of IL-1β are hematopoietic cells like monocytes, macrophages such as microglia or Kupffer cells and dendritic cells upon activation of PRR by PAMP or DAMP [21]. Furthermore, alpha cells of pancreas secrete IL-1β, and this can be studied in diabetic and obese patients [67]. Recent trials imply the contribution of epithelium and endothelial IL-1β to cardiovascular disease [68].
In contrast to IL-1α, IL-1β precursor is not biologically active, as its activation requires a proteolytic step by the IL-1β converting enzyme, e.g., caspase-1 within the multiprotein inflammasome complex [69]. Upon activation of myeloid differentiation primary response (MyD)88 in case of hypoxia, complement activation, or even IL-1β itself, pro-IL-1β mRNA is induced [21,70]. The translation occurs in the cytosol, however, it is discussed if a second signal is required for pro-IL-1β to be cleaved to IL-1β, and thereby activated [71]. Second signals can include DAMP or alarmin molecules, such as ATP, which binds to P2 × 7 receptors, thereby providing a signal to open potassium channels lowering intracellular potassium levels [72]. In consequence, the formation of NOD-like receptor (NLR) sensor molecule, such as NOD-, leucine-rich repeat (LRR)- and pyrin domaine-containing protein (NLRP)3 [73], and apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) to the inflammasome complex occurs [21]. As a result, pro-caspase-1 is activated into caspase-1 and cleaves, e.g., pro-IL-1β or pro-IL-18 cytokine precursors to their active forms, thereby initiating or enhancing the pro-inflammatory response [74].
In the last decades huge advances have been made in understanding the role of inflammasomes in the pathogenesis of infectious, autoinflammatory and autoimmune diseases. Familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS) and neonatal-onset multisystem inflammatory disorder (NOMID, also known as chronic infantile neurologic, cutaneous, and articular (CINCA) syndrome) have been shown to be caused by gain of function mutations in the NLPR3 gene encoding for cryopyrin, leading to increased caspase-1 and IL-1β activity [75,76,77,78]. Due to their similar etiology these diseases are today recognized as a group of diseases named pryopyrin-associated periodic syndrome (CAPS). Current treatment of CAPS is successfully and safely based upon three different medications named rilonacept (captures IL-1β as a decoy receptor), anakinra (IL-1R antagonist), and canakinumab (monoclonal antibody against IL-1β) [79,80]. Regarding the role of NRLP3 in the development of atherosclerosis Duewell et al. reported decreased development of atherosclerotic lesions in mice lacking inflammasome related molecules NLRP3, ASC or IL-1α/β and showed that cholesterol crystals as a possible DAMP strongly activated NLRP3 inflammasomes in macrophages [81]. Further studies in similar mouse models as well showed decreased severity of atherosclerosis in mice lacking NLRP3, caspase-1 or IL-1β [82,83,84], while in another study NLRP3 inflammasomes were not critically implicated in atherosclerosis progression [85]. Furthermore, the NLPR3 inflammasome is activated by oxidized low-density lipoprotein (LDL) and high levels of triglyceride, both being major risk factors for atherosclerosis [81,86,87].
The role of inflammasomes in the pathogenesis of rheumatoid arthritis has been studied extensively in the past decades in humans as well as in specific animal models. Guo et al. recently showed that the NLRP3 inflammasome was highly activated in synovia from patients with rheumatoid arthritis and in an animal model with collagen-induced arthritis (CIA) in mice [88]. Furthermore, treatment with MCC950, a selective NLRP3 inhibitor, in the animal model resulted in significantly less severe joints inflammation and bone destruction [88]. In a study by Ippagunta et al. the authors investigated the role of different components of the NLRP3 inflammasome and showed that NLRP3(−/−) and caspase-1(−/−) mice were predisposed to collagen-induced arthritis while ASC(−/−) mice were protected from arthritis [89]. Another study by Joosten et al. investigated the role of caspase-1, the downstream effector of inflammasomes, in the development of rheumatoid arthritis and obtained conflictive results showing no effect of caspase 1 deficiency in a model of acute (neutrophil-dominated) arthritis but reduced joint inflammation and cartilage destruction in a mouse model of chronic arthritis [90]. The crucial role of NRP3 inflammasomes in the development of rheumatoid arthritis was investigated by Vande Walle et al. showing that knock out of A20, a rheumatoid arthritis susceptibility gene, in mice led to increased expression of NLRP3 and pro-IL-1β genes and resulted in induction of NLRP3 inflammasome-mediated caspase-1 activation, pyroptosis, and IL-1β secretion [91].
Furthermore, deletion of NLRP3, caspase-1 and the interleukin-1 receptor markedly protected against rheumatoid-arthritis-associated inflammation and cartilage destruction in A20myel-KO mice and the authors depicted A20 as a novel negative regulator of NLRP3 inflammasome activation in rheumatoid arthritis [91]. Patients with active rheumatoid arthritis have higher intracellular levels of NLRP3 inflammasome components (including NLRP3, ASC, active caspase-1, and pro-IL-1β) as well as increased secretion of IL-1β [92] and monocytes from patients with rheumatoid arthritis show increased IL-1β production mediated by activation of NLRP3 inflammasome [93]. Shin et al. investigated the role of human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSCs) as a treatment for rheumatoid arthritis in mice with collagen-induced arthritis (CIA) and observed a reduced severity of CIA mediated by a downregulation of the NRLP3 inflammasome [94].
While a huge part of current and past research focused on NLRP3 inflammasomes, it could be shown that the G allele of a polymorphism (rs878329) in the NLRP1 promoter in the Chinese population up-regulates gene transcription and puts patients at risk for developing rheumatoid arthritis [95]. Treatment of CIA mice with BVT-2733, a selective inhibitor of 11β-hydroxysteroid dehydrogenase 1, attenuated arthritis severity by inhibition of the NF-κB and NLRP1 inflammasome signaling pathways [96]. Investigation of treatment with P2X4 antisense oligonucleotide (asODN) in the same CIA model indicated significantly reduced synovial inflammation and joint destruction by inhibition of NRLP1 inflammasome as the underlying mechanism [97].
Recently, mutations in the NLRP1 gene were shown to cause a novel autoinflammatory disorder that the authors proposed to call NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis) which causes arthritis and dyskeratosis [98]. Unfortunately, there are only a few studies analyzing the importance of inflammasomes in the pathogenesis of osteoporosis. IL-18BP, the natural antagonist of proinflammatory IL-18, was shown to be reduced in osteoporotic women [99]. Animal experiments from the same group showed that mIL-18BPd enhances osteoblast differentiation and inhibits the activation of NLRP3 inflammasome and caspase-1 in vitro [99]. In vivo mIL-18BPd treatment restored trabecular microarchitecture, preserved cortical bone parameters and reduced osteoclastogenesis [99].
Xu et al. investigated melatonin treatment in ovariectomized C57BL/6J mice and demonstrated that melatonin improved osteoporosis and impaired osteogenic differentiation potential by suppressing activation of the NLRP3 inflammasome via mediating the wingless-related integration site (Wnt)/β-catenin pathway [100]. Humanized mice carrying an NLRP3 mutation (D305N/D305N mice) developed arthritis and osteoporosis shown by increased radiolucency and thinner cortices in all bones of the lower hindlimb compared to control animals [101]. Kim et al. investigated auranofin, a gold-based compound approved in 1975 for the treatment of rheumatic diseases and found that auranofin suppresses inflammasome mediated IL-1β secretion in mouse bone marrow-derived macrophages (BMDMs) and J774.A1 cells [102]. Furthermore, administration of auranofin in ovariectomized mice led to recovery of bone mass [102].
In in vitro studies with human mesenchymal stem cells (MSCs), activation of NLRP3 inflammasome by lipopolysaccharide and palmitic acid (LPS/PA) treatment led to increased adipogenesis of MSCs and suppressed osteogenesis [103]. The role of inflammasomes in the pathogenesis of age-related diseases especially of the eyes (e.g., glaucoma or age-related macula degeneration) has been extensively studied in the past years and is reviewed profoundly elsewhere. In a mouse model of acute glaucoma the role of HMGB1 has been investigated and it was shown that HMGB1 activates the canonical NLRP3 and non-canonical caspase-8 inflammasomes and production of IL-1β during acute glaucoma development [104]. In a previous study by the same group was shown that inhibition of caspase-8 activation significantly attenuates retinal ganglion cell death by down-regulating the activation of NLRP1 and NLRP3 [105].
Age-related macular degeneration (AMD) is the leading cause of central vision loss worldwide [106] and huge progress has been made in the last decades to understand the role of inflammasomes in the pathogenesis of the disease. Doyle et al. showed that drusen, which are the major pathological hallmark of AMD, isolated from donor AMD eyes activate the NLRP3 inflammasome leading to secretion of IL-1β and IL-18 [107]. Interestingly in a mouse model of wet AMD in NLRP3(−/−) mice laser-induced choroidal neovascularization (CNV) was exacerbated so the authors concluded that NLRP3 and IL-18 might have a protective role in the progression of AMD [107]. The latest findings with regard to the connection between inflammasomes and AMD are thoroughly reviewed elsewhere [108,109]. Trauma is one of the leading causes for death worldwide and although it is indisputable that trauma-injury is closely associated with inflammasomes, there is no clear hypothesis whether the activation of inflammasomes is harmful or beneficial after trauma.
In an ex vivo in vitro experiment with LPS stimulation of CD14+-isolated monocytes from trauma patients (TP), gene expression of NLRP1 was markedly reduced compared to healthy controls [110]. Furthermore, transfected monocytes from TP, which expressed the lacking components, were recovered in their LPS-induced IL-1β release, and thus, the authors concluded that lacking NLRP1 is responsible for the suppressed monocyte activity after trauma [110]. NLRP1 has been shown to be an important component of the innate central nervous system inflammatory response after traumatic brain injury (TBI) as its neutralization reduced the innate immune response and improved histopathology after TBI in a mouse model [111]. Furthermore, the NLRP1 inflammasome was found to cause lung injury in a mouse model while lung damage was rather caused by pyroptosis of resident lung macrophages and not by caspase-1 or IL-1β [112]. NLRP1−/− mice were protected from these detrimental effects, indicating the pivotal role of NLRP1 in lung injury [112].
Recent investigations regarding inflammasome proteins as potential biomarker for TBI determined that apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) in serum and cerebral spinal fluid (CSF) as well as IL-18 in CSF are promising biomarkers of TBI pathology [113]. Moreover, higher protein levels of ASC were consistent with poorer outcomes after TBI [113]. Zhang et al. reported that genetic variations in the NLRP3-gene predict the development of sepsis and multi organ dysfunction syndrome (MODS) in trauma patients [114]. Continuous injury caused by mechanical ventilation, which is common in severely injured trauma patients, is supposed to be mediated by an increase in serum levels of DAMP (e.g., ATP or reactive oxygen species (ROS)), followed by activation of the NLRP3 inflammasome [115,116,117]. To investigate the underlying mechanism of why up to 30% of patients with TBI develop acute lung injury (ALI) or acute respiratory distress syndrome (ARDS), Kerr et al. studied extracellular vesicle (EV)-mediated inflammasome signaling in male C57BL/6 mice [118].
TBI leads to the release of EVs containing inflammasome proteins into serum that target the lung to cause ALI and administration of a blocker of EV uptake (enoxaparin) or monoclonal antibody against ASC improved ALI scores, thus, the authors concluded that neural-respiratory-inflammasome axis is an important part of the innate inflammatory response in lung pathology after TBI [118]. In an animal model of TBI, resveratrol was indicated to attenuate the inflammatory response and relieve TBI by reducing ROS production and inhibiting NLRP3 activation [119]. In burn-injured mice blocking of caspase-1, the downstream effectors of inflammasomes, caused significantly higher mortality, thus, Osuka et al. concluded that inflammasome activation plays a protective role in the host response to severe injury [120]. In contrast, treatment with MCC950, an inhibitor of the NLRP3 inflammasome, led to a better neurological outcome after TBI by alleviating brain edema, reducing lesion volume, and improving long-term motor and cognitive functions in a mouse model with TBI [121].
Inhibition of the NLRP3 inflammasome by treatment with BAY 11-7082 or A438079 alleviated the severity of spinal cord damage and improved neurological recovery after in a mouse model of spinal cord injury [122]. Another recent study showed protective effects in cholestatic liver injury and liver fibrosis by blocking NLRP3 inflammasome activation by treatment with MCC950 [123]. As these are still preclinical studies the value for clinical treatment has to be investigated intensively. In a rat model of subarachnoid hemorrhage minocycline protected against NLRP3 inflammasome-induced inflammation and p53-associated apoptosis, and therefore, the authors concluded that treatment with minocycline treatment may exhibit important clinical potentials in the management of subarachnoid hemorrhage [124]. Denes et al. investigated the role of NLRC4 and AIM2 in a rodent model of stroke and showed that that ischemic brain injury has been reduced in ASC−/− and NLRC4−/− mice without seeing such protective effects, in mice deficient for NLRP3 [125]. Although huge advances have already been made in recent decades, the specific role of the inflammasome in the development of several diseases and therapeutic options still has to be investigated intensively in future.
