Measuring the Thickness of Metal Films: A Selection Guide to the Most Suitable Technique



1
Department of Chemistry “Ugo Schiff”, Università degli Studi di Firenze, via della Lastruccia 3, 50019 Sesto Fiorentino (FI), Italy
2
Institute of Chemistry of Organometallic Compounds (ICCOM)—National Research Council (CNR), Via Madonna del Piano 10, 50019 Sesto Fiorentino (FI), Italy
*
Authors to whom correspondence should be addressed.
Mater. Proc. 2020, 2(1), 12; https://0-doi-org.brum.beds.ac.uk/10.3390/CIWC2020-06823
Published: 13 May 2020
(This article belongs to the Proceedings of 2nd Coatings and Interfaces Web Conference (CIWC-2 2020))
Abstract
:The determination of thickness has a fundamental importance in all fields in which the implementation of films and coatings are required and takes a crucial role in the electroplating sector. The thickness influences many aspects of the coatings such as electrical, mechanical, corrosion protection, and even aesthetic properties. In the multitude of applications of thin layer coatings, the variability of thicknesses and materials is very high, as well as the variability of possible techniques that can be used to determine the characteristics of the layers of interest. The first distinction that can be made between these techniques is that which divides destructive techniques from non-destructive ones, in which, however, the semi- or micro-destructive techniques are immediately difficult to place. Other important parameters to consider are the cost, both for the purchase of the instrumentation and for each single analysis, the difficulties in preparing and measuring the sample, data processing, and obviously the detectable thickness ranges, the possible measurable materials, and precision and accuracy. The purpose of this work is to compare the characteristics of the various investigation methods, with a particular focus on metal film applications, so that it will be easier to choose the most suitable technique for each purpose.
1. Introduction
The use of thin films has become a ubiquitous practice in many scientific and industrial sectors. Coatings are widely used to obtain a synergistic action between the characteristics of the substrate and the covering material to improve the physical, chemical, and aesthetic properties and to lower the cost of the final product. For this reason, the measurement of thickness in composite materials is mandatory both to obtain the right characteristics in the final artefact as well as to keep the costs under control. The composition of the films could be extremely vast: dielectrics (organic, such as polymers and self-assembled monolayers (SAM), or inorganic, like metal oxides), semiconductors, and metals are all used in the form of films, obtaining a composite material with combined characteristics. In this work, we focused on the metal film characterization obtained through electrodeposition or vapour-phase deposition, but in most of the cases the same principles can be applied to films of different materials. As far as the film dimension is concerned, the thinnest measurable thickness coincides with an atomic monolayer (ML), while the thicker layers could reach hundreds of microns of thickness in electroforming. Therefore, in this review, we made an overview of all the techniques that allow us to investigate in this range.
According to the type, the composition, and the thickness itself of the film and the substrate, various techniques could be employed to investigate the sizes of the layers [1], but all the methods could be classified into two different categories: destructive and non-destructive techniques. Within the various techniques, both the methods of sample preparation and the actual analysis methods are distinguished. In fact, a sample can be prepared with a certain procedure, for example by making a cross-section, and then analysed with different instrumentations such as optical or electronic microscopy. Of course, there are also self-consistent techniques which require no sample preparation, or minimal preparation.
A destructive technique is a method that alters the sample, generally by scratching it, at a macroscopic or microscopic level; therefore, the analysed piece cannot be put on the market and it must be destroyed instead. This means that we must be sure about the conformity with the sale of the unmeasured objects. On the other hand, a non-destructive technique permits us to measure the sample without damaging it but, generally, if it belongs to the group of indirect measurements, requires some assumption and calculation to be performed to obtain a numerical value of the thickness.
The preparation methods, covered in this review, are the cross-sectioning, TEM lamella preparation, angle lapping, and Calo test. Cross-sectioning and the Calo test are macroscopic methods, while TEM lamella preparation and angle lapping are microscopic procedures performed with a focused ion beam (FIB). In any case, all of them are destructive and are needed before any microscopic analysis, whether it exploits light, such as optical microscopy; electrons, in the case of the scanning electron microscope (SEM) (with both backscattered (BSE) and secondary (SE) electrons), transmission electron microscope (TEM) and scanning transmission electron microscope (STEM); or ions, e.g., scanning ion microscopy (SIM).
Another destructive preparation method involves masking the sample to leave the substrate partially uncovered to analyse the height profile of the sample with a profilometer [2] or atomic force microscopy (AFM) profilometry [3].
Other analytical techniques are self-consistent, and they only require that the sample surface be properly cleaned. These techniques analyse the sample along the direction perpendicular to the surface instead of using a lateral view of the cross-section, and even in this case there are representatives of both the two groups of destructive as well as non-destructive methods. With respect to the microscopy techniques, these ones do not give direct information about the thickness and the signal must be analysed to deconvolute all the information. For this reason, we must know as much information as possible about the sample to make the right assumptions on its nature and obtain reliable results. The X-ray fluorescence spectroscopy (XRF), in addition to being an excellent technique for compositional determination, is by far the most common technique used in the industrial field for thickness measurement and quality control as it allows fast, non-destructive analysis and with minimal sample preparation [4]. In addition to that, with XRF, practically all the elements of the periodic table can be analyzed, except for the lighter ones (if not with some precautions), whether conductive or not. The thickness can be extrapolated from XRF data by means of standards [5], using the analytical equations of the fundamental parameter method (FP) [6] or through Monte Carlo (MC) simulations [7,8]. Other non-destructive techniques, more commonly used for research purposes, are ellipsometry [9], which however is rarely applicable to metallic coatings are generally not optically transparent, and X-ray reflectivity (XRR) [10,11] that can detect thicknesses from tens of nanometres to some micrometres. Although they are commonly used for quantification purposes, even electron probe microanalysis (EPMA) [12,13] and X-ray Photoemission Spectroscopy (XPS) [14,15,16] could be employed to determinate the thickness information up to the atomic scale. The methods used for the deconvolution of the signal range from the use of standards [12,17], Monte Carlo (MC) simulations [18,19,20], attenuation length [21,22], and three-layer-model or multilayer model analysis [23,24,25,26,27,28]. Also, Rutherford backscattering spectroscopy can be used for the determination of film thickness [29,30]. Furthermore, the thickness of a coating can be determined using destructive sputtering techniques [31] with which the sample is bombarded with ions, the surface is slowly removed, and the elements emitted by it analysed by carrying out a depth profiling analysis. Also part of this group are secondary ion mass spectrometry (SIMS) [32,33] and XPS sputtering using a fixed-angle or an angle-resolved XPS (ARXPS), performing the reconstruction of the thickness with the Laplace transform or maximum entropy method [34,35,36,37,38].
2. Destructive Techniques
2.1. Preparation Methods
2.1.1. Mechanical Cross-Sectioning
The cross-section technique is very widespread and relatively simple to use, even if it requires particular attention and the manual skill of the operator. In fact, if not executed with attention, it can lead to inaccurate measurements. It consists of cutting the sample in half and observing it transversely along the profile of the layers, whose thickness must be measured. This technique, therefore, allows us to directly measure the thickness of the film through a ruler. The entire analysis process consists of three steps: sample preparation (cutting, embedding and lapping); microscopic analysis, through optical or electronic microscopy; and data processing, through dedicated software to convert the image dimensions from pixels into a unit of length through a scale.
The sample preparation process is the most critical and time-consuming step; it is during this stage that artefacts that could invalidate the subsequent analysis could be generated. Usually, the sample is sawn using a disc cutting machine with the use of an abrasive resin disc or a diamond disc, but if the sample allows it, it can also be cut in other ways, such as with a cutter or scissors. The cut must be made perpendicular to the surface, otherwise the thickness analysed will be overestimated due to the parallax error. In addition, the cut must neither be too fast nor overheat the sample in order to avoid damaging or detaching the film; for this reason, many saws are equipped with a liquid cooling system with a direct jet on the sample.
If our interest is to accurately measure the thickness of the outermost film, and this is very thin, the cut could compromise the analysis. To avoid this problem, if the sample is conductive, it can be covered with a galvanic deposition of a few microns in order to protect the layer of interest. Alternatively, the sample can first be incorporated into resin and then cut later, but in this case, it is more complex to perform a cut perpendicular to the surface. Moreover, the hardened resin tends to shrink, remaining not perfectly adherent to the sample and reducing the protection of the external film.
The incorporation can be performed with both hot melt and cold resins. The sample is placed in a housing ensuring that it is perfectly level, and the resin is poured, being careful not to leave air bubbles. The resin used can be both conductive and non-conductive. If the sample will be analysed under an optical microscope, there is no difference, and therefore non-conductive resins are preferred because they are cheaper. On the other hand, if it will be carried out with electronic microscopic analysis, a conductive resin is preferable to avoid polarization phenomena, but a non-conductive resin can also be used if it is possible to graphitize the sample.
The last step in sample preparation is lapping. Near the cut the sample will inevitably be damaged and the films will be altered: there could be both peeling, meaning the detachment of the less adhered films, and a spreading of the softer films. The lapping process is the longest phase, and it can take several hours and consists of polishing the section of the sample with gradually finer abrasives until a mirror surface is obtained. The surface roughness and scratches must be minimized in order to accurately measure the thicknesses. As a rule of the thumb, the surface roughness, and consequently the abrasive grain size, must be in the order of magnitude of the thickness to be analysed or less, generally 1–0.3 microns. The lapping process can be carried out by hand or using special automatic lapping machines. A wet abrasion is carried out first using sandpaper, gradually becoming finer, and then with a cloth soaked in an abrasive suspension. The suspended particles can be alumina or diamond dust. Initially, with sufficiently coarse sandpaper, the part that was damaged during cutting is removed, about 1 mm in depth; then, the mash size of the sandpaper is decreased. A fine grain size is not used immediately because, otherwise, it would not be possible to remove the deepest scratches. In the event of lapping by hand, the sample must not be pressed too hard, and care must be taken not to consume one side more than the other to avoid introducing a parallax error. For this reason, it is advisable to rotate the sample in the same direction as the lapping machine disk and occasionally turn it 90° with respect to its normal axis. When the sample is considered ready, it is washed and inspected visually or, if available, under an optical microscope to check that all the scratches have been flattened. If the sample is sufficiently smooth, the next phase of microscopic analysis can be carried out. As an example, a microscopic analysis of the same cross-section is shown in Figure 1, where comparisons between optical and electron microscopes have been carried out. The multilayer sample is made of brass/Cu/bronze/Pd/Au/Ni. With the optical microscope, it is possible to distinguish most of the layers. Even the thin gold layer is visible, although not quantifiable, but it is not possible to differentiate between the bronze and palladium layers. The layer with thickness over 500 nm can be measured with the optical microscope, but for thinner coatings, and to distinguish brass and palladium, the electron image is necessary.