As the biological activities of both IL-1α and IL-1β are rather similar, this review will refer to them as IL-1 in the following paragraph. Models with IL-1 deficient mice displayed no difference to control mice in terms of growth, homeostasis or fertility, however, they were rather prone to bacterial, mycotic and protozoa infections [21,126]. The ability of IL-1 to stimulate synthesis of inducible nitric oxide synthase (iNOS), Cyclooxygenase (COX)-2 and phospholipase (PL)A2 results in enhanced production of nitric oxide (NO), platelet activating factor as well as prostaglandin (PG)E2 in ex vivo in vitro analyses of chondrocytes from patients with osteoarthritis [127]. Accordingly, the patients in an inflammatory state experienced vasodilation and hypotension, fever and heightened pain sensitivity [128]. To raise systemic dissemination and infiltration of immune cells, chemokine production is upregulated as well as expression of ICAM-1 and VCAM-1 in mesenchymal stem cell models in vitro [129].
Another trait is the augmented permeability of the intestinal barrier and a blood-brain barrier to simplify neutrophil recruitment in these compartments as observed in in vitro models with Caco2-monolayers [130]. A mice model with LPS challenge revealed the angiogenic potential of IL-1 to contribute to blood vessel formation under hypoxic conditions [131]. However, modulation of lymphocytic response like B-cell proliferation is strictly seen as an effect of IL-6 that is inducted by IL-1 as seen in an animal model with either IL-6 or IL-1 knockout mice [132]. In fact, 1 ng/kg bodyweight IL-1 is enough to ensure high systemic levels of IL-6 in mice [21]. Thus, this stresses the important role of IL-1 in disease rather than in healthy individuals. The diagnostic value of IL-1 is rather limited due to its half-life of around 10 min [133]. However, some clinical studies determined IL-1 levels in sera via ELISA. One of those is a work by a Turkish group in neonatal sepsis (n = 50) that revealed significantly enhanced levels of IL-1 in septic patients [134]. Another study displayed significant levels of IL-1 in sera of malaria patients compared to control in a cohort of 60 patients [135]. Interestingly, IL1-Ra was not only significantly increased in patients with septic shock on admission day and day 28 but was also a predictor of mortality [136].
Emerging evidence highlights the role of these inflammatory cytokines in the regulation of bone homeostasis. Chronic inflammation is often characterized by an imbalance between bone formation and bone resorption. Here, a net prevalence of osteoclastogenesis has been described, which is an important determinant of bone loss in rheumatic diseases [137]. Yet, the totality of evidence is limited and provides no clear indication of which cytokine is the most important for bone biology. The link between osteoclasts and pro-inflammatory cytokines, especially IL-1, provides an explanation for the association between inflammation and osteoporosis. For inflammatory diseases, bisphosphonates may be chosen as therapy, however specific medications such as denosumab, IL-1 receptor antagonists, or TNF-α antibodies are targeted treatment strategies for osteoporosis secondary to inflammation [138].
In a meta-analysis to examine the efficacy and safety of denosumab in postmenopausal women with osteoporosis by Gu et al., adverse events between verum and placebo group were similar (pooled odds ratio = 1.04, p = 0.625) [139]. In a large cohort study Choi et al. observed comparable clinical safety and effectiveness with regard to the risk of serious infection, cardiovascular disease, and osteoporosis fracture within 365 days after initiation of medications between denosumab and zolendronic acid (an established standard of therapy) [140]. Kullenberg et al. investigated the safety of treatment with anakinra, a IL-1 receptor antagonist, in 43 patients for up to five years and observed 24 serious AEs (SAEs), all of which resolved during the study period, in 14 patients with the most common SAEs being infections such as pneumonia and gastroenteritis [141]. The authors concluded treatment with anakinra of patients with severe CAPS for up to 5 years was safe and well tolerated, both in pediatric and adult patients [141]. To assess the safety of treatment with TNF-α antibodies is difficult due to the many different antibodies which are authorized for treatment of e.g., rheumatoid arthritis. Hernández et al. observed a reasonable safety profile for TNF-α antibodies and argue that the benefits far outweigh the possible risk of adverse events [142]. There are several studies and meta-analysis dealing with these TNF-α antibodies and which are extensively reviewed elsewhere.
Interleukin-33 is the newest member of the IL-1 family and located on chromosome 9 [143]. Human IL-33 is located in the cell nucleus but is also found outside the cell as an alarmin [144]. Furthermore, it is synthesized as a 31 kDa protein [143]. The main sources of IL-33 are non-hematopoietic cells such as endothelial and epithelial cells [145]. IL-33 is a ligand to orphan receptor ST2 [146] of the TLR/IL1R superfamily of receptors, thereby potentially activating canonical NF-κB pathway via MyD88 [147]. Nevertheless, it was first described as a nuclear factor from high endothelial venules (NF-HEV) [148]. The name accurately describes its properties being both, a membrane receptor ligand and a nuclear factor for transcription [144]. Furthermore, distinct to other members of the IL-1 family such as IL-1β or IL-18, n-terminal end of IL-33 does not necessitate processing to be active [143]. Nonetheless, IL-33 lacks a secretory sequence for conventional pathways to be secreted into extracellular space [149]. One would suspect necrosis as a primary form causing its release, however, in vivo and in vitro models indicate that living cells secrete IL-33 as well [150]. Recent research of inflammation models like post-viral mice with chronic lung disease and in patients with chronic lung disease indicate that extracellular ATP may play a role in IL-33 expression [151]. Ex vivo analysis of airway basal cells of the mice revealed significant IL-33 secretion upon ATP exposure [151].
Its biological impact is associated with the type 2 immune response, mainly reliant on the activation of Th2 cells, eosinophils, mast cells, basophils and group 2 innate lymphoid cells (ILC-2) [143,149]. These cell populations express ST2 and show the importance of IL-33 in allergic and autoimmune disease [145,152]. For instance, chronic exposure of cigarette smoke to mice leads to enhanced systemic IL-33 levels [153]. Additionally, IL-33 skews T-cells toward Th2 differentiation, and high concentrations of this cytokine act as a chemoattractant for Th2 cells [154,155]. There is growing evidence that cells such as Th1, neutrophils, macrophages and natural killer cells (NK) express little ST2 in physiological conditions [156]. Yet, after priming with IL-12 in case of infection, the expression of the ST2 receptor, and thus susceptibility for IL-33 is highly increased [143,157]. This is emphasized by the ability of IL-33 to induce IFN-γ protein expression by aforementioned cells [158]. Subsequently, this mechanism is protective for the host as Bonilla et al. showed that IL-33 is needed for antiviral responses of CD8 cells in mice [159].
As briefly described above, there are several cytokines involved in the pathogenesis of osteoporosis. Yet, the involvement of IL-33 in osteoporotic patients has been studied well. Recently, IL-33 levels in the serum of 50 postmenopausal osteoporotic patients and 28 healthy postmenopausal control women were measured [160]. In postmenopausal osteoporotic women IL-33 was lower compared to controls and positively correlated respectively with serum levels of parathyroid hormone, while an inverse correlation was observed between IL-33 and C-terminal telopeptide of type 1 collagen levels. The authors suggest that IL-33 may represent an important bone-protecting cytokine which may hide therapeutic benefits for treating bone resorption.
2.3. Tumor Necrosis Factor-Alpha
Tumor necrosis factor-alpha was first described in 1975 by Carswell et al. for its cytotoxic activity to tumor cells via immune cells and thus was named TNF [161]. It is expressed as a type II transmembrane protein (mbTNFα) but can be cut to its soluble form (sTNFα) with increased biological activity [162]. The enzyme responsible for its cutting is TNF converting enzyme (TACE) or ADAM17 [163]. The membrane-bound mbTNFα has a 233 amino acid sequence, weighs 26 kDa and forms homotrimers [164]. This mbTNFα complex is cut to 51 kDa by TACE [165]. The main supply of TNF-α are macrophages and T-cells, yet many other cells such as B-cells, neutrophils, and endothelial cells have been described to produce TNF-α [165]. Targets for TNF-α include two type I transmembrane receptors, TNF receptor I (TNFR-I or CD120a) and TNF receptor II (TNFR-II or CD120b) [166]. Whereas TFNR-I is expressed on every cell except erythrocytes, TNFR-II is found only on endothelial and immune cells and can be activated by mbTNF [167].
The functional relevance is broad, and one prominent trait is the mediation of cell survival and pro-inflammatory response by TNFR-I via NF-κB and activator protein (AP)-1 [168]. Additionally, TNF-α instigates signaling pathways of cell death via Fas and Caspases [162,167]. For instance, this was demonstrated in in vitro ex vivo analyses of hematopoietic stem and progenitor cells of TNF-α knockout mice [169]. In a clinical study including 34 patients with at least 20% of total burn surface area, it was shown that systemic TNF levels correlated with burn severity and predicted a susceptibility to infection [170]. Just like in the case of IL-1, the determination of TNF-α levels can be very tricky because of a half-life of only 14 min [133]. Therefore, fast acquisition of blood samples to quantify TNF-α is imperative to use it as a potential biomarker. A German group reported significant levels of TNF-α and sTNF-α in blood samples taken 4, 12, and 24 h after admission to hospital as compared to control in patients with traumatic injury (n = 47) [171].
Adjacent to triggering the release of acute phase proteins after trauma, burns or infarction inter alia, TNF-α can initiate blood clotting [172]. Clinically, this can lead to a disseminated intravascular coagulation in case of severe inflammation like sepsis, cutting vital organs from blood perfusion, and thus driving them to failure [165,167]. To ensure necessary infiltration of immune cells to the local site of inflammation, e.g., in case of traumatic injury, vasodilation is essential [173]. Potent vasodilators are NO and prostaglandins like prostaglandin (PG)I2 or PGE2, which can be induced by TNF-α via iNOS and COX-2 upregulation [174]. In addition, expressions of adhesion molecules like E-selectin or ICAM-1 are upregulated by TNF-α aiding extravasation of monocytes and neutrophils in an endothelial cell model [175]. Yet, to effectively abolish bacterial infection, PMNLs use ROS as a means to destruct pathogens [176]. The essential protein for production of ROS is the nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase [176]. In an endothelial cell model, TNF-α was shown to be a potent inductor of NADPH oxidase (NOX) proteins gp91phox, p22phox and p67phox that are needed for NADPH oxidase activation [177].
In the late eighties, the first in vivo studies with TNF-α antagonists were carried out showing promising results. In 1985, Beutler et al. showed that mice treated with anti-TNF-α serum had higher survival rates after LPS administration compared to control group mice [178]. Shortly after, Tracey et al. infused female baboons with anti-TNF-α antibodies, and injected them with a lethal dose of Escherichia coli (LD100) [173]. Baboons with antibodies were protected against shock, vital organ dysfunction, persistent stress hormone release and death as compared to control animals [173]. Yet, other studies with TNF-α knockout mice have provided evidence that, while animals may be protected against shock, they were far more susceptible to bacterial challenge [179]. Subsequent clinical studies with septic and shock patients have uncovered that there was no significant benefit for critically ill and septic patients treated with experimental TNF-α and sTNF-α antagonists [180]. It has to be considered that the used substances were not the known modern TNF-α antagonists. However, TNF-α antagonists such as etanercept, infliximab, or adalimumab proved to be a highly effective treatment for auto-inflammatory diseases like psoriasis, Crohn’s disease, or rheumatoid arthritis [168].
The role of the immune system in the onset of osteoporosis, a serious worldwide public health concern, is an area of current research. In a panel including 10 cytokines obtained from postmenopausal women, with either normal or low bone mineral density IL-23, IL-12, IL-4, IL-6, and also TNF-α levels were the most important differentiating cytokines [181]. However, no significant difference between the osteopenic and osteoporotic groups were found [181]. In general TNF-α suppresses osteoblasts activity at some stages of differentiation and stimulates osteoclast proliferation and differentiation [182]. Similar to IL-6, TNF-α can regulate bone metabolism through the endocrine way [183]. In a retrospective cohort analysis including a total of 199 rheumatoid arthritis patients, who were newly diagnosed with osteoporosis and receiving bisphosphonate changes in bone mineral density after one year were compared between patients treated with and without TNF inhibitors [184]. The therapy did not influence bone mineral density improvement in rheumatoid arthritis patients with osteoporosis receiving bisphosphonate [184]. However, although this data suggested that TNF inhibition cannot be considered as a preferred therapeutic option for increasing bone mineral density, conflictive findings have been reported showing that the use of bisphosphonate might be important to improve bone mineral density in patients with rheumatoid arthritis even under tight control [185]. Recently it was shown that altered T-cell activity and a different composition such as the CD14+CD16+ vs. CD14+CD16- monocytes and priming of osteoclast precursors with increased macrophage colony-stimulating factor (M-CSF), receptor activator of NF-κB ligand (RANKL), and TNF-α levels in peripheral blood play a role in increased osteoclast formation and activity [186]. In summary, these findings may help the development of cytokine therapies for osteoporosis, and propose that the use of certain cytokine profiles as biomarkers for osteoporosis risk factors, may quantify the progress of therapies.
2.4. Interleukin-10
In 1989, IL-10 was first described by Fiorentino et al. as a cytokine synthesis inhibitory factor (CSIF) [187]. It is made up as a homodimer with each unit having a 178 amino acid sequence [188]. Interestingly, IL-10 is one of few anti-inflammatory cytokines next to IL-2, TGF, and the more recently discovered IL-25, IL-35, and IL-37 [189].
Biologically, IL-10 is usually found as a dimer and shares some structural and functional properties of interferon (IFN)-γ [190]. It is produced by almost all leukocytes including macrophages, dendritic cells, neutrophils, NK cells, B-cells, and CD8+ T-cells, however, CD4+ T-cells are the major producers [19,191]. For instance, FoxP3+ regulatory CD4+ T-cells (Tregs, thymus, and periphery-derived) and Foxp3- regulatory CD4+ T-cells (Tr1 cells) attenuate T-cells and Th17 cell response in particular via IL-10 [19,192]. This review, however, will focus on the contribution of the cells of innate immunity.
Interestingly, some viruses like Epstein–Barr or Human Cytomegalovirus among others produce IL-10 homologs, which are almost identical to human IL-10 [193]. The receptor responsible for downstream signaling of IL-10 is the IL-10 receptor (IL10R), which is made up of dimers IL10R1 and IL10R2 [194]. The former is an IL-10 specific receptor and the primary binding site for IL-10, while the latter enhances the affinity of IL-10 to bind to IL10R1 [194]. In fact, IL10R2 cannot associate with IL-10 independently, and is expressed on many tissue cells [195], while IL10R1 is mostly expressed on immune cells such as T-cells [192], neutrophils upon LPS administration in vitro [196] or monocytes in a LPS endotoxemia mouse model [197]. Nevertheless, IL10R2 is a co-ligand to many other molecules like IL-22, IL-26, or IL-29, thereby playing a role in various biological pathways [193,198].
Downstream mediators of the IL-10 receptor are mainly STAT molecules and Janus Kinases (JAK) [199]. As a matter of fact, IL-10R1 is associated with JAK1 and IL10R2 with Tyk2 [19,200]. After activation by JAK, STAT dimer molecules like STAT1 and STAT5 in cytoplasm undergo a conformational switch, relocate to the nucleus and bind to DNA as transcription factors to IL10-responsive genes [201,202]. The number of genes regulated by IL-10 is numbering up to thousands with new genes discovered each year [19,203].
The biological effects of IL-10 on innate immune cells suppress the release of immune mediators, antigen expression and phagocytosis [193]. Indeed, in vitro inflammation models show that IL-10 prevents PMNL activation and TNF-α as well as IL-8 release after LPS administration [204]. Studies with human umbilical vein epithelial cells (HUVEC) display that IL-10 attenuates TNF-α induced ROS production, ICAM-1 expression, and leukocyte adhesion to HUVEC [205]. Recent research of monocyte models in vitro designate upregulation of ubiquitin ligases by IL-10 as the mechanism to sequestrate major histocompatibility complex (MHC) complexes and thus inhibit antigen presentation by antigen presenting cells (APCs) [206]. The overall influence on chemotaxis of monocytes is, however, rather low [207]. What is more, IL-10 knockout mice suffer from cardiac and vascular dysfunction due to an upsurge of COX-2 activity and production of prostaglandins indicating an important role in the suppression of COX by IL-10 [208]. Additionally, phagocytic cells are protected against complement lysis infused by an anti-MHC antibody or binding of zymosan when administered IL-10 compared to control cells in vitro [209]. Nevertheless, IL-10 plays an important role in suppressing inflammation in mucosa cells evident by IL-10 knockout mice that will develop severe colitis [210]. To limit its own properties, IL-10R activation also triggers the transcription of suppressor of cytokine signaling (SOCS)3, thus limiting its own release [211]. Albeit, systemic levels of IL-10 were significantly increased in patients with severe sepsis and linked to mortality as compared to patients with moderate sepsis [212]. A smaller study comparing 16 septic shock patients with 11 shock patients without sepsis supports the predictive value of systemic IL-10 levels in the first days after admission [213]. In case of trauma, a Swiss study reported elevated systemic IL-10 levels in patients with injury (n = 417) and a correlation of IL-10 levels to the severity of the injury as compared to healthy volunteers [214]. The development of sepsis in trauma patients was also linked to elevated systemic IL-10 levels on admission in an American study (n = 66) [215]. With regard to bone biology, loss of IL-10 exacerbated early Type-1 diabetes-induced bone loss [216]. Serum IL-10 levels in systemic lupus erythematosus patients with osteonecrosis were higher than that in those without osteonecrosis [217].
2.5. Interleukin-8
Interleukin-8 was first observed for its trait as a chemoattractant for granulocytes, primarily neutrophils in vitro [218]. It is sometimes called chemokine (C-X-C motif) ligand 8 or CXCL-8 and encoded by the CXCL-8 gene [219]. Through transfected cell culture models, NF-κB and JNK, as well as AP-1, have been identified as vital pathways for inducible IL-8 expression [220]. Every cell with TLR can produce and secrete IL-8 including macrophages and smooth muscle cells [221], while endothelial cells accumulate IL-8 in vesicles known as Weibel–Palade bodies [222]. Indeed, IL-8 is translated as a 99 amino acid long precursor peptide and cleaved into two active isoforms; one being 77 amino acids long and secreted by endothelial cells in cell culture [223]. While the other has a 72 amino acid sequence and is produced by monocytes and other leukocytes [224].
The main targets for IL-8 are G-protein coupled receptors CXCR1 and CXCR2, though the latter has a weaker affinity for IL-8 [225]. Furthermore, IL-8 guides neutrophils to the direction of inflammation (chemotaxis), which is evident in increased concentrations of this cytokine in lungs of patients with ARDS [226]. However, high IL-8 levels are not correlated with the probability of development of ARDS [227]. Additionally, IL-8 does not activate NAPDH oxidase directly with in vitro, yet, it enhances the respiratory burst activity by the recruitment of NAPDH oxidase components, N-formyl-methionyl-leucyl-phenylalanine (fMLP) receptor, and P-selectin ligands into lipid drafts [228]. In vitro studies with colon cancer cells transfected with IL-8 cDNA displayed a significant rise in cell proliferation, migration, and invasion by these cells [229]. This is supported by earlier in vitro investigations with HUVEC where recombinant IL-8 induced endothelial cell proliferation and capillary tube organization [230]. In conclusion, IL-8 is a very potent trigger to cell migration and proliferation, and thus should always be considered in inflammation models. A study analyzing systemic IL-8 levels for 60 days after a burn injury in children (n = 468) provided interesting insights [231]. The IL-8 levels correlated with the percent of burned total body surface area and were predictive for multiple organ failure and mortality [231].
It has to be considered that systemic IL-8 levels do not only provide prognostic value by reading the absolute levels, but rather implicate the duration of sustained high IL-8 levels for diagnosis. For instance, in a study with 27 patients, those with severe sepsis (n = 17) presented high IL-8 plasma levels steadily for 24 h after admission, whereas those with uncomplicated sepsis (n = 10) did not [232]. Furthermore, a smaller study with 24 subjects with traumatic brain injury (TBI) linked elevated systemic IL-8 levels upon admission to the worsened outcome [233]. This was first reported in a similar Croatian study with 20 TBI patients, with elevated IL-8 plasma levels in the non-survivor group [234].
Postmenopausal osteoporosis is characterized by rapid bone loss and IL-8 has been implicated among other pro-inflammatory cytokines to play a role in bone remodeling. There was a significant IL-8 increase in post-menopausal women with osteoporosis and bone loss [235]. Atorvastatin, which is known for its pleiotropic effects on bone tissue, decreased IL-8 levels and bone loss of rats subjected to glucocorticoid-induced osteoporosis [236]. RANKL-expressing neutrophils are increased in male patients with Chronic obstructive pulmonary disease (COPD), and furthermore, associated with bone mineral density and lung function, suggesting that these cells play a role in osteoclastogenesis in COPD [237]. Plasma levels of IL-8 were increased in COPD patients and correlated with RANKL expression by neutrophils [237].
2.6. Limitations
This review only provides a short overview of selected cytokines that are important for inflammatory reactions of the body. However, one should note that hundreds of other cytokines, hormones, and proteins mediate the immune dysregulation as seen in many patients with inflammatory states like sepsis or after trauma. A more comprehensive overview about immune dysregulation in shock/sepsis is given by Angus and van der Poll, as well as Rittirsch et al. [238,239]. Also, large randomized controlled trials or meta-analysis for clinical value of the mentioned cytokines are still missing. The overall data is still too weak to give a definite clinical evaluation.
3. Materials and Methods
For this review, existing literature was screened through Pubmed and Google Scholar. After careful consideration, cytokines IL-6, IL-1, IL-33, TNF-α, IL-10, and IL-8 were chosen as focus points for further research. We used respective names of the mentioned cytokines followed by terms “shock”, “inflammation”, “trauma”, “severe injury”, “immune dysregulation”, “osteoporosis”, “inflammasome”, and “critical illness” in various combinations. For each cytokine, we briefly described the biology by using the search engines described further above. Then, a selection of clinical studies for value as biomarkers is presented. We did not limit the time frame although more recent studies were favored.
4. Conclusions
New insights into molecular mechanisms provide a new perspective for finding appropriate biomarkers that may be helpful to predict severe or complicated cases upon clinical presentation. The constant discovery of, not only cytokines but other proteins, as well as receptors will pave the way for future diagnostics. The aforementioned cytokines are among the best studied. However, one should note that except for IL-6, none has really found its way from bench to bedside. Interestingly, they may be promising in the experimental environment, but further clinical research is needed.

{kind=link}
Table 1.
Brief overview of cytokines that are involved in osteoporosis. ↑: upregulation.