2.1.2. Ion Beam Cross-Sectioning
The cross-sectioning procedure can be performed also using focused ion beams. By exploiting the sputtering effect of FIBs, it is possible to raster a surface producing precise trenches, which can be used to observe the in-depth evolution of the sample [39]. The trench dimensions can vary from mm (for a Xe or Ar plasma FIBS) to less than 1 µm, with a maximum depth in the range of hundreds of microns. Due to the small dimensions of the holes produced on the surface, this process (unlike mechanical cross-sectioning) can be considered as semi-destructive. This process is also fast with respect to mechanical cross-sectioning, because it permits us to produce clear cuts from which it is possible to characterize the profile of a sample in 20–60 min (depending on the size of the hole). For these reasons, it is particularly well suited for the characterization of thin films with thicknesses below 10 nm. The subsequent characterization step can be performed using SEM or SIM microscopy, or EDX analysis due to the small hole dimensions.
Even if the cross-sectioning process can be performed only by using a FIB for a coarse determination of thick films, films down to 10 µm will require the use of a FIB/SEM equipped with a Gas Injection System (GIS). This because, in order to avoid FIB-induced surface degradation, a thin protective layer above the surface is needed. This can be achieved only by using a beam deposition process achievable with the presence of a GIS. In FIB/SEMs, the protective layer is produced by a two-step deposition process. First, a thin layer of metal is deposited on the surface using the electron beam (e-beam deposition). Then, a thicker metallic layer is deposited using the ionic beam (i-beam deposition). The e-beam deposition avoids surface degradation (and thus loss of thickness information on the topmost layer) due to direct impingement of the ionic beam on the surface [40]. The full workflow for the preparation of a cross-section using a FIB is visible in Figure 2.
2.1.3. Angle Lapping
Angle lapping is a sample preparation method used to increase the resolution in thin-film thickness determination of the adopted microscopy characterization technique; it is based on a change in cutting geometry with respect to cross-sectioning (Section 2.1). During traditional preparation procedures, in order to unshed the stratigraphic information from the sample, the cutting plane is perpendicular to the surface (θcut = 90°) [41]. The uncovered section gives direct stratigraphic information on the displacement and thicknesses of the layers above the substrate. Instead, in angle lapping, the sectioning cut is performed at very low angles with respect to the sample surface (θcut < 10°). This produces a “magnification effect” on the newly created section surface, on which all the layers appear stretched (Figure 3). Knowing the cutting angle and a bit of trigonometry, it is consequently easy to derive the film thickness. The main advantage of this method is the magnification effect, which allows us to overcome the resolution limits of the microscopic technique adopted for the quantification, even for very thin films. As a prerequisite, a very flat film surface is mandatory for a precise determination of the thicknesses of the layers underneath; moreover, particular care must be put in the preparation of the surface after the cut. It is uncommon, especially for mechanically machined soft materials, to result in thin-film deformation on the cutting surface [42]. The origin of this sample preparation technique comes from the first metallographic studies, and it is still considered as a valuable method to overcome the resolution limits of the adopted microscopic characterization methods. Its adoption has shifted to FIB cross-sectioning or lamella preparation, enabling the fine characterization of ultrathin films. It can be used before both optical microscopy and SEM characterization.
2.1.4. Calo Tester
The Calo tester, also known as a ball craterer or crater grinding, is a semi-destructive technique that is not very widespread but extremely practical in some cases; in addition, it is regulated in ISO 26423 [43] (Ex EN-1071 and VDI 3198 [44]). Compared to the cross-section, it has the advantage of being only locally destructive, and therefore the sample, instead of having to be cut in half, is excavated in an area with a diameter of about one millimetre [45,46]. Furthermore, the whole analysis is much faster as the sample does not have to be incorporated and lapped.
This technique consists in fixing the sample on a variable angle support; on it is placed a steel sphere covered by an abrasive suspension; the sphere is in contact also with a rotating cylinder that makes it roll (Figure 4). Within a few minutes, depending on the hardness of the sample and its angle of inclination that is translated to the weight that the sphere impresses on it, a circular crater will form, revealing all of the layers [47]. Since the abrasion angle is very low (due to the diameter of the sphere), layers of even a few microns will have a much greater apparent size and can be appreciable under a normal optical microscope. Obviously, as in the case of the cross-section, only the layers with different colours can be distinguished. Once the diameters of the concentric circles that have been created on the sample have been measured, using a dedicated formula (Figure 4) that takes into account the diameter of the abrasion sphere, the real thickness of the films can be obtained. Manufacturers ensure that the range of thickness that can be measured is between 50 and 0.1 microns.
2.1.5. TEM Lamella Preparation
The ultimate procedure for the thickness determination of thin films is represented by the TEM lamella preparation process. It is in fact possible to extract a small portion of the sample surface, usually a 10 × 5 × 1 micron solid, containing the surface cross-section. This lamella can then be thinned down to less than 100 nm in order to be observed transversally using TEM or STEM [48,49]. This preparation process is important for the characterization of very thin films (below 1 µm) using SEM and TEM. The small thickness of the lamella decreases abruptly the interaction volume, cutting down the signal coming from in-depth SE and BSE. To prepare a TEM lamella, a FIB/SEM equipped with a GIS and a nanomanipulator is required. Moreover, the lamella preparation process is quite complex, and is constituted by numerous steps that can vary depending on the load-out of the adopted machine and the geometry of the sample chamber. Generally, the workflow can be divided into three main stages: in the first stage, the lamella is shaped (carved) directly onto the surface of the sample. In the second stage, called lift-out, the lamella is detached from the sample and is mounted on a TEM support grid. In the third stage, the lamella is finally thinned down to enable transmission electron analysis. This last process is crucial for the obtainment of defect-free lamellas. In Figure 5, an example of a workflow for a Tescan GAIA 3 FIB/SEM is shown: (a) a raw lamella (about 10 × 5 × 1 microns) is carved on the surface; (b) an undercut is performed in order to detach the lamella; (c) the nanomanipulator is moved onto a side of the lamella and soldered to the body, then the lamella is detached from the surface; (d–f) the lamella is moved from the surface of the sample to the TEM support; (g) the lamella is soldered to the support; (h) the nanomanipulator is detached from the lamella; (i) the lamella is thinned down until electrontransparence, and (j) in the end, the lamella quality is tested using the microscope’s built-in STEM detector.
2.2. Microscopic Analysis
2.2.1. Optical Microscopy
In order to measure a cross-section of a sample, it is necessary to use the microscope in reflection mode; additionally, to be able to recognize the different layers, it is necessary that they have sufficiently high contrast, or more simply different colours. Distinguishing different layers made, for example, of the same silvery metals, is impossible, while it is very simple to measure, for example, a silver film on copper or brass.
The lateral resolution, i.e., the minimum distance between two resolved points, is defined by the Abbe principle. The resolution is related to the wavelength of the source used, in this case the visible photons. The theoretical resolution of an optical microscope, not taking into account optical aberrations, using white light, is about 0.2 microns. However, more realistically, the measurement of films with a thickness of less than one micron is difficult and features consistent uncertainty [50,51,52,53].
2.2.2. Electron Microscopy
Scanning electron microscopy (SEM) is used when the thicknesses are too small to be analysed with the optical microscope or if the layers have too low contrast among them. As a general rule, if the cross-section of a sample can be adequately analysed with an optical microscope, there are no valid reasons to use an electron microscope; in fact, the sample needs some characteristics in order to be measured with an SEM. Moreover, SEM analysis is intrinsically more expensive than a simple optical analysis. Despite this, for most coatings, optical microscopy is not enough to obtain adequate results. A sample that is analysed using SEM must be stable under a high vacuum (about 10−7 bar), must be stable when irradiated by an electron beam, and must be conductive. These limitations can be circumvented by means of some tricks: there are specific environmental or low-vacuum SEM techniques which allow analysis to be carried out under pressure in the order of one Pascal; the acceleration potential of the electron beam can be reduced to a few kV in order to not damage the sample, but decreasing the signal-to-noise ratio; non-conductive samples can be graphitized to avoid the accumulation of surface charges that in this way are dispersed to the ground. However, these problems arise when biological, organic, or polymeric samples are analysed; for metallographic cross-sections, there are rarely complications of this type.
Numerous advances have been made over the last few decades in the field of electron microscopy, and the resolution of these instruments is quite variable and ranges from about 20 nm for older instruments with thermionic emission up to falling below the nanometre scale for new instruments with a field emission source (FEG-SEM). In SEM, the main limit to the resolution is not so much the wavelength of the probe or the electrons as the diameter of the beam, and therefore its focus and collimation. For this reason, a strategy to improve the resolution consists of bringing the sample closer to the source, reducing the working distance, and consequently the opening of the electronic cone. The SEM images can be acquired using secondary electrons (SE), backscattered electrons (BSE), or through a microanalysis map (EPMA). Because of the different natures of the signal, they differ in the depth from which the signal comes (Table 1) and in their volume of interaction (Figure 6). A higher volume of interaction results in a lower lateral resolution, translating to a less-clear separation of the edges between films that have to be analysed [1,13].
SEs are produced when a primary electron from the beam excites an electron of the atoms of the sample to the point of tearing it from the nucleus. These electrons are low in energy (<50 eV) and only those generated most superficially on the surface are detected. SEs carry the morphological information with them, so the contrast in the image is based on the heights of the sample. Since our sample has been levelled, the contrast produced can only be due to the different nature of the material. The SEs in small parts also contain information on the composition since the quantity of electrons emitted is proportional to the atomic number of the element under the electron beam. Thus, layers of elements with significantly different atomic numbers can be distinguished [54,55]. A trick that allows us to observe similar elements with the SE is to chemically etch the sample before the analysis to slightly corrode one material more than another. In this way, morphological differences, clearly visible to the SE, are reintroduced between the different layers. However, the etching must be studied appropriately with respect to the samples that have to be analysed.
BSEs are the primary electrons which, interacting with the positive nuclei of the atoms of the sample, are back-scattered towards the source. For this reason, they are very energetic and penetrating. Therefore, BSEs carry mainly compositional information: heavier elements generate more BSE. By sacrificing part of the lateral resolution, the image obtained will be much more contrasted by highlighting differences between the layers that can be below an atomic number unit for more modern systems, allowing us in some cases to distinguish even deposits of a metal from their alloy (for example copper on brass).