| Species | Study | Cell Type/Organ | Major Finding | Reference |
|---|---|---|---|---|
| IL (Interleukin)-6 | ||||
| human | in vivo | bone | IL-6 is a predictor of postmenopausal bone loss | [240] |
| meta-analysis | bone | GG genotype of IL-6-634C/G polymorphism seems to play a role in reducing bone mineral density | [47] | |
| meta-analysis | bone | IL-6-634C/G and IL-6-174G/C polymorphisms lead to modest effects on bone mineral density | [48] | |
| meta-analysis | bone | CC genotype of IL-6 G-174C polymorphism may be associated with high bone mineral density at femoral neck and distal radius and decreased risk of osteoporosis in the Caucasian population | [49] | |
| in vivo | bone | IL-6 G-174C promoter polymorphism may be a genetic marker for bone loss and wrist fracture among older women | [241] | |
| meta-analysis | bone | IL6-174 G/C gene polymorphism positively correlated with osteoporosis risk | [242,243] | |
| in vivo | bone | Variation within the low levels of IL-6 predicts bone loss and resorption | [244] | |
| in vitro | whole blood cells | Increased IL-6 production by whole blood cells from postmenopausal women with osteoporosis compared to controls | [245] | |
| in vivo | bone | IL-6 is upregulated in postmenopausal women with low bone mineral density compared to postmenopausal women with normal bone mineral density | [181] | |
| Chronic obstructive pulmonary disease (COPD) patients | bone | RANKL (Receptor activator of NF-κB ligand)-expressing neutrophils correlate negatively with bone marrow density. Plasma levels of IL-6 are increased in COPD patients and correlate with RANKL expression by neutrophils | [237] | |
| mouse | in vivo | osteoclasts | IL-6 mediates stimulation of osteoclastogenesis after estrogen loss | [246] |
| IL-1 | ||||
| human | in vitro | whole blood cells | Increased IL-1 beta production by whole blood cells from postmenopausal women with osteoporosis compared to controls | [245] |
| COPD patients | bone | RANKL-expressing neutrophils correlate negatively with bone marrow density. Plasma levels of IL-1 beta are increased in COPD patients and correlate with RANKL expression by neutrophils | [237] | |
| in vivo | bone | IL-1β (-511C/T) polymorphism is associated with pathogenesis of osteoporosis in postmenopausal women | [247] | |
| in vivo | bone | Serum IL-1β significantly higher in women with osteoporosis than controls | [248] | |
| in vivo | bone | Serum IL-1 is significantly reduced after treatment of postmenopausal with calcitriol | [249] | |
| in vitro | osteoblasts | IL-1beta and Tumor necrosis factor-alpha (TNF-α) regulate osteoblast cell number by up-regulating the Fas-mediated apoptosis of osteoblasts | [250] | |
| rat | in vivo | bone | Interleukin-1 receptor antagonist decreases bone loss and bone resorption in a rat model of postmenopausal osteoporosis | [251] |
| IL-33 | ||||
| human | in vivo | bone | IL-33 levels in postmenopausal women significantly lower compared to healthy controls, positively correlated with serum parathyroid hormone and inverse correlated with C-terminal telopeptide of type 1 collagen | [160] |
| TNF-α | ||||
| human | in vitro | whole blood cells | Increased TNF-α production by whole blood cells from postmenopausal women with osteoporosis compared to controls | [245] |
| in vivo | bone | TNF-α is upregulated in postmenopausal women with low bone mineral density compared to postmenopausal women with normal bone mineral density | [181] | |
| in vivo + in vitro | osteoclasts | Estrogen deficiency → TNF-α and RANKL ↑ → osteoclast formation and number of osteoclast precursors ↑ | [252] | |
| in vivo | bone | Association between TNF-α-308G>A polymorphism and postmenopausal osteoporosis | [253] | |
| in vivo | bone | Serum TNF-α is significantly reduced after treatment of postmenopausal with calcitriol | [249] | |
| in vivo in vitro | TNF-α increased in postmenopausal women with osteoporosis and highly correlated with the RANK and estrogen levels TNF-α synergistically promotes RANKL-induced osteoclasts formation through activation of Phosphoinositide 3-kinases (PI3K)/Akt signaling | [254] | ||
| in vitro | mesenchymal stem cells (MSC) | TNF-α suppresses osteogenic differentiation of MSCs by accelerating P2Y2 receptor in estrogen-deficiency induced osteoporosis | [255] | |
| in vitro | osteoblasts | IL-1beta and TNF-α regulate osteoblast cell number by up-regulating the Fas-mediated apoptosis of osteoblasts | [250] | |
| mouse | in vivo | bone marrow-derived mesenchymal stem cells (BMMSCs) | TNF-α inhibits Forkhead box protein O1 (FoxO1) and thereby aggravates oxidative damage in BMMSCs during osteoporosis | [256] |
| rat | in vitro | bone cultures of fetal rat calvariae | TNF-α causes osteoclastic bone resorption and inhibits bone collagen synthesis | [257] |
| IL-10 | ||||
| human | patients with systemic lupus erythematosus | bone | IL-10 level is elevated in systemic lupus erythematosus patients with osteoporosis | [217] |
| in vivo | bone | IL-10 gene-597 C>A polymorphism is associated with higher risk for osteoporosis | [258] | |
| in vivo | bone | Lower levels of IL-10 in postmenopausal women with osteoporosis | [259,260] | |
| in vivo | bone | Association between IL10-1082G>A polymorphism and postmenopausal osteoporosis | [253] | |
| mouse | in vitro | RAW264.7 monocytes | IL-10 directly inhibits osteoclastogenesis is by suppressing Nuclear factor of activated T-cells, cytoplasmic 1 activity | [261] |
| in vivo | bone | IL-10 is important for promoting osteoblast maturation and reducing bone loss during early stages of type-1 diabetes | [216] | |
| in vivo | bone | IL-10−/− mice develop the reduced bone mass, increased mechanical fragility, and suppressed bone formation (hallmarks of osteoporosis) | [262] | |
| IL-8 | ||||
| human | in vivo | bone | IL-8 significantly increase in post-menopausal women with osteoporosis and bone loss | [235] |
| COPD patients | bone | RANKL-expressing neutrophils correlate negatively with bone marrow density, plasma levels of IL-8 are increased in COPD patients and correlate with RANKL expression by neutrophils | [237] | |
| in vitro | osteoblasts | IL-8 may contribute to osteoporosis in rheumatoid arthritis by enhanced osteoblast-mediated osteoclastogenesis (partly via IL-6 production) | [263] | |
| rats | in vivo | bone | IL-8 and bone loss reduction after atorvastatin treatment of rats with glucocorticoid-induced osteoporosis | [236] |
Author Contributions
Conceptualization, methodology, S.K. and B.R.; writing—original draft preparation, S.K.; writing—review and editing, J.T.V. and B.R.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ADAM | A disintegrin and metalloproteinase |
| AP | Activator protein |
| APC | Antigen presenting cells |
| ARDS | Acute respiratory distress syndrome |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| ATP | Adenosine triphosphate |
| CARD | Caspase activation and recruitment domain |
| CD | Cluster of differentiation |
| COPD | Chronic obstructive pulmonary disease |
| COX | Cyclooxygenase |
| CRP | C-reactive protein |
| CSIF | Cytokine synthesis inhibitory factor |
| DAMP | Damage-associated molecular patterns |
| DNA | Deoxyribonucleic acid |
| fMLP | Formyl-methionyl-leucyl-phenylalanine |
| FoxO1 | Forkhead box protein O1 |
| gp | Glycoprotein |
| HMGB1 | High-mobility group box 1 |
| HSP | Heat shock proteins |
| ICAM | Intercellular cell adhesion molecules |
| IFN | Interferon |
| IL | Interleukin |
| ILC | Innate lymphoid cells |
| IL-1R | Interleukin-1 receptor |
| iNOS | Inducible nitric oxide synthase |
| kDa | Kilo Dalton |
| LPS | Lipopolysaccharide |
| LRR | Leucine-rich repeat |
| MAP | Mitogen activated protein |
| mbIL6R | membrane-bound IL-6 receptor |
| MBL | Mannose-binding lectin |
| MCP | Monocyte chemotactic protein |
| M-CSF | Macrophage-colony stimulating factor |
| MHC | Major histocompatibility complex |
| MODS | Multi organ dysfunction syndrome |
| MyD88 | Myeloid differentiation primary response 88 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-HEV | Nuclear factor from high endothelial venules |
| NF-κB | nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells |
| NLR | NOD-Like Receptors |
| NLRP | NOD-, LRR- and pyrin domaine-containing protein |
| NO | Nitric oxide |
| NOD | Nucleotide-binding oligomerization domain |
| NPC | Nasopharyngeal carcinoma |
| PAMP | Pathogen-associated molecular patterns |
| PBMC | Peripheral blood mononuclear cells |
| PG | Prostaglandin |
| PGN | Peptidoglycan |
| PI3K | Phosphoinositide 3-kinases |
| PLA | Phospholipase A |
| PMNL | Polymorphonuclear leukocytes |
| PRR | Pattern recognition receptors |
| R, r | Receptor |
| RANKL | Receptor activator of NF-κB ligand |
| ROS | Reactive oxygen species |
| s | Soluble |
| sIL6R | IL-6 receptor |
| STAT | Signal transducer and activator of transcription |
| TACE | TNF converting enzyme |
| TBI | Traumatic brain injury |
| Th | T helper cells |
| TLR | Toll-like receptors |
| TNF-α | Tumor necrosis factor-alpha |
| VCAM | Vascular cell adhesion molecule |
| Wnt | Wingless-related integration site |
References
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef]
- Häcker, G.; Redecke, V.; Häcker, H. Activation of the immune system by bacterial CpG-DNA. Immunology 2002, 105, 245–251. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Seong, S.Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef]
- Frank, M.G.; Adhikary, S.; Sobesky, J.L.; Weber, M.D.; Watkins, L.R.; Maier, S.F. The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 2016, 51, 99–108. [Google Scholar] [CrossRef]
- Zong, M.; Bruton, J.D.; Grundtman, C.; Yang, H.; Li, J.H.; Alexanderson, H.; Palmblad, K.; Andersson, U.; Harris, H.E.; Lundberg, I.E.; et al. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann. Rheum. Dis. 2013, 72, 1390–1399. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 1–18. [Google Scholar] [CrossRef]
- Gay, N.J.; Packman, L.C.; Weldon, M.A.; Barna, J.C. A leucine-rich repeat peptide derived from the Drosophila Toll receptor forms extended filaments with a beta-sheet structure. FEBS Lett. 1991, 291, 87–91. [Google Scholar] [CrossRef]
- Creagh, E.M.; O’Neill, L.A.J. TLRs, NLRs and RLRs: A trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Tang, D.; Shi, Y.; Kang, R.; Li, T.; Xiao, W.; Wang, H.; Xiao, X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 2007, 81, 741–747. [Google Scholar] [CrossRef]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Geginat, J.; Larghi, P.; Paroni, M.; Nizzoli, G.; Penatti, A.; Pagani, M.; Gagliani, N.; Meroni, P.; Abrignani, S.; Flavell, R.A. The light and the dark sides of Interleukin-10 in immune-mediated diseases and cancer. Cytokine Growth Factor Rev. 2016, 30, 87–93. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.N.; Jéru, I.; Lecron, J.C.; Medlej-Hashim, M. Cytokine signatures in hereditary fever syndromes (HFS). Cytokine Growth Factor Rev. 2017, 33, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Rincón, M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002, 39, 531–536. [Google Scholar] [CrossRef]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.I.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef]
- Hibi, M.; Murakami, M.; Saito, M.; Hirano, T.; Taga, T.; Kishimoto, T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 1990, 63, 1149–1157. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Radtke, S.; Wüller, S.; Yang, X.P.; Lippok, B.E.; Mütze, B.; Mais, C.; de Leur, H.S.V.; Bode, J.G.; Gaestel, M.; Heinrich, P.C.; et al. Cross-regulation of cytokine signalling: Pro-inflammatory cytokines restrict IL-6 signalling through receptor internalisation and degradation. J. Cell Sci. 2010, 123, 947–959. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science (N.Y.) 2003, 300, 2101–2104. [Google Scholar] [CrossRef]
- Franchimont, N.; Lambert, C.; Huynen, P.; Ribbens, C.; Relic, B.; Chariot, A.; Bours, V.; Piette, J.; Merville, M.P.; Malaise, M. Interleukin-6 receptor shedding is enhanced by interleukin-1beta and tumor necrosis factor alpha and is partially mediated by tumor necrosis factor alpha-converting enzyme in osteoblast-like cells. Arthritis Rheum. 2005, 52, 84–93. [Google Scholar] [CrossRef]
- Walev, I.; Vollmer, P.; Palmer, M.; Bhakdi, S.; Rose-John, S. Pore-forming toxins trigger shedding of receptors for interleukin 6 and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1996, 93, 7882–7887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lust, J.A.; Donovan, K.A.; Kline, M.P.; Greipp, P.R.; Kyle, R.A.; Maihle, N.J. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4, 96–100. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Moon, Y.P.; Liu, K.M.; Spitalnik, S.; Paik, M.C.; Cheung, K.; Sacco, R.L.; Elkind, M.S.V. High-sensitivity C-reactive protein and interleukin-6-dominant inflammation and ischemic stroke risk: The northern Manhattan study. Stroke 2014, 45, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mroczko, B.; Groblewska, M.; Gryko, M.; Kędra, B.; Szmitkowski, M. Diagnostic usefulness of serum interleukin 6 (IL-6) and C-reactive protein (CRP) in the differentiation between pancreatic cancer and chronic pancreatitis. J. Clin. Lab. Anal. 2010, 24, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Panichi, V.; Maggiore, U.; Taccola, D.; Migliori, M.; Rizza, G.M.; Consani, C.; Bertini, A.; Sposini, S.; Perez-Garcia, R.; Rindi, P.; et al. Interleukin-6 is a stronger predictor of total and cardiovascular mortality than C-reactive protein in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Fisher, D.T.; Clancy, K.A.; Gauguet, J.M.M.; Wang, W.C.; Unger, E.; Rose-John, S.; von Andrian, U.H.; Baumann, H.; Evans, S.S. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat. Immunol. 2006, 7, 1299–1308. [Google Scholar] [CrossRef]
- Jenkins, B.J.; Grail, D.; Inglese, M.; Quilici, C.; Bozinovski, S.; Wong, P.; Ernst, M. Imbalanced gp130-dependent signaling in macrophages alters macrophage colony-stimulating factor responsiveness via regulation of c-fms expression. Mol. Cell. Biol. 2004, 24, 1453–1463. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, R.M.; Jenkins, B.J.; Grail, D.; Williams, A.S.; Fielding, C.A.; Parker, C.R.; Ernst, M.; Topley, N.; Jones, S.A. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9589–9594. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luig, M.; Kluger, M.A.; Goerke, B.; Meyer, M.; Nosko, A.; Yan, I.; Scheller, J.; Mittrucker, H.W.; Rose-John, S.; Stahl, R.A.K.; et al. Inflammation-Induced IL-6 Functions as a Natural Brake on Macrophages and Limits GN. J. Am. Soc. Nephrol. 2015, 26, 1597–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Neuhöfer, P.; Song, L.; Rabe, B.; Lesina, M.; Kurkowski, M.U.; Treiber, M.; Wartmann, T.; Regnér, S.; Thorlacius, H.; et al. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J. Clin. Investig. 2013, 123, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan, M.; Bataille, R.; Seguin, J.; Zhang, X.G.; Chaptal, P.A.; Klein, B. Constitutive production of interleukin-6 and immunologic features in cardiac myxomas. Arthritis Rheum. 1990, 33, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.A.; Taylor, J.M.G.; Terrell, J.E.; Islam, M.; Li, Y.; Fowler, K.E.; Wolf, G.T.; Teknos, T.N. Interleukin-6 predicts recurrence and survival among head and neck cancer patients. Cancer 2008, 113, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Tsang, C.M.; Deng, W.; Yip, Y.L.; Lui, V.W.Y.; Wong, S.C.C.; Cheung, A.L.M.; Hau, P.M.; Zeng, M.; Lung, M.L.; et al. Enhanced IL-6/IL-6R Signaling Promotes Growth and Malignant Properties in EBV-Infected Premalignant and Cancerous Nasopharyngeal Epithelial Cells. PLoS ONE 2013, 8, e62284. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Hu, R.; Tu, S.; Cheng, W.J.; Zheng, Q.; Wang, J.W.; Kan, W.S.; Ren, Y.J. Meta-analysis of association between IL-6 -634C/G polymorphism and osteoporosis. Genet. Mol. Res. 2015, 14, 19225–19232. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Y.; He, M.; Wang, R.; Ma, J.; Zhang, Y.; Zhao, L.; Yu, K. Association between interleukin-6 gene polymorphisms and bone mineral density: A meta-analysis. Genet. Test. Mol. Biomark. 2013, 17, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Li, H.; Zhang, Y.; Zhang, H.; Pan, Y.; Ma, J.; Wang, L. Association of IL-6 G-174C polymorphism with bone mineral density. J. Bone Min. Metab. 2014, 32, 167–173. [Google Scholar] [CrossRef]
- Reinhart, K.; Meisner, M.; Brunkhorst, F.M. Markers for Sepsis Diagnosis: What is Useful? Crit. Care Clin. 2006, 22, 503–519. [Google Scholar] [CrossRef]
- Cruickshank, A.M.; Fraser, W.D.; Burns, H.J.G.; Van Damme, J.; Shenkin, A. Response of Serum Interleukin-6 in Patients Undergoing Elective Surgery of Varying Severity. Clin. Sci. 1990, 79, 161–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, L.F.; Cuenca, A.G.; Vanzant, E.L.; Efron, P.A.; McKinley, B.; Moore, F.; Moldawer, L.L. Is there value in plasma cytokine measurements in patients with severe trauma and sepsis? Methods 2013, 61, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitanovski, L.; Jazbec, J.; Hojker, S.; Gubina, M.; Derganc, M. Diagnostic accuracy of procalcitonin and interleukin-6 values for predicting bacteremia and clinical sepsis in febrile neutropenic children with cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 413–415. [Google Scholar] [CrossRef]
- Haasper, C.; Kalmbach, M.; Dikos, G.D.; Meller, R.; Müller, C.; Krettek, C.; Hildebrand, F.; Frink, M. Prognostic value of procalcitonin (PCT) and/or interleukin-6 (IL-6) plasma levels after multiple trauma for the development of multi organ dysfunction syndrome (MODS) or sepsis. Technol. Health Care 2010, 18, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Pallás Beneyto, L.A.; Rodríguez Luis, O.; Saiz Sánchez, C.; Cotell Simón, O.; Bautista Rentero, D.; Miguel Bayarri, V. Valor pronóstico de la interleucina 6 en la mortalidad de pacientes con sepsis. Med. Clínica 2016, 147, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Goldin, N.P.; Wolff, S.M. Demonstration and characterization of two distinct human leukocytic pyrogens. J. Exp. Med. 1974, 139, 1369–1381. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1α and the inflammatory process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Van de Veerdonk, F.L.; Van der Meer, J.W.M.; Dinarello, C.A.; Joosten, L.A.B. Inflammasome-Independent Regulation of IL-1-Family Cytokines. Annu. Rev. Immunol. 2015, 33, 49–77. [Google Scholar] [CrossRef]
- Bersudsky, M.; Luski, L.; Fishman, D.; White, R.M.; Ziv-Sokolovskaya, N.; Dotan, S.; Rider, P.; Kaplanov, I.; Aychek, T.; Dinarello, C.A.; et al. Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut 2014, 63, 598–609. [Google Scholar] [CrossRef]
- McCarthy, D.A.; Ranganathan, A.; Subbaram, S.; Flaherty, N.L.; Patel, N.; Trebak, M.; Hempel, N.; Melendez, J.A. Redox-control of the alarmin, Interleukin-1α. Redox Biol. 2013, 1, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Lee, Y.; Kim, E.; Kwak, A.; Ryoo, S.; Bae, S.H.; Azam, T.; Kim, S.; Dinarello, C.A. The Interleukin-1α Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Inukai, Y.; Takii, T.; Furutani, Y.; Shibata, Y.; Hayashi, H.; Sakurada, S.; Okamoto, T.; Inoue, J.; Oomoto, Y.; et al. Molecular analysis of constitutive IL-1alpha gene expression in human melanoma cells: Autocrine stimulation through NF-kappaB activation by endogenous IL-1alpha. Cytokine 1998, 10, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Groß, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.X.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome Activators Induce Interleukin-1α Secretion via Distinct Pathways with Differential Requirement for the Protease Function of Caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef] [Green Version]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science (N. Y.) 1995, 267, 2000–2003. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Muzio, M.; Bertini, R.; Polentarutti, N.; Sironi, M.; Giri, J.G.; Dower, S.K.; Sims, J.E.; Mantovani, A. Interleukin-1 type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science (N. Y.) 1993, 261, 472–475. [Google Scholar] [CrossRef]
- Anquetil, F.; Sabouri, S.; Thivolet, C.; Rodriguez-Calvo, T.; Zapardiel-Gonzalo, J.; Amirian, N.; Schneider, D.; Castillo, E.; Lajevardi, Y.; von Herrath, M.G. Alpha cells, the main source of IL-1β in human pancreas. J. Autoimmun. 2017, 81, 68–73. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF- B Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1 Secretion Stimulated by P2X7 Receptors Is Dependent on Inflammasome Activation and Correlated with Exosome Release in Murine Macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattorno, M.; Tassi, S.; Carta, S.; Delfino, L.; Ferlito, F.; Pelagatti, M.A.; D’Osualdo, A.; Buoncompagni, A.; Alpigiani, M.G.; Alessio, M.; et al. Pattern of interleukin-1β secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients withCIAS1 mutations. Arthritis Rheum. 2007, 56, 3138–3148. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Zhen, J.; Zhang, L.; Pan, J.; Ma, S.; Yu, X.; Li, X.; Chen, S.; Du, W. AIM2 Mediates Inflammation-Associated Renal Damage in Hepatitis B Virus-Associated Glomerulonephritis by Regulating Caspase-1, IL-1 β, and IL-18. Mediat. Inflamm. 2014, 2014, 190860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, J.; Prieur, A.M.; Quartier, P.; Berquin, P.; Certain, S.; Cortis, E.; Teillac-Hamel, D.; Fischer, A.; de Saint Basile, G. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 2002, 71, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Aksentijevich, I.; Nowak, M.; Mallah, M.; Chae, J.J.; Watford, W.T.; Hofmann, S.R.; Stein, L.; Russo, R.; Goldsmith, D.; Dent, P.; et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002, 46, 3340–3348. [Google Scholar] [CrossRef] [Green Version]
- Cuisset, L.; Jeru, I.; Dumont, B.; Fabre, A.; Cochet, E.; Le Bozec, J.; Delpech, M.; Amselem, S.; Touitou, I.; French, C.S.G. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: Epidemiological study and lessons from eight years of genetic analysis in France. Ann. Rheum. Dis. 2011, 70, 495–499. [Google Scholar] [CrossRef]
- ter Haar, N.M.; Oswald, M.; Jeyaratnam, J.; Anton, J.; Barron, K.S.; Brogan, P.A.; Cantarini, L.; Galeotti, C.; Grateau, G.; Hentgen, V.; et al. Recommendations for the management of autoinflammatory diseases. Ann. Rheum. Dis. 2015, 74, 1636–1644. [Google Scholar] [CrossRef] [Green Version]
- Landmann, E.C.; Walker, U.A. Pharmacological treatment options for cryopyrin-associated periodic syndromes. Expert Rev. Clin. Pharm. 2017, 10, 855–864. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arter. Thromb/Vasc/Biol/ 2003, 23, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Gijbels, M.J.; Walenbergh, S.M.; Houben, T.; van Gorp, R.; Pottgens, C.C.; Stienstra, R.; Netea, M.G.; et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 2015, 282, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediat. Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef] [Green Version]
- Menu, P.; Pellegrin, M.; Aubert, J.F.; Bouzourene, K.; Tardivel, A.; Mazzolai, L.; Tschopp, J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011, 2, e137. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1beta by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Son, S.J.; Rhee, K.J.; Lim, J.; Kim, T.U.; Kim, T.J.; Kim, Y.S. Triglyceride-induced macrophage cell death is triggered by caspase-1. Biol. Pharm. Bull. 2013, 36, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Fu, R.; Wang, S.; Huang, Y.; Li, X.; Zhou, M.; Zhao, J.; Yang, N. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 2018, 194, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Ippagunta, S.K.; Brand, D.D.; Luo, J.; Boyd, K.L.; Calabrese, C.; Stienstra, R.; Van de Veerdonk, F.L.; Netea, M.G.; Joosten, L.A.; Lamkanfi, M.; et al. Inflammasome-independent role of apoptosis-associated speck-like protein containing a CARD (ASC) in T cell priming is critical for collagen-induced arthritis. J. Biol. Chem. 2010, 285, 12454–12462. [Google Scholar] [CrossRef] [Green Version]