Finally, the EPMA signal can be used. This is the signal with the highest volume of interaction, and for this reason the lateral resolution is very poor when compared with the previous ones. In this case, the subjects to be analysed are not the electrons but the X photons that are emitted from the sample after the interaction with the primary electrons, which contain detailed information on the composition. With EPMA, alloys can be distinguished which vary in composition by a few percentage points. Then the different layers can be highlighted by making a map or a linear scan perpendicular to the layers. However, given the low lateral resolution, films just below the micron limit are difficult to quantify.
Another trick to increase lateral resolution is to reduce the volume of interaction, and this is possible by decreasing the energy of the electrons by lowering their acceleration potential. This greatly reduces the signal, but with modern instruments it is possible to obtain good images using acceleration potentials of only a few kV. This strategy is very limited, however, in the case of the EPMA, since in order to have a good emission of the X photons, the electron beam must have indicatively an acceleration potential one and a half times the energy of the emission peak of interest.
2.2.3. Focused Ion Beam Methods
Another series of techniques enabling the fast characterization of thin metallic films rely on the use of focused beams of ions. Ionic probes display different beam-matter interactions with respect to electrons, resulting in different effects of the impinging beam on the surface [56,57,58,59,60,61]. When a focused ion beam (FIB) hits a surface, it can generate a series of different effects with respect to an electron beam; all these effects are a consequence of elastic or inelastic collisions between the charged particles and the atoms forming the surface. The most important interactions can be listed: (a) surface sputtering and secondary ion emission; (b) deformation of the surface reticule; (c) ion implantation; (d) emission of the electrons; (e) emission of the electromagnetic waves, and (f) heating. All these effects manifest at the same time, but their ratio is strongly dependent on the physicochemical nature of both the probe and targeted surface, and on the apparatus setup. For a certain ion source, parameters such as beam energy, beam current, and impact angle can be tailored to favour one phenomenon with respect to the others. Among all the ion beam-matter interactions, two are widely used for the characterization of thin metallic films: (a) the production of electrons [62] and (c) the sputtering and secondary ion emission [63]. The first exploits a signal which is similar to the one responsible for SEM images, while the second permits us to perform subtractive manufacturing on the surface. The FIB apparatus could in fact perform both sample preparation and analysis in a single workflow by preparing the surface cross-section and acquiring its images, though not all FIB columns are fitted to perform well in both processes. As already mentioned, different elements could be used as ion sources; their charge/mass ratio could modify deeply the ratios at which high sputtering and SE production manifest. Today, the main commercial FIB machines are engineered to work using He, Ga, Ar, or Xe ions [64]. Smaller ions, like He, are well suited for imaging purposes, while bigger ions like Ar and Xe are used for fast sputtering; Ga FIB machines are considered a good trade-off between the two effects and are still considered as the best choice for all-purpose tasks.
From a technical point of view, FIB machines are very similar to SEMs. In both of them, we have a column that can be divided into: (a) a top part composed of the source, responsible for the ion/electron generation, and (b) a bottom part, the focusing apparatus, a series of electrostatic/electromagnetic lenses, condensers, and apertures to focalize and control the beam [57]. In FIBs, the working principle exploited for ion emission varies depending on the physicochemical properties of the element of choice; we could divide the sources into three main branches: Gaseous Field Ionization Sources (GFISs) for He [65], Liquid Metal Ion Sources (LMISs) for Ga [57], and Plasma sources for Ar and Xe [66,67]. Despite SEMs, in FIBs, the focusing apparatus is composed of electrostatic elements (lenses, condensers, etc.) rather than electromagnetic. Ions, in fact, suffer weakness from Lorentz forces due to their slower travelling speeds (with respect to electrons) in the column, meaning also that these instruments are less prone to suffer from stray external magnetic fields. Ion sources are not interchangeable; the focusing apparatus must be finely engineered to suit the particular physical properties of the source element. This means also that the choice between different FIB machines, exploiting different sources, must be carefully planned with respect to the need. Historically, industrial FIB columns were intended as standalone instruments, and these devices were diffused especially in the semiconductor/electronic industry, where their use both for quality checks and prototyping dates back to the ‘70s. Today FIBs can be found easily when paired with an SEM column [68]. This double column configuration allows a vast array of different procedures for the characterization of materials in the range of mm down to the nm for all R&D fields. FIB/SEMs can be equipped with a vast array of accessories and sensors to enable different characterization techniques. Among the available accessories, the gas injection system (GIS) has a particular relevance because it permits additive manufacturing in the range of tens of nm using both the ionic and the electronic beams. The GIS is constituted by a series of external reservoirs containing the precursors of the elements we want to deposit onto surfaces, which are heated to produce a reactive gas. This gas is injected using a hollow needle in the vicinity of the surface of the sample, where it forms a cloud between the beam and the surface. The beam particles hitting the precursor produce the degradation of the molecules, followed by the precipitation of the elements onto the surface. It is possible to use both the electron beam (e-beam deposition) and the ionic beam (i-beam deposition) [69] to achieve deposition. In the following paragraphs, the main techniques which can be exploited for metallic layer thickness determination will be presented: an analytical technique (Scanning Ion Microscopy) and two preparation techniques (TEM lamella preparation and angle lapping). All the FIB methods are well suited for the characterization of metallic films of thicknesses ranging from 50 µm to 10 nm.
2.2.4. Scanning Ion Microscopy
Scanning Ion Microscopy (SIM) can be used as an alternative to SEM for the study of cross-sections obtained using mechanical methods. The main advantage in using charged atoms instead of electrons is related to the smaller ion mean free path inside the matter [65,70]. This effect is responsible for a smaller beam-surface interaction volume to a higher number of surface SEs, thus leading to crisper images. Moreover, the number of SEs produced per impacting Ion is much bigger with respect to the number of SEs produced by the impact of an electron, greatly enhancing the brilliance of the signal [71]. Ions are also particularly sensitive to crystal orientation; images acquired using this method tend to possess strong crystalline contrast due to the enhanced channeling effect [72]. Ionic imaging can be performed using the same SEM SE detector, with a lesser lateral resolution with respect to electrons.
2.2.5. Data Analysis
Once the image is obtained with a microscopic technique, the pixels must be converted into a unit of length. Most of the software that allows us to acquire microscopic images (both optical and electronic) commonly have a tool for the extraction of this information, otherwise there is free or paid software that allows us to do the same job as ImageJ, Gimp, and Adobe Photoshop, to name the most well-known. In order to convert the pixels to a length on the image, a reference scale must be printed on it. Through that scale, we can get all the dimensions of interest in the image. The thickness of the films can therefore be measured, being careful to carry out the measurement perpendicular to the film. By making the measurement on several points, a statistic of the thickness can also be carried out. However, in the event that the edges of the film in the image are not very defined, finding the limits using the eye could be complex, as shown in the blurred image of the Au film reported in Figure 7. In these cases, instead of analysing the image, it is more practical to observe the profile graph in which the greyscale values are reported. If the different layers have a contrast between them, they also have different grey values; the delimitation between one layer and another can be defined as the point where the value is intermediate between the two layers or, more rigorously, at the inflection points of the graph. In this case, the spatial resolution, and therefore the uncertainty of the measurement, is defined as the distance between the points where the variation of the grey value is in the range of 20%–80% [73], as required by ISO 18516 [74]. On the other hand, an incorrect method used to make the separation of the layers more defined is digital post-processing of the images via acting on brightness and contrast. In fact, contrary to varying these parameters during the measurement, by performing software alterations of the image, the edge of the film can move as the different shades of grey are processed, as demonstrated in Figure 7.
3. Non-Destructive Techniques
3.1. X-ray Fluorescence Spectroscopy
X-ray fluorescence spectroscopy (XRF) is an analysis tool widely used for the elemental analysis and chemical analysis of materials. When materials are exposed to high-energy X-rays, ionization of their component atoms may take place, exciting them, and during the relaxation process characteristic X photons are emitted and detected for analysis. Due to the incident of high-energy X-rays of the inner shell (K, L, M, etc.), transition phenomena occur within 100 fs, producing characteristic fluorescence radiation. Ionization consists of the ejection of one or more electrons from the atom and may occur if the atom is exposed to radiation with energy greater than its ionization energy. X-rays and gamma rays can be energetic enough to eject tightly held electrons from the inner orbitals of the atom. The removal of an electron in this way makes the electronic structure of the atom unstable, and electrons in higher orbitals “fall” into the lower orbital to fill the holes left behind. In falling, energy is released in the form of photons with an amount of energy equal to the difference between the two orbitals involved. Thus, materials emit radiations of the characteristic energies of the present atoms. A variety of samples in different states, such as solids, powders, and liquids, can be analysed using this technique. It can also be used to measure the composition, the thickness of the coating, and the layers. The characteristic photons of the sample are collected by a detector that uses the same working principle as EPMA. Both the source photons as well the emitted ones could pass through an analysing crystal that acts as monochromator differentiating between energy dispersive (ED) XRF without an analysing crystal, wavelength-dispersive (WD) XRF in which the emitted photons are selected with a monochromator, and monochromatic wavelength dispersive (MWD) XRF, in which two optics are used—one for the source and one for the emitted photons. For reasons of cost and ease of use, energy dispersion instruments are the most widely used. The incoming high-energy beam is very penetrating in Figure 8; for this reason, the maximum detectable thickness is related to the energy of the emitted X-rays.
XRF is the most common instrument used by industries for film thickness investigation since it is fast, non-destructive, and relatively simple to use, making it perfect for the quality control of the products [75,76], and for this reason there are present also standard procedures to perform the measurement like ISO 3497 [77] and ASTM B568 [78]. Commercial instruments can measure easily the thicknesses of almost every material (with some restriction for lighter elements), whether conductive or not, in the range of 10 nm to 100 µm [79,80]; nevertheless, depending on the materials under investigation and the instrumental settings, the limits of measurement could be extended from less than 1 nm [81] to a few centimetres [82]. The lateral resolution of XRF is very low, and spot size commonly ranges from 0.1 to 15 mm. The relative intensity (normalized respect to the bulk element) emitted from a film follows an exponential trend [18], but could be approximated to a second-order curve for small films far from the saturation thickness [7]. The emission of a gold film on Cu substrate, as a function of the thickness, is reported in Figure 9 using a log-log scale. In this case, the range of thicknesses between the relative intensities of 0.9 (semi-infinite thickness [83]) and 0.1 (infinitely small) of gold is between 0.2 µm and 50 µm.