- Joosten, L.A.; Netea, M.G.; Fantuzzi, G.; Koenders, M.I.; Helsen, M.M.; Sparrer, H.; Pham, C.T.; van der Meer, J.W.; Dinarello, C.A.; van den Berg, W.B. Inflammatory arthritis in caspase 1 gene-deficient mice: Contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009, 60, 3651–3662. [Google Scholar] [CrossRef] [Green Version]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; Van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choulaki, C.; Papadaki, G.; Repa, A.; Kampouraki, E.; Kambas, K.; Ritis, K.; Bertsias, G.; Boumpas, D.T.; Sidiropoulos, P. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res. 2015, 17, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscitti, P.; Cipriani, P.; Di Benedetto, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Ciccia, F.; Alvaro, S.; Triolo, G.; Giacomelli, R. Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1beta via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: A possible implication for therapeutic decision in these patients. Clin. Exp. Immunol. 2015, 182, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, T.H.; Kim, H.S.; Kang, T.W.; Lee, B.C.; Lee, H.Y.; Kim, Y.J.; Shin, J.H.; Seo, Y.; Won Choi, S.; Lee, S.; et al. Human umbilical cord blood-stem cells direct macrophage polarization and block inflammasome activation to alleviate rheumatoid arthritis. Cell Death Dis. 2016, 7, e2524. [Google Scholar] [CrossRef]
- Sui, J.; Li, H.; Fang, Y.; Liu, Y.; Li, M.; Zhong, B.; Yang, F.; Zou, Q.; Wu, Y. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han chinese. Arthritis Rheum. 2012, 64, 647–654. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Y.; Zou, F.; Wu, M.; Fan, C.; Ding, Y. 11beta-Hydroxysteroid dehydrogenase 1 inhibition attenuates collagen-induced arthritis. Int. Immunopharmacol. 2013, 17, 489–494. [Google Scholar] [CrossRef]
- Li, F.; Guo, N.; Ma, Y.; Ning, B.; Wang, Y.; Kou, L. Inhibition of P2X4 suppresses joint inflammation and damage in collagen-induced arthritis. Inflammation 2014, 37, 146–153. [Google Scholar] [CrossRef]
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Tran Mau-Them, F.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum. Dis. 2017, 76, 1191–1198. [Google Scholar] [CrossRef]
- Mansoori, M.N.; Shukla, P.; Kakaji, M.; Tyagi, A.M.; Srivastava, K.; Shukla, M.; Dixit, M.; Kureel, J.; Gupta, S.; Singh, D. IL-18BP is decreased in osteoporotic women: Prevents Inflammasome mediated IL-18 activation and reduces Th17 differentiation. Sci. Rep. 2016, 6, 33680. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhang, L.; Wang, Z.; Li, C.; Li, S.; Li, L.; Fan, Q.; Zheng, L. Melatonin Suppresses Estrogen Deficiency-Induced Osteoporosis and Promotes Osteoblastogenesis by Inactivating the NLRP3 Inflammasome. Calcif. Tissue Int. 2018, 103, 400–410. [Google Scholar] [CrossRef]
- Snouwaert, J.N.; Nguyen, M.; Repenning, P.W.; Dye, R.; Livingston, E.W.; Kovarova, M.; Moy, S.S.; Brigman, B.E.; Bateman, T.A.; Ting, J.P.; et al. An NLRP3 Mutation Causes Arthropathy and Osteoporosis in Humanized Mice. Cell Rep. 2016, 17, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, K.S.; Kim, M.J.; Kim, H.S.; Lee, K.Y.; Kang, K.W. Auranofin Inhibits RANKL-Induced Osteoclastogenesis by Suppressing Inhibitors of kappaB Kinase and Inflammasome-Mediated Interleukin-1beta Secretion. Oxid. Med. Cell Longev. 2019, 2019, 3503912. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, K.; Wan, X.; Wang, F.; Guo, Z.; Mo, Z. NLRP3 inflammasome activation in mesenchymal stem cells inhibits osteogenic differentiation and enhances adipogenic differentiation. Biochem. Biophys. Res. Commun. 2017, 484, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.; Doyle, S.L. An eye on the future of inflammasomes and drug development in AMD. J. Mol. Med. (Berl.) 2013, 91, 1059–1070. [Google Scholar] [CrossRef]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef]
- Ildefonso, C.J.; Biswal, M.R.; Ahmed, C.M.; Lewin, A.S. The NLRP3 Inflammasome and its Role in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2016, 854, 59–65. [Google Scholar] [CrossRef]
- Yerramothu, P.; Vijay, A.K.; Willcox, M.D.P. Inflammasomes, the eye and anti-inflammasome therapy. Eye (Lond.) 2018, 32, 491–505. [Google Scholar] [CrossRef]
- Relja, B.; Horstmann, J.P.; Kontradowitz, K.; Jurida, K.; Schaible, A.; Neunaber, C.; Oppermann, E.; Marzi, I. Nlrp1 inflammasome is downregulated in trauma patients. J. Mol. Med. (Berl) 2015, 93, 1391–1400. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovarova, M.; Hesker, P.R.; Jania, L.; Nguyen, M.; Snouwaert, J.N.; Xiang, Z.; Lommatzsch, S.E.; Huang, M.T.; Ting, J.P.; Koller, B.H. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J. Immunol. 2012, 189, 2006–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, N.; Lee, S.W.; Perez-Barcena, J.; Crespi, C.; Ibanez, J.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome proteins as biomarkers of traumatic brain injury. PLoS ONE 2018, 13, e0210128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Q.; Zeng, L.; Gu, W.; Zhang, L.Y.; Zhou, J.; Jiang, D.P.; Du, D.Y.; Hu, P.; Yang, C.; Yan, J.; et al. Clinical relevance of single nucleotide polymorphisms within the entire NLRP3 gene in patients with major blunt trauma. Crit. Care 2011, 15, R280. [Google Scholar] [CrossRef] [Green Version]
- Hosseinian, N.; Cho, Y.; Lockey, R.F.; Kolliputi, N. The role of the NLRP3 inflammasome in pulmonary diseases. Adv Respir. Dis. 2015, 9, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.D.; Crother, T.R.; Gonzalez-Villalobos, R.A.; Jupelli, M.; Chen, S.; Dagvadorj, J.; Arditi, M.; Shimada, K. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am. J. Respir. Cell Mol. Biol. 2014, 50, 270–280. [Google Scholar] [CrossRef]
- Kuipers, M.T.; Aslami, H.; Janczy, J.R.; van der Sluijs, K.F.; Vlaar, A.P.; Wolthuis, E.K.; Choi, G.; Roelofs, J.J.; Flavell, R.A.; Sutterwala, F.S.; et al. Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology 2012, 116, 1104–1115. [Google Scholar] [CrossRef] [Green Version]
- Kerr, N.A.; De Rivero Vaccari, J.P.; Abbassi, S.; Kaur, H.; Zambrano, R.; Wu, S.; Dietrich, W.D.; Keane, R.W. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J. Neurotrauma 2018, 35, 2067–2076. [Google Scholar] [CrossRef]
- Zou, P.; Liu, X.; Li, G.; Wang, Y. Resveratrol pretreatment attenuates traumatic brain injury in rats by suppressing NLRP3 inflammasome activation via SIRT1. Mol. Med. Rep. 2018, 17, 3212–3217. [Google Scholar] [CrossRef]
- Osuka, A.; Hanschen, M.; Stoecklein, V.; Lederer, J.A. A protective role for inflammasome activation following injury. Shock 2012, 37, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yin, D.; Ren, H.; Gao, W.; Li, F.; Sun, D.; Wu, Y.; Zhou, S.; Lyu, L.; Yang, M.; et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol. Dis. 2018, 117, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, M.; He, F.; Zhou, S.; Zhu, L. Targeting the NLRP3 inflammasome to attenuate spinal cord injury in mice. J Neuroinflamm. 2017, 14, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Yuan, Z.; Wang, G.; Wang, X.; Li, K. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharmacol. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Mo, H.; Chen, J.; Qian, C.; Yan, F.; Gu, C.; Hu, Q.; Wang, L.; Chen, G. Minocycline Protects Against NLRP3 Inflammasome-Induced Inflammation and P53-Associated Apoptosis in Early Brain Injury After Subarachnoid Hemorrhage. Mol. Neurobiol. 2016, 53, 2668–2678. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Coutts, G.; Lenart, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horai, R.; Asano, M.; Sudo, K.; Kanuka, H.; Suzuki, M.; Nishihara, M.; Takahashi, M.; Iwakura, Y. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 1998, 187, 1463–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.; Wu, D.; Zuo, Q.; Wang, Z.; Fan, W. Ginsenoside Rb1 prevents interleukin-1 beta induced inflammation and apoptosis in human articular chondrocytes. Int. Orthop. 2013, 37, 2065–2070. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A.; Van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef]
- Al-Sadi, R.M.; Ma, T.Y. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J. Immunol. 2007, 178, 4641–4649. [Google Scholar] [CrossRef] [Green Version]
- Carmi, Y.; Voronov, E.; Dotan, S.; Lahat, N.; Rahat, M.A.; Fogel, M.; Huszar, M.; White, M.R.; Dinarello, C.A.; Apte, R.N. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J. Immunol. 2009, 183, 4705–4714. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Oleinika, K.; Tonon, S.; Doyle, R.; Bosma, A.; Carter, N.A.; Harris, K.A.; Jones, S.A.; Klein, N.; Mauri, C. Regulatory B cells are induced by gut microbiota-driven interleukin-1β and interleukin-6 production. Nat. Med. 2014, 20, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Van Griensven, M. Zytokine als Marker bei Polytrauma. Der. Unf. 2014, 117, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Kurt, A.N.C.; Aygun, A.D.; Godekmerdan, A.; Kurt, A.; Dogan, Y.; Yilmaz, E. Serum IL-1beta, IL-6, IL-8, and TNF-alpha levels in early diagnosis and management of neonatal sepsis. Mediat. Inflamm. 2007, 2007, 31397. [Google Scholar] [CrossRef] [PubMed]
- Al-Fadhli, M.A.; Saraya, M.A.; Qasem, J.A. Evaluation of leptin, interleukin-1 beta and tumor necrosis factor alpha in serum of malaria patients as prognostic markers of treatment outcome. Asian Pac. J. Trop. Biomed. 2014, 4, 441–445. [Google Scholar] [CrossRef] [Green Version]
- De Pablo, R.; Monserrat, J.; Reyes, E.; Diaz-Martin, D.; Rodriguez Zapata, M.; Carballo, F.; De la Hera, A.; Prieto, A.; Alvarez-Mon, M. Mortality in Patients With Septic Shock Correlates With Anti-Inflammatory But not Proinflammatory Immunomodulatory Molecules. J. Intensive Care Med. 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Maruotti, N.; Corrado, A.; Cantatore, F.P. Osteoporosis and rheumatic diseases. Reumatismo 2014, 66, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Lacativa, P.G.; Farias, M.L. Osteoporosis and inflammation. Arq. Bras. Endocrinol. Metab. 2010, 54, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.F.; Gu, L.J.; Wu, Y.; Zhao, X.H.; Zhang, Q.; Xu, Z.R.; Yang, Y.M. Efficacy and Safety of Denosumab in Postmenopausal Women With Osteoporosis: A Meta-Analysis. Medicine (Baltim.) 2015, 94, e1674. [Google Scholar] [CrossRef]
- Choi, N.K.; Solomon, D.H.; Tsacogianis, T.N.; Landon, J.E.; Song, H.J.; Kim, S.C. Comparative Safety and Effectiveness of Denosumab Versus Zoledronic Acid in Patients With Osteoporosis: A Cohort Study. J. Bone Min. Res. 2017, 32, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Kullenberg, T.; Lofqvist, M.; Leinonen, M.; Goldbach-Mansky, R.; Olivecrona, H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology (Oxf.) 2016, 55, 1499–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, M.V.; Sanmarti, R.; Canete, J.D. The safety of tumor necrosis factor-alpha inhibitors in the treatment of rheumatoid arthritis. Expert Opin. Drug Saf. 2016, 15, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA. 2007, 104, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.P. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: In situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [Green Version]
- Palmer, G.; Lipsky, B.P.; Smithgall, M.D.; Meininger, D.; Siu, S.; Talabot-Ayer, D.; Gabay, C.; Smith, D.E. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine 2008, 42, 358–364. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Baekkevold, E.S.; Roussigné, M.; Yamanaka, T.; Johansen, F.E.; Jahnsen, F.L.; Amalric, F.; Brandtzaeg, P.; Erard, M.; Haraldsen, G.; Girard, J.P. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am. J. Pathol. 2003, 163, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, R.; Hei, H.; Dobner, S.; Lee, R.T. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 2012, 287, 6941–6948. [Google Scholar] [CrossRef] [Green Version]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.P.; et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.H.; Ward, C.K.; Criner, G.J.; et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komai-Koma, M.; Xu, D.; Li, Y.; McKenzie, A.N.J.; McInnes, I.B.; Liew, F.Y. IL-33 is a chemoattractant for human Th2 cells. Eur. J. Immunol. 2007, 37, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Rank, M.A.; Kobayashi, T.; Kozaki, H.; Bartemes, K.R.; Squillace, D.L.; Kita, H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009, 123, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.C.; Ma, A.; Lanier, L.L. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J. Immunol. 2009, 183, 2911–2914. [Google Scholar] [CrossRef]
- Bourgeois, E.; Van, L.P.; Samson, M.; Diem, S.; Barra, A.; Roga, S.; Gombert, J.M.; Schneider, E.; Dy, M.; Gourdy, P.; et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur. J. Immunol. 2009, 39, 1046–1055. [Google Scholar] [CrossRef]
- Villarreal, D.O.; Weiner, D.B. Interleukin 33: A switch-hitting cytokine. Curr. Opin. Immunol. 2014, 28, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, W.V.; Frohlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The Alarmin Interleukin-33 Drives Protective Antiviral CD8+ T Cell Responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Saitta, S.; Sirufo, M.M.; Mannucci, C.; Casciaro, M.; Ciccarelli, F.; Gangemi, S. Interleukin-33 serum levels in postmenopausal women with osteoporosis. Sci. Rep. 2019, 9, 3786. [Google Scholar] [CrossRef] [Green Version]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Mountz, J.D.; Kimberly, R.P. Immunobiology of tumor necrosis factor receptor superfamily. Immunol. Res. 2002, 26, 323–336. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Grell, M. Tumor necrosis factor (TNF) receptors in cellular signaling of soluble and membrane-expressed TNF. J. Inflamm. 1995, 47, 8–17. [Google Scholar] [PubMed]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, H.; Zhang, J.; Volk, A.; Zhang, S.; Wei, W.; Zhang, S.; Breslin, P.; Zhang, J. TNF-/Fas-RIP-1-induced cell death signaling separates murine hematopoietic stem cells/progenitors into 2 distinct populations. Blood 2011, 118, 6057–6067. [Google Scholar] [CrossRef]
- Tsurumi, A.; Que, Y.A.; Ryan, C.M.; Tompkins, R.G.; Rahme, L.G. TNF-α/IL-10 Ratio Correlates with Burn Severity and May Serve as a Risk Predictor of Increased Susceptibility to Infections. Front. Public Health 2016, 4, 216. [Google Scholar] [CrossRef] [Green Version]
- Spielmann, S.; Kerner, T.; Ahlers, O.; Keh, D.; Gerlach, M.; Gerlach, H. Early detection of increased tumour necrosis factor alpha (TNFalpha) and soluble TNF receptor protein plasma levels after trauma reveals associations with the clinical course. Acta Anaesthesiol. Scand. 2001, 45, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Reikerås, O.; Borgen, P. Activation of markers of inflammation, coagulation and fibrinolysis in musculoskeletal trauma. PLoS ONE 2014, 9, e107881. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Fong, Y.; Hesse, D.G.; Manogue, K.R.; Lee, A.T.; Kuo, G.C.; Lowry, S.F.; Cerami, A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987, 330, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.S.; Trickler, W.J.; Miller, D.W. Tumor necrosis factor-alpha induces cyclooxygenase-2 expression and prostaglandin release in brain microvessel endothelial cells. J. Pharmacol. Exp. Ther. 2001, 297, 1051–1058. [Google Scholar] [PubMed]
- Van Kampen, C.; Mallard, B.A. Regulation of bovine intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) on cultured aortic endothelial cells. Vet. Immunol. Immunopathol. 2001, 79, 129–138. [Google Scholar] [CrossRef]
- Stoecklein, V.M.; Osuka, A.; Lederer, J.A. Trauma equals danger--damage control by the immune system. J. Leukoc. Biol. 2012, 92, 539–551. [Google Scholar] [CrossRef]
- Yoshida, L.S.; Tsunawaki, S. Expression of NADPH oxidases and enhanced H(2)O(2)-generating activity in human coronary artery endothelial cells upon induction with tumor necrosis factor-alpha. Int. Immunopharmacol. 2008, 8, 1377–1385. [Google Scholar] [CrossRef]
- Beutler, B.; Milsark, I.W.; Cerami, A.C. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science (N. Y.) 1985, 229, 869–871. [Google Scholar] [CrossRef]
- Pfeffer, K.; Matsuyama, T.; Kündig, T.M.; Wakeham, A.; Kishihara, K.; Shahinian, A.; Wiegmann, K.; Ohashi, P.S.; Krönke, M.; Mak, T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993, 73, 457–467. [Google Scholar] [CrossRef]
- Eichacker, P.Q.; Parent, C.; Kalil, A.; Esposito, C.; Cui, X.; Banks, S.M.; Gerstenberger, E.P.; Fitz, Y.; Danner, R.L.; Natanson, C. Risk and the efficacy of antiinflammatory agents: Retrospective and confirmatory studies of sepsis. Am. J. Respir. Crit. Care Med. 2002, 166, 1197–1205. [Google Scholar] [CrossRef]
- Dingle, K.; Azizieh, F. Multivariate Comparison of Cytokine Profiles for Normal- and Low-Bone-Density Subjects. Diagnostics (Basel) 2019, 9, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; He, C. TNF-alpha and IL-6: The link between immune and bone system. Curr Drug Targets 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C.; Yu, X. Pro-Inflammatory Cytokines: New Potential Therapeutic Targets for Obesity-Related Bone Disorders. Curr. Drug Targets 2017, 18, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lim, D.H.; Oh, J.S.; Kim, Y.G.; Lee, C.K.; Yoo, B.; Hong, S. Effect of TNF inhibitors on bone mineral density in rheumatoid arthritis patients receiving bisphosphonate: A retrospective cohort study. Rheumatol. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Inui, K.; Sugioka, Y.; Mamoto, K.; Okano, T.; Anno, S.; Koike, T. Use of bisphosphonate might be important to improve bone mineral density in patients with rheumatoid arthritis even under tight control: The TOMORROW study. Rheumatol. Int. 2017, 37, 999–1005. [Google Scholar] [CrossRef]
- De Vries, T.J.; El Bakkali, I.; Kamradt, T.; Schett, G.; Jansen, I.D.C.; D’Amelio, P. What Are the Peripheral Blood Determinants for Increased Osteoclast Formation in the Various Inflammatory Diseases Associated With Bone Loss? Front. Immunol. 2019, 10, 505. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Bond, M.W.; Mosmann, T.R. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 1989, 170, 2081–2095. [Google Scholar] [CrossRef]
- Zdanov, A.; Schalk-Hihi, C.; Gustchina, A.; Tsang, M.; Weatherbee, J.; Wlodawer, A. Crystal structure of interleukin-10 reveals the functional dimer with an unexpected topological similarity to interferon γ. Structure 1995, 3, 591–601. [Google Scholar] [CrossRef]
- Banchereau, J.; Pascual, V.; O’Garra, A. From IL-2 to IL-37: The expanding spectrum of anti-inflammatory cytokines. Nat. Immunol. 2012, 13, 925–931. [Google Scholar] [CrossRef]
- Savan, R.; Ravichandran, S.; Collins, J.R.; Sakai, M.; Young, H.A. Structural conservation of interferon gamma among vertebrates. Cytokine Growth Factor Rev. 2009, 20, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Siewe, L.; Bollati-Fogolin, M.; Wickenhauser, C.; Krieg, T.; Müller, W.; Roers, A. Interleukin-10 derived from macrophages and/or neutrophils regulates the inflammatory response to LPS but not the response to CpG DNA. Eur. J. Immunol. 2006, 36, 3248–3255. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Gagliani, N.; Esplugues, E.; O’Connor, W.; Huber, F.J.; Chaudhry, A.; Kamanaka, M.; Kobayashi, Y.; Booth, C.J.; Rudensky, A.Y.; et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3- and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011, 34, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotenko, S.V.; Krause, C.D.; Izotova, L.S.; Pollack, B.P.; Wu, W.; Pestka, S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J. 1997, 16, 5894–5903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.I.; Logsdon, N.J.; Sheikh, F.; Donnelly, R.P.; Walter, M.R. Conformational changes mediate interleukin-10 receptor 2 (IL-10R2) binding to IL-10 and assembly of the signaling complex. J. Biol. Chem. 2006, 281, 35088–35096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crepaldi, L.; Gasperini, S.; Lapinet, J.A.; Calzetti, F.; Pinardi, C.; Liu, Y.; Zurawski, S.; de Waal Malefyt, R.; Moore, K.W.; Cassatella, M.A. Up-regulation of IL-10R1 expression is required to render human neutrophils fully responsive to IL-10. J. Immunol. (Baltimore Md. 1950) 2001, 167, 2312–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pils, M.C.; Pisano, F.; Fasnacht, N.; Heinrich, J.-M.; Groebe, L.; Schippers, A.; Rozell, B.; Jack, R.S.; Müller, W. Monocytes/macrophages and/or neutrophils are the target of IL-10 in the LPS endotoxemia model. Eur. J. Immunol. 2010, 40, 443–448. [Google Scholar] [CrossRef]
- Sheikh, F.; Baurin, V.V.; Lewis-Antes, A.; Shah, N.K.; Smirnov, S.V.; Anantha, S.; Dickensheets, H.; Dumoutier, L.; Renauld, J.C.; Zdanov, A.; et al. Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J. Immunol. 2004, 172, 2006–2010. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.R.; Quinton, L.J.; Simms, B.T.; Lupa, M.M.; Kogan, M.S.; Mizgerd, J.P. Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J. Infect. Dis. 2006, 193, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Finbloom, D.S.; Winestock, K.D. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J. Immunol. 1995, 155, 1079–1090. [Google Scholar]
- Kretzschmar, A.K.; Dinger, M.C.; Henze, C.; Brocke-Heidrich, K.; Horn, F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem. J. 2004, 377, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 Receptor Signaling. In Advances in Immunology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 122, pp. 177–210. [Google Scholar]
- Jung, M.; Sabat, R.; Krätzschmar, J.; Seidel, H.; Wolk, K.; Schönbein, C.; Schütt, S.; Friedrich, M.; Döcke, W.-D.; Asadullah, K.; et al. Expression profiling of IL-10-regulated genes in human monocytes and peripheral blood mononuclear cells from psoriatic patients during IL-10 therapy. Eur. J. Immunol. 2004, 34, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Martire-Greco, D.; Rodriguez-Rodrigues, N.; Landoni, V.I.; Rearte, B.; Isturiz, M.A.; Fernández, G.C. Interleukin-10 controls human peripheral PMN activation triggered by lipopolysaccharide. Cytokine 2013, 62, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Huet, O.; Laemmel, E.; Fu, Y.; Dupic, L.; Aprico, A.; Andrews, K.L.; Moore, X.L.; Harrois, A.; Meikle, P.L.; Vicaut, E.; et al. IL-10 anti oxidant effect decreases leukocytes/endothelial interaction induced by TNF-α. Shock 2013, 39, 83–88. [Google Scholar] [CrossRef]
- Thibodeau, J.; Bourgeois-Daigneault, M.-C.; Huppé, G.; Tremblay, J.; Aumont, A.; Houde, M.; Bartee, E.; Brunet, A.; Gauvreau, M.-E.; de Gassart, A.; et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur. J. Immunol. 2008, 38, 1225–1230. [Google Scholar] [CrossRef] [Green Version]
- Vicioso, M.A.; Garaud, J.J.; Réglier-Poupet, H.; Lebeaut, A.; Gougerot-Pocidalo, M.A.; Chollet-Martin, S. Moderate inhibitory effect of interleukin-10 on human neutrophil and monocyte chemotaxis in vitro. Eur. Cytokine Netw. 1998, 9, 247–253. [Google Scholar]
- Sikka, G.; Miller, K.L.; Steppan, J.; Pandey, D.; Jung, S.M.; Fraser, C.D.; Ellis, C.; Ross, D.; Vandegaer, K.; Bedja, D.; et al. Interleukin 10 knockout frail mice develop cardiac and vascular dysfunction with increased age. Exp. Gerontol. 2013, 48, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Koch, N.; Jung, M.; Sabat, R.; Krätzschmar, J.; Döcke, W.D.; Asadullah, K.; Volk, H.-D.; Grütz, G. IL-10 protects monocytes and macrophages from complement-mediated lysis. J. Leukoc. Biol. 2009, 86, 155–166. [Google Scholar] [CrossRef]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Ito, S.; Ansari, P.; Sakatsume, M.; Dickensheets, H.; Vazquez, N.; Donnelly, R.P.; Larner, A.C.; Finbloom, D.S. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 1999, 93, 1456–1463. [Google Scholar] [CrossRef]
- Heper, Y.; Akalin, E.H.; Mistik, R.; Akgöz, S.; Töre, O.; Göral, G.; Oral, B.; Budak, F.; Helvaci, S. Evaluation of serum C-reactive protein, procalcitonin, tumor necrosis factor alpha, and interleukin-10 levels as diagnostic and prognostic parameters in patients with community-acquired sepsis, severe sepsis, and septic shock. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2006, 25, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Marchant, A.; Alegre, M.L.; Hakim, A.; Piérard, G.; Marécaux, G.; Friedman, G.; De Groote, D.; Kahn, R.J.; Vincent, J.L.; Goldman, M. Clinical and biological significance of interleukin-10 plasma levels in patients with septic shock. J. Clin. Immunol. 1995, 15, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, R.; Keel, M.; Steckholzer, U.; Safret, A.; Ungethuem, U.; Trentz, O.; Ertel, W. Relationship of Interleukin-10 Plasma Levels to Severity of Injury and Clinical Outcome in Injured Patients. J. Trauma Inj. Infect. Crit. Care 1997, 42, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Sherry, R.M.; Cue, J.I.; Goddard, J.K.; Parramore, J.B.; DiPiro, J.T. Interleukin-10 is associated with the development of sepsis in trauma patients. J. Trauma 1996, 40, 613–616. [Google Scholar] [CrossRef]