The output of the instrument is a spectrum in which the position of the peaks corresponds to the spectroscopic emission of the elements present in the sample, while the intensity is correlated with the sample composition in the volume of the interaction of the incident beam. For this reason, there is no direct information on the thicknesses, but the intensity of the peaks in the spectra will be a function of the thickness. In fact, a sample with a thicker coating will emit more photons from the film and less from the substrate than a thinner one. Since no information about the thickness can be extracted a priori from the spectra, only with the right assumptions of the nature of the sample and the use of an adequate calibration curve can the thickness information be deconvoluted. This complication could yield high uncertainties or even wrong results.
Deriving the coating’s thickness from the X-ray spectrum requires an experimental calibration curve that employs standards; however, due to the large dependence of the X-ray spectrum on the nature of the coating and the substrate, standards are not always available. The variability of thickness, layer composition, multilayer architectures, and substrate chemical nature creates difficulties in producing certified standards. This issue is critical in industrial applications; indeed, the determination of precious metal coatings in the fashion industry is a major one, where the products are made with many coatings and substrates with extreme variability in the system. The calibration curve obtained with standards of known thickness was used to measure vanadium(V) oxide nanometric films on glass, with portable XRF measuring the attenuation of the Ca emission [5]. Hamann [84] was able to detect a fraction up to 1% of a monolayer of over 20 samples without the use of standards or models, combining WD-XRF and XRD measurements to obtain the proportionality constant between X-ray emitted intensity and the number of atoms per unit area.
Nowadays, the most common approach is the use of the fundamental parameter (FP) method [6,79,85,86]. FP relies on theoretical equations that consider the composition and thickness of the sample to evaluate the XRF intensity. Practically, the FP method is combined with a few pure element empirical standards to correct unpredicted deviations due to matrix effects [87,88]. With the FP method, it is possible to determine the film thickness of single- and even multilayer samples if the structure and the composition are known exactly; nevertheless, the error correlated with the measurement is significant. The typical accuracy for single-layer samples is ±5%, while for multiple-layer samples this value grows to ±10% for the upper layer and ±37% for the first underlayer [89,90,91] due to the inaccuracy in the method for complex samples. Additionally, very often, the thickness and composition of the underlying layers in multilayer architectures are not exactly known, and they are introduced in the measurement software using an initial estimation [92]. The FP method was investigated by many authors for multi-layered samples in the micron range (Au/Ni/Cu [89]), as well as in the nanometre range (Ni/Cu/Si [93]). This method is very useful when it is difficult to obtain accurate certified reference materials for layer thickness calibration, such as in the case of semiconductor research [94]. Vrilink [90] showed a good correlation between FP and SEM and profilometry measurement of multilayer samples with different compositions (Rh, Ta, W, Ti, Pd, Pt, Ni, Au, Cr) between 20 and 250 nm, considering the density variation for thin film. Ager [92] highlights the discrepancy between SEM and non-destructive techniques like RBS and XRF measurement due to differences in density between bulk metal and thin films due to porosity; in the paper, comparisons between references and electroplated samples were performed to prove the hypothesis for ancient gildings, but his considerations are also valid in many other fields. When exploiting the FP, both the emission line of the top layer as well the reduction in the intensity of the underlying layer can be used for thickness determination, as shown in a 2017 study in which the results of ALD oxides samples are tested [83].
An alternative to the use of standards and the FP method consists of a semiquantitative approach based on calibration curves obtained with a simulation software using Monte Carlo (MC) algorithms. During the simulation, when the materials and the architecture to simulate are chosen, it is also possible to specify the density of the materials; in this way, the user can decide to simulate materials that have a porosity different from the nominal one due to the deposition method, as for example, happens during electroplating in which the density of the coatings is often lower than that of the bulk material. Moreover, the MC method simulates X-ray spectra using a statistical approach that counts the photon interactions in the sample. With this approach, inhomogeneities of the sample, spectral and spatial distributions of the beam, polarization effects, photo-absorption, multiple fluorescence, and scattering effects, which are difficult to model with the FP method, can be considered. The simulation approach is not very common, probably because the FP was preferred for many years since it was computationally favourable, but with the latest technological developments even a personal computer can obtain a good simulation in a relatively small amount of time. The two main pieces of software that provide simulated spectra with the MC approach are XRMC [95] and XMI-MSIM [96]. Both codes use the Xraylib database [97,98]. XRMC is generally used for complex 3D geometries, while XMI-MSIM can only simulate samples composed of parallel layers, but for simple geometries, XMI-MSIM is currently superior to XRMC in simulating XRF experiments [99]. Thickness evaluation using the MC method is diffuse in the field of cultural heritage applications. Schiavon [100] uses the XRMC code to obtain the thickness and composition of Nuragic manufacts, comparing the simulations with the experimental measurements to confirm hypotheses based on bulk chemical composition, structural observations, and historical information. A similar approach was used by Brunetti [101] and Bottaini [102] for Peruvian and Portuguese manufacts: a MC simulation is performed defining the experimental setup and the sample, then the simulated spectrum is compared to the measured one visually and with the chi-squared test. If differences are found, the model is corrected until the two spectra match, determining both the composition and structures. Besides the comparative method, MC simulation can be also employed to obtain a calibration curve based on a simulated standard. XMI-MSIM has been successfully used for this purpose for electroplated samples, normalizing the result with respect to a semi-infinite bulk element, with even better results than with the FP semi-empirical method [7]. A similar approach was used by Pessanha [8] for cultural heritage gildings on Pb using the PENELOPE code, exploiting the ratio between two lines of the same element for normalization, as if it were an internal standard. This latter method of data processing has been widely used by Cesareo in the last decade [103,104,105,106,107,108], exploiting the differential attenuation (or self-attenuation) of the substrate (or coating) of two lines of the same element. The curves of the X lines ratios over the thickness can be obtained by knowing the value of these ratios for an infinitely thin layer and a semi-infinite one, and these values are tabulated.
XRF techniques are not commonly used for organic films since they do not provide fluorescent radiation detectable in air. Porcinai [109] used the X-ray attenuation of the substrate, calculating the ratio between two emission lines of the same element, to evaluate the thicknesses of polymers for protective purposes; empirical, semi-empirical, and analytical (FP) methods were compared. Recently, De Almeida [110] used a multivariate approach to evaluate multiple regions in the XRF spectra and obtain the thicknesses of polymeric films. To keep up with all the innovations in this field, every year a review [111,112,113] on the latest advances in XRF group techniques is published to highlight developments in instrumentation, methodologies, and data handling.
3.2. Electron Probe Microanalysis
The electron probe microanalysis (EPMA) was first developed in 1951 by Casting [114]. EPMA permits us to analyse the compositions of homogeneous materials in a region of few microns from the surface. The EPMA can be conducted using two different approaches: wavelength dispersive X-ray spectroscopy (WDS) [115] or energy-dispersive X-ray spectroscopy (EDS) [116,117,118,119,120]. WDS is generally considered an excellent method for microanalysis because it is more sensitive and has a higher resolution than EDS, but it is more expensive and needs a dedicated device. EDS, on the other hand, can be conducted by simply coupling a detector to the SEM, a widespread instrument in the academic and industrial sphere, especially due to the recent spread of inexpensive benchtop instruments.
EPMA was mentioned before in the destructive section for mapping the cross-sectioned samples. Here, it is used as a non-destructive technique measuring the sample perpendicularly to the surface [121]. This technique interprets every sample as homogeneous since the output information is a spectrum. For this reason, there is no direct information on the thicknesses, but the intensities of the peaks in the spectra are a function of the thickness, as is the case for XRF. EPMA is not known much for thickness measurements, but it is an attractive candidate because it enables fast, quantitative [122,123], and non-destructive [124] analysis with the additional benefit of having a lateral resolution in the micron range [125]. In addition to that, the probe (electrons) is not very penetrating (Figure 10), and for this reason it is possible (by adjusting the beam energy) to analyse ultrathin films or just the top layer of a film to obtain its composition [126,127,128,129,130].
The EPMA detector is present as an upgrade to conventional SEM, but most of the instruments come at least with the EDS detector by default. The electron bombardment of the beam excites the atoms in the sample, knocking out the electrons from the inner shells. Such a state is unstable, and the resulting electron hole is immediately filled by a higher-energy electron from a higher atomic orbital. The energy difference is released in the form of an X-ray quantum. The resulting X-ray radiation is characteristic of the transition and the atom. For a single element, different transitions are allowed, depending on which shell the higher-energy electron comes from and which shell the hole must be filled in. This results in X-ray quanta, which are marked with Kα, Kβ, Lα, etc. The energy of an X-ray line (the position of the lines in the spectrum) is an indicator of which element is under investigation. The intensity of the line depends on the concentration of the element within the sample. Furthermore, the electrons, slowing down in the electric field of the atomic nuclei, generate X-ray braking radiation, called bremsstrahlung, which constitutes the continuous background of the EPMA spectrum. The EPMA detector exploits the energy interaction between X-rays and a suitable material, generally represented by a single silicon crystal doped with lithium, coated at both ends with a gold conductive layer at a temperature of −192 °C with liquid nitrogen. Other variants are the high purity germanium detectors and silicon drift detector (SSD) with Peltier cooling. When an X-ray photon is absorbed in the sensitive area of the detector, then electron-hole pairs are produced, and this causes the production of an electric current which is then sensitively amplified. In WDS instruments, a diffracting crystal is present that selects the photons to be sent to the detector, which measures only the number of pulses, i.e., photons. In the EDS system, there is not a photon selector, and thus the signal of each photon is processed to obtain its energy value; during this time, the system rejects every other signal, resulting in a dead time. A high dead time produces high spectra with high resolutions but low signals because many photons are rejected. On the other hand, a low dead time produces high signals but wide peaks. A longer process time is needed for quantitative analysis where spectral resolution is important, whereas if maximizing the number of X-rays in a spectrum or map is most important, a shorter process time can be used. Si(Li) detectors operate at count rates of about 1 to 20 kCPS with optimal dead times of 20–30%. The reason why SDDs are now preferred to Si(Li) detectors is that they can handle much higher count rates of >100 kCPS and dead times of 50%. The count rate can be optimized by adjusting the beam current (probe current or spot size) and the processing time. It is important to select a processing time and beam current that will give an acceptable X-ray count rate and detector dead time for the analysis, as well as the desired spectral resolution. The typical energy resolution of an EDS detector is 130–140 eV, while it is of about only 10 eV for WDS systems. Moreover, EDS systems have much lower count rates and poor reproducibility, generally of a factor of ten with respect to the WDS detectors. The beam energy can be varied to increase the sensitivity for thinner or thicker coatings (Figure 11).
The film thickness can be obtained from the measured spectra through various approaches. The calibration curve can be obtained using standards of known thickness [131] or Monte Carlo simulations [19,20]. In terms of the quantification method, the quantity used in the calibration curves, multiple alternatives were evaluated: the K-ratio [127,132,133,134], which is the ratio between the intensity in the sample and the intensity in a standard with known composition, commonly used in que quantification analysis; the ratio of intensities [135], and the atomic ratio [131], obtained performing the ZAF correction algorithm on the K-ratios. Both the absolute thickness [136,137] as well as the mass thickness [138,139,140,141] were taken into consideration for the quantification.
In the last fifty years, many pieces of software were written to simulate EDS spectra [142]; many of them are written by researchers and some were commercial: MAGIC [143,144], STRATAGEM [145,146,147], GMRFILM [122], Electron Flight Simulator [148,149], ThinFilmID [150] and LayerProbe [150,151], pyPENELOPE [152,153], Win X-Ray [154,155] and MC X-Ray [154,156], XFilms [157], CASINO [124,158,159,160,161], CalcZAF [162,163] and DTSA-II [164,165,166]. Many of these pieces of software exploit the PENEPMA algorithm [153]. PENEPMA is a simplified version dedicated to EPMA, written to perform the simulation of X-ray spectra, and calculates different quantities of interest of another algorithm called PENELOPE. PENELOPE (Penetration and ENErgy LOss of Positrons and Electrons) is a general-purpose Monte Carlo code system for the simulation of coupled electron-photon transport in arbitrary materials. PENELOPE covers the energy range from 1 GeV down to, nominally, 50 eV. The physical interaction models implemented in the code are based on the most reliable information available at present, limited only by the required generality of the code. These models combine results from first-principles calculations, semi-empirical models, and evaluated databases. It should be kept in mind that, although PENELOPE can run particles down to 50 eV, the interaction cross-sections for energies below 1 keV may be affected by sizeable uncertainties; the results for these energies should be considered as semi-quantitative. PENELOPE incorporates a flexible geometry package called PENGEOM that permits automatic tracking of particles in complex geometries consisting of homogeneous bodies limited by quadratic surfaces. The PENELOPE code system is distributed by the OECD/NEA Data Bank. The distribution package includes a report [167] that provides detailed information on the physical models and random sampling algorithms adopted in PENELOPE, on the PENGEOM geometry package, and on the structure and operation of the simulation routines. PENELOPE is coded as a set of FORTRAN subroutines that perform the random sampling of interactions and the tracking of particles (either electrons, positrons or photons). In principle, the user should provide a main steering program to follow the particle histories through the material structure and to keep score of the quantities of interest. In PENEPMA, photon interactions are simulated in chronological succession, allowing the calculation of X-ray fluorescence in complex geometries. PENEPMA makes extensive use of interaction forcing (a variance-reduction technique which artificially increases the probability of occurrence of relevant interactions) to improve the efficiency. CalcZAF [162] simulation software is based on PENEPMA and is a general-purpose software package for the simulation of both relativistic and sub relativistic electron interactions with matter. Even in this case, the characteristics and the geometry of the detector are not taken into account, and the output consists of a line-like unconvoluted spectrum. DTSA-II [164] shares many physical models with PENEPMA, but was designed exclusively for the simulation of X-ray spectra generated by sub relativistic electrons. DTSA2 uses variance reduction techniques unsuited to general-purpose code. These optimizations help the program to be orders of magnitude more computationally efficient while retaining the detector position sensitivity. Simulations are executed in minutes rather than hours, and differences that result from varying the detector position can be modelled. It is possible to insert the characteristics and the geometry of the detector in DTSA2, which is capable of handling complex sample geometries. The primary and secondary bremsstrahlung and fluorescence can be calculated. The outputs result in a real-looking spectrum since it is deconvoluted considering the detector resolution; even the electron trajectories can ben visualized. The CASINO [158] is a single scattering Monte Carlo simulation software of electron trajectory in a solid, specifically designed for low beam interactions in bulk and thin foils. This software can be used to generate many of the recorded signals (X-rays and backscattered electrons) in a scanning electron microscope. This program can also be efficiently used for all the accelerated voltages found on a field emission scanning electron microscope (0.1 to 30 keV). The characteristics and the geometry of the detector are not taken into account, and the output is not a spectrum, but the characteristic emission lines intensities are shown as a function of the depth.
The X-ray depth distribution of the emissions is described by the φ(ρz) curve, which can be used for the determination of thin film thicknesses [12]. The thickness of the film can vary between two extremes relative to the curve: extremely thin or extremely thick [137]. In the first case, the emission corresponds to a bulk sample with the composition of the substrate, in the second case, to a bulk with the composition of the film. In the intermediate cases, the φ (ρz) curves vary between these two extremes. The maximum thickness that can be analysed with the EPMA method is of some microns: this is determined by the acceleration potential of the electrons together with the atomic number of the elements in the sample [168]. On the other hand, the minimum detectable thickness (lower detection limit) is given by the combination of the X-ray energy characteristics of the elements in the sample and the properties of the detector and can be as low as a few monolayers or less [169].
STRATAGEM software Kühn [146] was capable of obtaining both the elemental composition and thickness of a thin film ternary alloy Pd-Ni-Co co-deposited via magnetron sputtering on a silicon wafer using ED-EPMA in the range of 50 to 250 nm. The results were confirmed by AES and XPS measurement, for the composition, and by SEM imaging for the thickness. The volume of interanion was confirmed using CASINO simulations. A similar approach was used for the determination of electrodeposited Ni, Pd, and Au on Cu comparing the results of CASINO, CalcZAF, and DTSA [18]. A comparison between GRMfilm, DTSA-II, and PENEPMA was performed for very thin films (5–20 m) of Al and Cu on Bi. In this study, the variation in the film density with respect to the bulk material was also evaluated [170]. Ultra-thin films of Ge, Sn, Ag and Au on Si wafer were evaluated also by Campos, performing multiple analyses with different beam energies [171]. DTSA-II was used also to determine the sputter coater deposition of Ti and Ag on Si for medical applications [172]. Osada [136] developed its new MC simulation software to evaluate the thickness of aluminium oxide on aluminium sheets in the range of 5 nm to 50 nm. Recently, in 2018, Darznek performed thickness measurement tilting the sample off to the normal incidence angle to increase the signal of the superficial coatings, specifically to determine the thickness of chromium film on a silicon substrate. With this approach, he was able to determine up to 1014 atoms per square centimetre with an error of less than 10%, exploiting the K-ratio measurement with the MC simulation.
In 2016, Sokolov [124] measured the thickness of Silicon dioxide and silicon nitride thin films using EDS, varying the penetration depth of the analysis, changing the acceleration voltage of the beam, and correlating the thickness of the film with the signal of the substrate elements to the collected noise. Stanford [173] in 2020 measured the oxide layer formation on Pu from 35 nm to 400 nm using measured standards to build the k-ratio calibration curves of oxygen through FIM-SEM analysis. Previously, Bastin made a massive study collecting the K-ratios of Al [174] and Pd [175] of films from 10 to 320 nm in thickness at various beam energies between 3 kV and 30 kV on many substrates between Be and Bi.
Even the thickness of multi-layered samples can be measured using EPMA measurements [176,177]. In 2019, Pazzaglia [178] developed a new model for the standardless determination of mass thickness and composition using EDS for multilayer samples with an accuracy of 10 μg/cm2. Previously, Lesch used a sputtering method with EPMA, the signal deconvolution with the max entropy algorithm, to provide the thickness of Ti/Al/Ti layers deposited on Si.
EPMA was used by some authors to evaluate important information about layered samples besides their thickness: Christien [119] used the EDS measurement to determine the interdiffusion coefficient between thin films of miscible metals. Using various annealing temperatures and the Fick’s diffusion equations, he was able to estimate the coefficients for a Ni film on Pd. Darznek [179] proposed a method to evaluate the thickness uniformity of nanofilms by means MC simulations correlating the peak intensities in the EDS spectrum with the film thickness. In 2016, Ortel [116] developed a technique combining EPMA measurement for mass deposition determination and SEM analysis for thickness determination to obtain the change in density of the films with respect to the bulk materials and consequently to extrapolate the porosity of the coatings.
4. Conclusions
Thickness measurement is a challenge that affects many scientists and companies. The composite materials on which a coating is present are ubiquitous and allow us to obtain properties that a single element would not have. Thickness is decisive for obtaining these properties, and thus, its control and measurement are important. In this review, we describe the main techniques, both for preparation and analysis, which are used both in research and in the industrial sector for this purpose. In fact, there is no perfect technique suitable for any type of sample, but the most appropriate route must be chosen for every need. At the end of this work, it seems appropriate to report a rough comparison between the various methods with regards to the range of thickness that can be analyzed (Figure 12) and the time required to carry out the analysis (Figure 13). Obviously, the costs are also an important parameter to consider, and they are very variable: from a general point of view, mechanical and optical techniques are cheaper than the electronic, ionic, and spectroscopic ones. It should also be taken into account that some instruments are supplied with multiple combined analyzers, such as in the case of SE, BSE, and EDS or for FIB techniques.
Talking about the measurable ranges of thickness, microscopic techniques tend to have only a lower limit, dictated by the aberrations that the beam undergoes under certain dimensions, while it is possible to lower the magnifications until observing shapes above the millimeter level. Spectroscopic techniques instead suffer from the attenuation of the signal inside the sample and, therefore, cannot measure coatings beyond a certain size which appear as infinitely thick. Furthermore, for those analysis techniques that require sample preparation, the range that can be analyzed is the intersection of the ranges of the individual techniques.
Considering now the preparation and measurement time, it is highly dependent on the presence of automated systems and on the experience and manual skills of the operator, as well as on the degree of accuracy required for the result. In general, the XRF, in addition to being extremely versatile, is the fastest technique, not even requiring sample preparation. On the other hand, microscopic techniques coupled to cross-sectioning are extremely widespread as they allow us to obtain a result in which the thickness is directly visible. Furthermore, cross-sectioning, although time-consuming, is a procedure that is generally automated.
Some of the illustrated techniques are well established and have not undergone many innovations in recent years, or engineering optimization of performance and costs; for others, however, research is still very active, as we have shown in this work, and therefore they must be followed with interest in order to make the best use of them as powerful analytical tools.