- Rios-Arce, N.D.; Dagenais, A.; Feenstra, D.; Coughlin, B.; Kang, H.J.; Mohr, S.; McCabe, L.R.; Parameswaran, N. Loss of interleukin-10 exacerbates early Type-1 diabetes-induced bone loss. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Wang, T.; Jiang, C.; Zhao, P.; Chen, L.; Li, Z. Detection and analysis of serum IL-10 and TGF-beta1 in the SLE patients with osteoporosis or osteonecrosis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2018, 34, 1032–1035. [Google Scholar]
- Baggiolini, M.; Clark-Lewis, I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992, 307, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Bezzerri, V.; Borgatti, M.; Finotti, A.; Tamanini, A.; Gambari, R.; Cabrini, G. Mapping the Transcriptional Machinery of the IL-8 Gene in Human Bronchial Epithelial Cells. J. Immunol. 2011, 187, 6069–6081. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar]
- Lehner, M.; Morhart, P.; Stilper, A.; Petermann, D.; Weller, P.; Stachel, D.; Holter, W. Efficient chemokine-dependent migration and primary and secondary IL-12 secretion by human dendritic cells stimulated through Toll-like receptors. J. Immunother. 2007, 30, 312–322. [Google Scholar] [CrossRef]
- Hol, J.; Küchler, A.M.; Johansen, F.E.; Dalhus, B.; Haraldsen, G.; Oynebråten, I. Molecular requirements for sorting of the chemokine interleukin-8/CXCL8 to endothelial Weibel-Palade bodies. J. Biol. Chem. 2009, 284, 23532–23539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maheshwari, A.; Voitenok, N.N.; Akalovich, S.; Shaik, S.S.; Randolph, D.A.; Sims, B.; Patel, R.P.; Killingsworth, C.R.; Fallon, M.B.; Ohls, R.K. Developmental changes in circulating IL-8/CXCL8 isoforms in neonates. Cytokine 2009, 46, 12–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, I.H.K.; Marshall, L.; Lambert, P.A.; Chapple, I.L.C.; Matthews, J.B.; Griffiths, H.R. Gingipains from Porphyromonas gingivalis increase the chemotactic and respiratory burst-priming properties of the 77-amino-acid interleukin-8 variant. Infect. Immun. 2008, 76, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramjeesingh, R.; Leung, R.; Siu, C.H. Interleukin-8 secreted by endothelial cells induces chemotaxis of melanoma cells through the chemokine receptor CXCR1. FASEB J. 2003, 17, 1292–1294. [Google Scholar] [CrossRef]
- Allen, T.C.; Kurdowska, A. Interleukin 8 and Acute Lung Injury. Arch. Pathol. Lab. Med. 2014, 138, 266–269. [Google Scholar] [CrossRef]
- Kurdowska, A.; Noble, J.M.; Steinberg, K.P.; Ruzinski, J.T.; Hudson, L.D.; Martin, T.R. Anti-interleukin 8 autoantibody: Interleukin 8 complexes in the acute respiratory distress syndrome. Relationship between the complexes and clinical disease activity. Am. J. Respir. Crit. Care Med. 2001, 163, 463–468. [Google Scholar] [CrossRef]
- Kobayashi, Y. The role of chemokines in neutrophil biology. Front. Biosci. 2008, 13, 2400–2407. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Manegold, P.C.; Hong, Y.K.; Zhang, W.; Pohl, A.; Lurje, G.; Winder, T.; Yang, D.; LaBonte, M.J.; Wilson, P.M.; et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int. J. Cancer 2011, 128, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef]
- Kraft, R.; Herndon, D.N.; Finnerty, C.C.; Cox, R.A.; Song, J.; Jeschke, M.G. Predictive Value of IL-8 for Sepsis and Severe Infections After Burn Injury: A Clinical Study. Shock (AugustaGa.) 2015, 43, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, S.P.J.; Stone, S.F.; Neil, C.L.; van Eeden, P.E.; Fatovich, D.M.; Arendts, G.; Brown, S.G.A. Sustained elevation of resistin, NGAL and IL-8 are associated with severe sepsis/septic shock in the emergency department. PLoS ONE 2014, 9, e110678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.C.B.; Regner, A.; Miotto, K.D.L.; Moura, S.d.; Ikuta, N.; Vargas, A.E.; Chies, J.A.B.; Simon, D. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014, 28, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Gopcevic, A.; Mazul-Sunko, B.; Marout, J.; Sekulic, A.; Antoljak, N.; Siranovic, M.; Ivanec, Z.; Margaritoni, M.; Bekavac-Beslin, M.; Zarkovic, N. Plasma interleukin-8 as a potential predictor of mortality in adult patients with severe traumatic brain injury. Tohoku J. Exp. Med. 2007, 211, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Daghri, N.M.; Yakout, S.; Al-Shehri, E.; Al-Fawaz, H.A.; Aljohani, N.; Al-Saleh, Y. Inflammatory and bone turnover markers in relation to PTH and vitamin D status among saudi postmenopausal women with and without osteoporosis. Int. J. Clin. Exp. Med. 2014, 7, 3528–3535. [Google Scholar] [PubMed]
- Sousa, L.H.; Linhares, E.V.; Alexandre, J.T.; Lisboa, M.R.; Furlaneto, F.; Freitas, R.; Ribeiro, I.; Val, D.; Marques, M.; Chaves, H.V.; et al. Effects of Atorvastatin on Periodontitis of Rats Subjected to Glucocorticoid-Induced Osteoporosis. J. Periodontol. 2016, 87, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Sun, Y.; Xu, W.; Lin, T.; Zeng, H. Expression of RANKL by peripheral neutrophils and its association with bone mineral density in COPD. Respirology 2017, 22, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 2063. [Google Scholar] [CrossRef]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Scheidt-Nave, C.; Bismar, H.; Leidig-Bruckner, G.; Woitge, H.; Seibel, M.J.; Ziegler, R.; Pfeilschifter, J. Serum interleukin 6 is a major predictor of bone loss in women specific to the first decade past menopause. J. Clin. Endocrinol. Metab 2001, 86, 2032–2042. [Google Scholar] [CrossRef]
- Moffett, S.P.; Zmuda, J.M.; Cauley, J.A.; Stone, K.L.; Nevitt, M.C.; Ensrud, K.E.; Hillier, T.A.; Hochberg, M.C.; Joslyn, G.; Morin, P.; et al. Association of the G-174C variant in the interleukin-6 promoter region with bone loss and fracture risk in older women. J. Bone Min. Res. 2004, 19, 1612–1618. [Google Scholar] [CrossRef]
- Fajar, J.K.; Azharuddin, A. The association between interleukin 6 -174 G/C gene polymorphism and the risk of osteoporosis: A meta-analysis. J. Taibah. Univ. Med. Sci. 2017, 12, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czerny, B.; Kaminski, A.; Kurzawski, M.; Kotrych, D.; Safranow, K.; Dziedziejko, V.; Bohatyrewicz, A.; Pawlik, A. The association of IL-1beta, IL-2, and IL-6 gene polymorphisms with bone mineral density and osteoporosis in postmenopausal women. Eur. J. Obs. Gynecol. Reprod. Biol. 2010, 149, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Parameswaran, V.; Udayan, R.; Burgess, J.; Jones, G. Circulating levels of inflammatory markers predict change in bone mineral density and resorption in older adults: A longitudinal study. J. Clin. Endocrinol Metab 2008, 93, 1952–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.X.; Vrindts, Y.; Lopez, M.; De Groote, D.; Zangerle, P.F.; Collette, J.; Franchimont, N.; Geenen, V.; Albert, A.; Reginster, J.Y. Increase in cytokine production (IL-1 beta, IL-6, TNF-alpha but not IFN-gamma, GM-CSF or LIF) by stimulated whole blood cells in postmenopausal osteoporosis. Maturitas 1997, 26, 63–71. [Google Scholar] [CrossRef]
- Jilka, R.L.; Hangoc, G.; Girasole, G.; Passeri, G.; Williams, D.C.; Abrams, J.S.; Boyce, B.; Broxmeyer, H.; Manolagas, S.C. Increased osteoclast development after estrogen loss: Mediation by interleukin-6. Science 1992, 257, 88–91. [Google Scholar] [CrossRef]
- Chao, T.H.; Yu, H.N.; Huang, C.C.; Liu, W.S.; Tsai, Y.W.; Wu, W.T. Association of interleukin-1 beta (-511C/T) polymorphisms with osteoporosis in postmenopausal women. Ann. Saudi. Med. 2010, 30, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Al-Daghri, N.M.; Aziz, I.; Yakout, S.; Aljohani, N.J.; Al-Saleh, Y.; Amer, O.E.; Sheshah, E.; Younis, G.Z.; Al-Badr, F.B. Inflammation as a contributing factor among postmenopausal Saudi women with osteoporosis. Medicine (Baltim.) 2017, 96, e5780. [Google Scholar] [CrossRef]
- Inanir, A.; Ozoran, K.; Tutkak, H.; Mermerci, B. The effects of calcitriol therapy on serum interleukin-1, interleukin-6 and tumour necrosis factor-alpha concentrations in post-menopausal patients with osteoporosis. J. Int. Med. Res. 2004, 32, 570–582. [Google Scholar] [CrossRef]
- Tsuboi, M.; Kawakami, A.; Nakashima, T.; Matsuoka, N.; Urayama, S.; Kawabe, Y.; Fujiyama, K.; Kiriyama, T.; Aoyagi, T.; Maeda, K.; et al. Tumor necrosis factor-alpha and interleukin-1beta increase the Fas-mediated apoptosis of human osteoblasts. J. Lab. Clin. Med. 1999, 134, 222–231. [Google Scholar] [CrossRef]
- Kimble, R.B.; Vannice, J.L.; Bloedow, D.C.; Thompson, R.C.; Hopfer, W.; Kung, V.T.; Brownfield, C.; Pacifici, R. Interleukin-1 receptor antagonist decreases bone loss and bone resorption in ovariectomized rats. J. Clin. Investig. 1994, 93, 1959–1967. [Google Scholar] [CrossRef] [Green Version]
- D’Amelio, P.; Grimaldi, A.; Di Bella, S.; Brianza, S.Z.M.; Cristofaro, M.A.; Tamone, C.; Giribaldi, G.; Ulliers, D.; Pescarmona, G.P.; Isaia, G. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: A key mechanism in osteoporosis. Bone 2008, 43, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kotrych, D.; Dziedziejko, V.; Safranow, K.; Sroczynski, T.; Staniszewska, M.; Juzyszyn, Z.; Pawlik, A. TNF-alpha and IL10 gene polymorphisms in women with postmenopausal osteoporosis. Eur. J. Obs. Gynecol. Reprod. Biol. 2016, 199, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Zha, L.; He, L.; Liang, Y.; Qin, H.; Yu, B.; Chang, L.; Xue, L. TNF-alpha contributes to postmenopausal osteoporosis by synergistically promoting RANKL-induced osteoclast formation. Biomed. Pharm. 2018, 102, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Zhou, Z.; Zhu, L.; Hu, X.; Lu, J.; Shi, C.; Chen, F.; Chen, A. TNF-alpha suppresses osteogenic differentiation of MSCs by accelerating P2Y2 receptor in estrogen-deficiency induced osteoporosis. Bone 2018, 117, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Su, X.; Yang, X.; Hu, C.; Li, B.; Lv, Y.; Shuai, Y.; Jing, H.; Deng, Z.; Jin, Y. TNF-alpha Inhibits FoxO1 by Upregulating miR-705 to Aggravate Oxidative Damage in Bone Marrow-Derived Mesenchymal Stem Cells during Osteoporosis. Stem. Cells 2016, 34, 1054–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolini, D.R.; Nedwin, G.E.; Bringman, T.S.; Smith, D.D.; Mundy, G.R. Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 1986, 319, 516–518. [Google Scholar] [CrossRef]
- Tural, S.; Alayli, G.; Kara, N.; Tander, B.; Bilgici, A.; Kuru, O. Association between osteoporosis and polymorphisms of the IL-10 and TGF-beta genes in Turkish postmenopausal women. Hum. Immunol. 2013, 74, 1179–1183. [Google Scholar] [CrossRef]
- Azizieh, F.; Raghupathy, R.; Shehab, D.; Al-Jarallah, K.; Gupta, R. Cytokine profiles in osteoporosis suggest a proresorptive bias. Menopause 2017, 24, 1057–1064. [Google Scholar] [CrossRef]
- Gur, A.; Denli, A.; Nas, K.; Cevik, R.; Karakoc, M.; Sarac, A.J.; Erdogan, F. Possible pathogenetic role of new cytokines in postmenopausal osteoporosis and changes during calcitonin plus calcium therapy. Rheumatol. Int. 2002, 22, 194–198. [Google Scholar] [CrossRef]
- Evans, K.E.; Fox, S.W. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Dresner-Pollak, R.; Gelb, N.; Rachmilewitz, D.; Karmeli, F.; Weinreb, M. Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology 2004, 127, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Pathak, J.L.; Bakker, A.D.; Verschueren, P.; Lems, W.F.; Luyten, F.P.; Klein-Nulend, J.; Bravenboer, N. CXCL8 and CCL20 Enhance Osteoclastogenesis via Modulation of Cytokine Production by Human Primary Osteoblasts. PLoS ONE 2015, 10, e0131041. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic representation of various cells expressing different cytokines. Interleukin (IL), Natural killer cells (NK), Tumor necrosis factor-alpha (TNF-α).
Figure 1.
A schematic representation of various cells expressing different cytokines. Interleukin (IL), Natural killer cells (NK), Tumor necrosis factor-alpha (TNF-α).

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236008
AMA Style
Kany S, Vollrath JT, Relja B. Cytokines in Inflammatory Disease. International Journal of Molecular Sciences. 2019; 20(23):6008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236008
Chicago/Turabian StyleKany, Shinwan, Jan Tilmann Vollrath, and Borna Relja. 2019. "Cytokines in Inflammatory Disease" International Journal of Molecular Sciences 20, no. 23: 6008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236008
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.