Author Contributions
Writing—original draft preparation, W.G. and E.B.; writing—review and editing, W.G. and E.B.; supervision, A.L. and M.I.; project administration, A.L. and M.I.; funding acquisition, A.L. and M.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the PRIN (“Progetti di Ricerca di Rilevante Interesse Nazionale”), which made possible the project “Novel Multilayered and Micro-Machined Electrode Nano-Architectures for Electrocatalytic Applications (Fuel Cells and Electrolyzers)”, grant number 2017YH9MRK. The authors also acknowledge “Ente Cassa di Risparmio di Firenze” Grant Number n. 2013.0878, Regione Toscana POR FESR 2014–2020 for the project FELIX (Fotonica ed Elettronica Integrate per l’Industria), Grant Number 6455.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Whiteside, P.; Chininis, J.; Hunt, H. Techniques and Challenges for Characterizing Metal Thin Films with Applications in Photonics. Coatings 2016, 6, 35. [Google Scholar] [CrossRef]
- Nash, C.R.; Fenton, J.C.; Constantino, N.G.N.; Warburton, P.A. Compact chromium oxide thin film resistors for use in nanoscale quantum circuits. J. Appl. Phys. 2014, 116. [Google Scholar] [CrossRef]
- Lobo, R.F.M.; Pereira-da-Silva, M.A.; Raposo, M.; Faria, R.M.; Oliveira, O.N. In situ thickness measurements of ultra-thin multilayer polymer films by atomic force microscopy. Nanotechnology 1999, 10, 389–393. [Google Scholar] [CrossRef]
- Sitko, R. Quantitative X-ray fluorescence analysis of samples of less than “infinite thickness”: Difficulties and possibilities. Spectrochim. Acta Part B At. Spectrosc. 2009, 64, 1161–1172. [Google Scholar] [CrossRef]
- Lopes, F.; Cardozo Amorin, L.H.; da Silva Martins, L.; Urbano, A.; Roberto Appoloni, C.; Cesareo, R. Thickness Measurement of V2O5 Nanometric Thin Films Using a Portable XRF. J. Spectrosc. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Criss, J.W.; Birks, L.S. Calculation Methods for Fluorescent X-Ray Spectrometry: Empirical Coefficients vs. Fundamental Parameters. Anal. Chem. 1968, 40, 1080–1086. [Google Scholar] [CrossRef]
- Giurlani, W.; Berretti, E.; Innocenti, M.; Lavacchi, A. Coating Thickness Determination Using X-ray Fluorescence Spectroscopy: Monte Carlo Simulations as an Alternative to the Use of Standards. Coatings 2019, 9, 79. [Google Scholar] [CrossRef]
- Pessanha, S.; Manso, M.; Antunes, V.; Carvalho, M.L.; Sampaio, J.M. Monte Carlo simulation of portable X-ray fluorescence setup: Non-invasive determination of gold leaf thickness in indo-Portuguese panel paintings. Spectrochim. Acta Part B At. Spectrosc. 2019, 156, 1–6. [Google Scholar] [CrossRef]
- Malarde, D.; Powell, M.J.; Quesada-Cabrera, R.; Wilson, R.L.; Carmalt, C.J.; Sankar, G.; Parkin, I.P.; Palgrave, R.G. Optimized Atmospheric-Pressure Chemical Vapor Deposition Thermochromic VO2 Thin Films for Intelligent Window Applications. ACS Omega 2017, 2, 1040–1046. [Google Scholar] [CrossRef]
- Krumrey, M.; Gleber, G.; Scholze, F.; Wernecke, J. Synchrotron radiation-based x-ray reflection and scattering techniques for dimensional nanometrology. Meas. Sci. Technol. 2011, 22. [Google Scholar] [CrossRef]
- Serafińczuk, J.; Pietrucha, J.; Schroeder, G.; Gotszalk, T.P. Thin film thickness determination using X-ray reflectivity and Savitzky-Golay algorithm. Opt. Appl. 2011, 41, 315–322. [Google Scholar]
- Sokolov, S.A.; Milovanov, R.A.; Sidorov, L.N. Determination of the Thickness of Thin Films Based on Scanning Electron Microscopy and Energy Dispersive X-Ray Analysis. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2019, 13, 836–847. [Google Scholar] [CrossRef]
- Goldstein, J.I.; Newbury, D.E.; Michael, J.R.; Ritchie, N.W.M.; Scott, J.H.J.; Joy, D.C. Scanning Electron Microscopy and X-Ray Microanalysis; Springer Publishing: New York, NY, USA, 2018; ISBN 978-1-4939-6674-5. [Google Scholar]
- Jablonski, A. Evaluation of procedures for overlayer thickness determination from XPS intensities. Surf. Sci. 2019, 688, 14–24. [Google Scholar] [CrossRef]
- Walton, J.; Alexander, M.R.; Fairley, N.; Roach, P.; Shard, A.G. Film thickness measurement and contamination layer correction for quantitative XPS. Surf. Interface Anal. 2016, 48, 164–172. [Google Scholar] [CrossRef]
- Cumpson, P.J. The Thickogram: A method for easy film thickness measurement in XPS. Surf. Interface Anal. 2000, 29, 403–406. [Google Scholar] [CrossRef]
- Alexander, M.R.; Thompson, G.E.; Zhou, X.; Beamson, G.; Fairley, N. Quantification of oxide film thickness at the surface of aluminium using XPS. Surf. Interface Anal. 2002, 34, 485–489. [Google Scholar] [CrossRef]
- Giurlani, W.; Innocenti, M.; Lavacchi, A. X-ray Microanalysis of Precious Metal Thin Films: Thickness and Composition Determination. Coatings 2018, 8, 84. [Google Scholar] [CrossRef]
- Kyser, D.F.; Murata, K. Quantitative Electron Microprobe Analysis of Thin Films on Substrates. IBM J. RES. Dev. 1974, 92, 352–363. [Google Scholar] [CrossRef]
- Roming, A.D.; Plimpton, S.J.; Michael, J.R.; Myklebust, R.L. Newbury DE Microbeam Analysis; San Francisco Press: San Francisco, CA, USA, 1990. [Google Scholar]
- Hill, J.M.; Royce, D.G.; Fadley, C.S.; Wagner, L.F.; Grunthaner, F.J. Properties of oxidized silicon as determined by angular-dependent X-ray photoelectron spectroscopy. Chem. Phys. Lett. 1976, 44, 225–231. [Google Scholar] [CrossRef]
- Benoit, M.; Bataillon, C.; Gwinner, B.; Miserque, F.; Orazem, M.E.; Sánchez-Sánchez, C.M.; Tribollet, B.; Vivier, V. Comparison of different methods for measuring the passive film thickness on metals. Electrochim. Acta 2016, 201, 340–347. [Google Scholar] [CrossRef]
- Passiu, C.; Rossi, A.; Weinert, M.; Tysoe, W.; Spencer, N.D. Probing the outermost layer of thin gold films by XPS and density functional theory. Appl. Surf. Sci. 2020, 507, 145084. [Google Scholar] [CrossRef]
- Tougaard, S. Surface nanostructure determination by x-ray photoemission spectroscopy peak shape analysis. J. Vac. Sci. Technol. A Vac. Surfaces Film. 1996, 14, 1415–1423. [Google Scholar] [CrossRef]
- Matthew, J. Surface Analysis by Auger and x-ray Photoelectron Spectroscopy; Briggs, D., Grant, J.T., Eds.; IMPublications: Chichester, UK; SurfaceSpectra: Manchester, UK, 2003; p. 900. ISBN 1-901019-04-7, 900 pp. Surf. Interface Anal. 2004, 36, 1647. [Google Scholar] [CrossRef]
- Elsener, B.; Rossi, A. XPS investigation of passive films on amorphous FeCr alloys. Electrochim. Acta 1992, 37, 2269–2276. [Google Scholar] [CrossRef]
- Rossi, A.; Elsener, B. XPS analysis of passive films on the amorphous alloy Fe70Cr10P13C7: Effect of the applied potential. Surf. Interface Anal. 1992, 18, 499–504. [Google Scholar] [CrossRef]
- Asami, K.; Hashimoto, K. An XPS study of the surfaces on Fe-Cr, Fe-Co and Fe-Ni alloys after mechanical polishing. Corros. Sci. 1984, 24, 83–97. [Google Scholar] [CrossRef]
- Jeynes, C.; Colaux, J.L. Thin film depth profiling by ion beam analysis. Analyst 2016, 141, 5944–5985. [Google Scholar] [CrossRef]
- Ho, S.M. Investigation of the electrical properties of metal chalcogenide thin films: A review. Der Pharma Chem. 2016, 8, 17–20. [Google Scholar]
- Hofmann, S.; Zhou, G.; Kovac, J.; Drev, S.; Lian, S.Y.; Lin, B.; Liu, Y.; Wang, J.Y. Preferential sputtering effects in depth profiling of multilayers with SIMS, XPS and AES. Appl. Surf. Sci. 2019, 483, 140–155. [Google Scholar] [CrossRef]
- Bardi, U.; Caporali, S.; Chenakin, S.P.; Lavacchi, A.; Miorin, E.; Pagura, C.; Tolstogouzov, A. Characterization of electrodeposited metal coatings by secondary ion mass spectrometry. Surf. Coat. Technol. 2006, 200, 2870–2874. [Google Scholar] [CrossRef]
- Bardi, U.; Chenakin, S.P.; Ghezzi, F.; Giolli, C.; Goruppa, A.; Lavacchi, A.; Miorin, E.; Pagura, C.; Tolstogouzov, A. High-temperature oxidation of CrN/AlN multilayer coatings. Appl. Surf. Sci. 2005, 252, 1339–1349. [Google Scholar] [CrossRef]
- Kalina, L.; Bílek Jr., V.; Bušo, M.; Koplík, J.; Másilko, J. Thickness determination of corrosion layers on iron using XPS depth profiling. Mater. Tehnol. 2018, 52, 537–540. [Google Scholar] [CrossRef]
- Scorciapino, M.A.; Navarra, G.; Elsener, B.; Rossi, A. Nondestructive Surface Depth Profiles from Angle-Resolved X-ray Photoelectron Spectroscopy Data Using the Maximum Entropy Method. I. A New Protocol. J. Phys. Chem. C 2009, 113, 21328–21337. [Google Scholar] [CrossRef]
- Olla, M.; Navarra, G.; Elsener, B.; Rossi, A. Nondestructive in-depth composition profile of oxy-hydroxide nanolayers on iron surfaces from ARXPS measurement. Surf. Interface Anal. 2006, 38, 964–974. [Google Scholar] [CrossRef]
- Smith, G.C.; Livesey, A.K. Maximum entropy: A new approach to non-destructive deconvolution of depth profiles from angle-dependent XPS. Surf. Interface Anal. 1992, 19, 175–180. [Google Scholar] [CrossRef]
- Skilling, J.; Bryan, R.K. Maximum entropy image reconstruction: General algorithm. Mon. Not. R. Astron. Soc. 1984, 211, 111–124. [Google Scholar] [CrossRef]
- Ishitani, T.; Yaguchi, T. Cross-sectional sample preparation by focused ion beam: A review of ion-sample interaction. Microsc. Res. Tech. 1996, 35, 320–333. [Google Scholar] [CrossRef]
- Mohamad Rashid, N.N.; Ahmad Junaidi, N.H.; Rahmah Aid, S. Interface Damage of Protective Layer in TEM Lamella Preparation for Highly Doped Ge Substrate. IOP Conf. Ser. Mater. Sci. Eng. 2019, 522, 012003. [Google Scholar] [CrossRef]
- Khamsehpour, B.; Davies, S.T. Angle lapping of multilayer structures for thickness measurements using focused ion beam micromachining. Semicond. Sci. Technol. 1994, 9, 249–255. [Google Scholar] [CrossRef]
- Gao, S.; Kang, R.K.; Guo, D.M.; Huang, Q.S. Study on the subsurface damage distribution of the silicon wafer ground by diamond wheel. Adv. Mater. Res. 2010, 126–128, 113–118. [Google Scholar] [CrossRef]
- International Organization for Standardization. Fine Ceramics (Advanced Ceramics, Advanced Technical Ceramics)—Determination of Coating Thickness by Crater-Grinding Method; ISO 26423:2009; ISO: Geneva, Switzerland, 2009. [Google Scholar]
- Vidakis, N.; Antoniadis, A.; Bilalis, N. The VDI 3198 indentation test evaluation of a reliable qualitative control for layered compounds. J. Mater. Process. Technol. 2003, 143–144, 481–485. [Google Scholar] [CrossRef]
- Randall, N. Finer particle size allows better coating characterization with the Calotest. Appl. Bull. 1997, 3–6. [Google Scholar]
- Demas, N.G.; Lorenzo-Martin, C.; Ajayi, O.O.; Erck, R.A.; Shareef, I. Measurement of Thin-film Coating Hardness in the Presence of Contamination and Roughness: Implications for Tribology. Metall. Mater. Trans. A 2016, 47, 1629–1640. [Google Scholar] [CrossRef]
- Rupetsov, V.; Minchev, R. Experimental Calo Tester for the Coating Thickness Measurement. In Proceedings of the XV International Scientific Conference, Smolyan, Bulgaria, 10–11 June 2016; pp. 188–192. [Google Scholar]
- Mayer, J.; Giannuzzi, L.A.; Kamino, T.; Michael, J. TEM sample preparation and FIB-induced damage. MRS Bull. 2007, 32, 400–407. [Google Scholar] [CrossRef]
- Schaffer, M.; Schaffer, B.; Ramasse, Q. Sample preparation for atomic-resolution STEM at low voltages by FIB. Ultramicroscopy 2012, 114, 62–71. [Google Scholar] [CrossRef]
- Özkan, D.; Kaleli, H.; Yüksek, L. Quantitative comparison of tribological performance of chromium- and zinc-phosphate-coated piston rings in tribotest rig. Proc. Inst. Mech. Eng. Part J J. Eng. Tribol. 2017, 231, 75–92. [Google Scholar] [CrossRef]
- Škarohlíd, J.; Ashcheulov, P.; Škoda, R.; Taylor, A.; Čtvrtlík, R.; Tomáštík, J.; Fendrych, F.; Kopeček, J.; Cháb, V.; Cichoň, S.; et al. Nanocrystalline diamond protects Zr cladding surface against oxygen and hydrogen uptake: Nuclear fuel durability enhancement. Sci. Rep. 2017, 7, 6469. [Google Scholar] [CrossRef]
- Vieira Junior, L.E.; Bendo, T.; Nieto, M.I.; Klein, A.N.; Hotza, D.; Moreno, R.; Rodrigues Neto, J.B. Processing of Copper Based Foil Hardened with Zirconia by Non-Deformation Method. Mater. Res. 2017, 20, 835–842. [Google Scholar] [CrossRef]
- Nyyssonen, D.; Kirk, C.P. Optical microscope imaging of lines patterned in thick layers with variable edge geometry: theory. J. Opt. Soc. Am. A 1988, 5, 1270. [Google Scholar] [CrossRef]
- Sakai, Y.; Yamada, T.; Suzuki, T.; Ichinokawa, T. Contrast mechanisms of secondary electron images in scanning electron and ion microscopy. Appl. Surf. Sci. 1999, 144–145, 96–100. [Google Scholar] [CrossRef]
- Li, P.; Bao, S.X.; Zhang, D.Z.; Zhuang, L.B.; Ma, L.L. Application of Secondary Electron Composition Contrast Imaging Method in Microstructure Studies on Cathode Materials of TWT. Mater. Sci. Forum 2011, 689, 255–259. [Google Scholar] [CrossRef]
- Sohail, K.; Khan, I.U.; Shahzad, Y.; Hussain, T.; Ranjha, N.M. Ph-sensitive polyvinylpyrrolidone-acrylic acid hydrogels: Impact of material parameters on swelling and drug release. Brazilian J. Pharm. Sci. 2014, 50, 173–184. [Google Scholar] [CrossRef]
- Gierak, J. Focused ion beam technology and ultimate applications. Semicond. Sci. Technol. 2009, 24. [Google Scholar] [CrossRef]
- Kleyn, A.W. Ion-Surface Interactions--From Channeling to Soft-Landing. Science 1997, 275, 1440–1441. [Google Scholar] [CrossRef]
- Huh, Y.; Hong, K.J.; Shin, K.S. Amorphization Induced by Focused Ion Beam Milling in Metallic and Electronic Materials. Microsc. Microanal. 2013, 19, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S. Ion Implantation and Beam Processing; Elsevier: Amsterdam, The Netherlands, 1984; ISBN 9780127569802. [Google Scholar]
- MoberlyChan, W.J.; Adams, D.P.; Aziz, M.J.; Hobler, G.; Schenkel, T. Fundamentals of focused ion beam nanostructural processing: Below, at, and above the surface. MRS Bull. 2007, 32, 424–432. [Google Scholar] [CrossRef]
- Ishitani, T.; Tsuboi, H. Objective comparison of scanning ion and scanning electron microscope images. Scanning 1997, 19, 489–497. [Google Scholar] [CrossRef]
- Tseng, A.A. Recent developments in micromilling using focused ion beam technology. J. Micromech. Microeng. 2004, 14. [Google Scholar] [CrossRef]
- Van Kouwen, L. Introduction to focused ion beams, ion sources, and the nano-aperture ion source. In Advances in Imaging and Electron Physics; Elsevier: Amsterdam, The Netherlands, 2019; pp. 181–216. [Google Scholar]
- Joy, D.C. SEM for the 21st century-scanning ion microscopy. Electron. Device Fail. Anal. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Eder, K.; Bhatia, V.; Van Leer, B.; Cairney, J.M. Using a Plasma FIB Equipped with Xe, N 2, O 2 and Ar for Atom Probe Sample Preparation—Ion Implantation and Success Rates. Microsc. Microanal. 2019, 25, 316–317. [Google Scholar] [CrossRef]
- Ernst, A.; Wei, M.; Aindow, M. A Comparison of Ga FIB and Xe-Plasma FIB of Complex Al Alloys. Microsc. Microanal. 2017, 23, 288–289. [Google Scholar] [CrossRef]
- Kant, K.; Losic, D. FIB Nanostructures; Springer International Publishing: New York, NY, USA, 2013; Volume 20, pp. 1–22. [Google Scholar] [CrossRef]
- An, B.-S.; Kwon, Y.; Oh, J.-S.; Shin, Y.-J.; Ju, J.; Yang, C.-W. Evaluation of ion/electron beam induced deposition for electrical connection using a modern focused ion beam system. Appl. Microsc. 2019, 49. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Livengood, R.; Shima, D.; Notte, J.; McVey, S. Gas field ion source and liquid metal ion source charged particle material interaction study for semiconductor nanomachining applications. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 2010, 28, C6F15–C6F21. [Google Scholar] [CrossRef]
- Scipioni, L.; Stern, L.A.; Notte, J.; Sijbrandij, S.; Griffin, B. Helium ion microscope. Adv. Mater. Process. 2008, 166, 27–30. [Google Scholar] [CrossRef]
- Giannuzzi, L.A.; Michael, J.R. Comparison of channeling contrast between ion and electron images. Microsc. Microanal. 2013, 19, 344–349. [Google Scholar] [CrossRef]
- Passiu, C.; Rossi, A.; Bernard, L.; Paul, D.; Hammond, J.; Unger, W.E.S.; Venkataraman, N.V.; Spencer, N.D. Fabrication and Microscopic and Spectroscopic Characterization of Planar, Bimetallic, Micro- and Nanopatterned Surfaces. Langmuir 2017, 33, 5657–5665. [Google Scholar] [CrossRef]
- International Organization for Standardization. Surface Chemical Analysis—Auger Electron Spectroscopy and X-ray Photoelectron Spectroscopy—Determination of Lateral Resolution; ISO 18516; International Organization for Standardization: Geneva, Switzerland, 2006. [Google Scholar]
- Giurlani, W.; Zangari, G.; Gambinossi, F.; Passaponti, M.; Salvietti, E.; Di Benedetto, F.; Caporali, S.; Innocenti, M. Electroplating for Decorative Applications: Recent Trends in Research and Development. Coatings 2018, 8, 260. [Google Scholar] [CrossRef]
- Innocenti, M.; Giurlani, W.; Passaponti, M.; Luca, D.; Salvietti, E. Electrodeposition and innovative characterization of precious metal alloys for the Galvanic and Jewel industry. Substantia 2019, 3, 29–37. [Google Scholar] [CrossRef]
- International Organization for Standardization. Metallic Coatings—Measurement of Coating Thickness—X-ray Spectrometric Methods; ISO 3497:2000; International Organization for Standardization: Geneva, Switzerland, 2000. [Google Scholar]
- International ASTM. Standard Test Method for Measurement of Coating Thickness by X-Ray Spectrometry; ASTM B568-98; International ASTM: West Conshohocken, PA, USA, 1998. [Google Scholar]
- Takahara, H. Thickness and Composition Analysis of Thin Film Samples Using FP Method by XRF Analysis. Rigaku J. 2017, 33, 17–21. [Google Scholar]
- Boehm, S.; Bügler, M.J.L. Layer Thickness Analysis of Thin Metal Coatings with micro-XRF on SEM; Bruker Nano Analytics: Berlin, Germany, 2017. [Google Scholar]
- Axic Inc. Non-Destructive Composition and Thickness Analysis of Metal Films Using Combination EDS/WDS X-Ray Fluorescence Spectrometry. Axic Inc. application note, 1989. [Google Scholar]
- Brouwer, P. Theory of XRF—Getting Acquainted with the Principles; PANalytical BV: Almelo, The Netherland, 2010; ISBN 9090167587. [Google Scholar]
- Abdel-Fattah, T.M.; Wixtrom, A.; Arias, L.; Zhang, K.; Baumgart, H. Quantitative Analysis of X-ray Fluorescence Absorption and Emission for Thickness Determination of ALD-Grown Metal and Oxide Nanoscaled Films. J. Nanosci. Nanotechnol. 2017, 17, 5745–5750. [Google Scholar] [CrossRef]
- Hamann, D.M.; Bardgett, D.; Cordova, D.L.M.; Maynard, L.A.; Hadland, E.C.; Lygo, A.C.; Wood, S.R.; Esters, M.; Johnson, D.C. Sub-Monolayer Accuracy in Determining the Number of Atoms per Unit Area in Ultrathin Films Using X-ray Fluorescence. Chem. Mater. 2018, 30, 6209–6216. [Google Scholar] [CrossRef]
- Kataoka, Y. Standardless x-ray fluorescence spectrometry (Fundamental Parameter Method using Sensitivity Library). Rigaku J. 1989, 6, 33–40. [Google Scholar]
- Thomsen, V. Basic Fundamental Parameters in X-Ray Fluorescence. Spectroscopy 2007, 22, 46–50. [Google Scholar]
- Han, X.Y.; Zhuo, S.J.; Shen, R.X.; Wang, P.L.; Ji, A. Comparison of the quantitative results corrected by fundamental parameter method and difference calibration specimens in X-ray fluorescence spectrometry. J. Quant. Spectrosc. Radiat. Transf. 2006, 97, 68–74. [Google Scholar] [CrossRef]
- Pessanha, S.; Fonseca, C.; Santos, J.P.; Carvalho, M.L.; Dias, A.A. Comparison of standard-based and standardless methods of quantification used in X-ray fluorescence analysis: Application to the exoskeleton of clams. X-Ray Spectrom. 2018, 47, 108–115. [Google Scholar] [CrossRef]
- Nygård, K.; Hämäläinen, K.; Manninen, S.; Jalas, P.; Ruottinen, J.-P. Quantitative thickness determination using x-ray fluorescence: application to multiple layers. X-Ray Spectrom. 2004, 33, 354–359. [Google Scholar] [CrossRef]
- Vrielink, J.A.M.A.M.; Tiggelaar, R.M.M.; Gardeniers, J.G.E.G.E.; Lefferts, L. Applicability of X-ray fluorescence spectroscopy as method to determine thickness and composition of stacks of metal thin films: A comparison with imaging and profilometry. Thin Solid Films 2012, 520, 1740–1744. [Google Scholar] [CrossRef]
- Elam, W.T.T.; Shen, R.B.; Scruggs, B.; Nicolosi, J. Accuracy of Standardless FP Analysis of Bulk and Thin Film Samples Using a New Atomic Database. Adv. X-ray Anal. 2004, 47, 104–109. [Google Scholar]
- Ager, F.J.; Ferretti, M.; Grilli, M.L.; Juanes, D.; Ortega-Feliu, I.; Respaldiza, M.A.; Roldán, C.; Scrivano, S. Reconsidering the accuracy of X-ray fluorescence and ion beam based methods when used to measure the thickness of ancient gildings. Spectrochim. Acta Part B At. Spectrosc. 2017, 135, 42–47. [Google Scholar] [CrossRef]
- Kolbe, M.; Beckhoff, B.; Krumrey, M.; Ulm, G. Thickness determination for Cu and Ni nanolayers: Comparison of completely reference-free fundamental parameter-based X-ray fluorescence analysis and X-ray reflectometry. Spectrochim. Acta Part B At. Spectrosc. 2005, 60, 505–510. [Google Scholar] [CrossRef]
- Queralt, I.; Ibañez, J.; Marguí, E.; Pujol, J. Thickness measurement of semiconductor thin films by energy dispersive X-ray fluorescence benchtop instrumentation: Application to GaN epilayers grown by molecular beam epitaxy. Spectrochim. Acta Part B At. Spectrosc. 2010, 65, 583–586. [Google Scholar] [CrossRef]
- XRMC. Available online: https://github.com/golosio/xrmc/wiki (accessed on 30 July 2020).
- XMI-MSIM. Available online: https://github.com/tschoonj/xmimsim/wiki (accessed on 30 July 2020).
- Xraylib. Available online: https://github.com/tschoonj/xraylib/wiki (accessed on 30 July 2020).
- Schoonjans, T.; Brunetti, A.; Golosio, B.; Sanchez Del Rio, M.; Solé, V.A.; Ferrero, C.; Vincze, L. The xraylib library for X-ray-matter interactions. Recent developments. Spectrochim. Acta Part B At. Spectrosc. 2011, 66, 776–784. [Google Scholar] [CrossRef]
- Golosio, B.; Schoonjans, T.; Brunetti, A.; Oliva, P.; Masala, G.L. Monte Carlo simulation of X-ray imaging and spectroscopy experiments using quadric geometry and variance reduction techniques. Comput. Phys. Commun. 2014, 185, 1044–1052. [Google Scholar] [CrossRef]
- Schiavon, N.; de Palmas, A.; Bulla, C.; Piga, G.; Brunetti, A. An Energy-Dispersive X-Ray Fluorescence Spectrometry and Monte Carlo simulation study of Iron-Age Nuragic small bronzes (“Navicelle”) from Sardinia, Italy. Spectrochim. Acta Part B At. Spectrosc. 2016, 123, 42–46. [Google Scholar] [CrossRef]
- Brunetti, A.; Fabian, J.; La Torre, C.W.; Schiavon, N. A combined XRF/Monte Carlo simulation study of multilayered Peruvian metal artifacts from the tomb of the Priestess of Chornancap. Appl. Phys. A 2016, 122, 571. [Google Scholar] [CrossRef]
- Bottaini, C.E.; Brunetti, A.; Montero-Ruiz, I.; Valera, A.; Candeias, A.; Mirão, J. Use of Monte Carlo Simulation as a Tool for the Nondestructive Energy Dispersive X-ray Fluorescence (ED-XRF) Spectroscopy Analysis of Archaeological Copper-Based Artifacts from the Chalcolithic Site of Perdigões, Southern Portugal. Appl. Spectrosc. 2018, 72, 17–27. [Google Scholar] [CrossRef]
- Barcellos Lins, S.A.; Ridolfi, S.; Gigante, G.E.; Cesareo, R.; Albini, M.; Riccucci, C.; di Carlo, G.; Fabbri, A.; Branchini, P.; Tortora, L. Differential X-Ray Attenuation in MA-XRF Analysis for a Non-invasive Determination of Gilding Thickness. Front. Chem. 2020, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cesareo, R. Gold, gildings, and tumbaga from the Moche tomb of the Lady of Cao: An EDXRF test for the internal ratio method. X-Ray Spectrom. 2019, 48, 202–207. [Google Scholar] [CrossRef]
- Cesareo, R.; Franco Jordan, R.; Fernandez, A.; Bustamante, A.; Fabian, J.; del Pilar Zambrano, S.; Azeredo, S.; Lopes, R.T.; Ingo, G.M.; Riccucci, C.; et al. Analysis of the spectacular gold and silver from the Moche tomb ‘Señora de Cao’. X-Ray Spectrom. 2016, 45, 138–154. [Google Scholar] [CrossRef]
- Cesareo, R.; de Assis, J.T.; Roldán, C.; Bustamante, A.D.; Brunetti, A.; Schiavon, N. Multilayered samples reconstructed by measuring Kα/Kβ or Lα/Lβ X-ray intensity ratios by EDXRF. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2013, 312, 15–22. [Google Scholar] [CrossRef]
- Cesareo, R.; Bustamante, A.; Fabian, J.; Calza, C.; Dos Anjos, M.; Lopes, R.T.; Elera, C.; Shimada, I.; Curay, V.; Rizzutto, M.A. Energy-dispersive X-ray fluorescence analysis of a pre-Columbian funerary gold mask from the Museum of Sicán, Peru. X-Ray Spectrom. 2010, 39, 122–126. [Google Scholar] [CrossRef]
- Cesareo, R. Non-destructive EDXRF-analysis of the golden haloes of Giotto’s frescos in the Chapel of the Scrovegni in Padua. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2003, 211, 133–137. [Google Scholar] [CrossRef]
- Porcinai, S.; Ferretti, M. X-ray fluorescence-based methods to measure the thickness of protective organic coatings on ancient silver artefacts. Spectrochim. Acta Part B At. Spectrosc. 2018, 149, 184–189. [Google Scholar] [CrossRef]
- de Almeida, E.; Melquiades, F.L.; Marques, J.P.R.; Marguí, E.; de Carvalho, H.W.P. Determination of the polymeric thin film thickness by energy dispersive X-ray fluorescence and multivariate analysis. Spectrochim. Acta Part B At. Spectrosc. 2020, 167, 105818. [Google Scholar] [CrossRef]
- West, M.; Ellis, A.T.; Streli, C.; Vanhoof, C.; Wobrauschek, P. 2017 atomic spectrometry update—A review of advances in X-ray fluorescence spectrometry and its special applications. J. Anal. At. Spectrom. 2017, 32, 1629–1649. [Google Scholar] [CrossRef]
- Vanhoof, C.; Bacon, J.R.; Ellis, A.T.; Vincze, L.; Wobrauschek, P. 2018 atomic spectrometry update—A review of advances in X-ray fluorescence spectrometry and its special applications. J. Anal. At. Spectrom. 2018, 33, 1413–1431. [Google Scholar] [CrossRef]
- Vanhoof, C.; Bacon, J.R.; Ellis, A.T.; Fittschen, U.E.A.; Vincze, L. 2019 atomic spectrometry update—A review of advances in X-ray fluorescence spectrometry and its special applications. J. Anal. At. Spectrom. 2019, 34, 1750–1767. [Google Scholar] [CrossRef]
- Castaing, R. Application of Electron Probes to Local Chemical and Crystallographic Analysis. Ph.D. Thesis, University of Paris, Paris, France, 1951. [Google Scholar]
- Richter, S.; Pinard, P.T. Combined EPMA, FIB and Monte Carlo simulation: A versatile tool for quantitative analysis of multilayered structures. IOP Conf. Ser. Mater. Sci. Eng. 2016, 109, 012014. [Google Scholar] [CrossRef]
- Ortel, E.; Hertwig, A.; Berger, D.; Esposito, P.; Rossi, A.M.A.M.; Kraehnert, R.; Hodoroaba, V.D.V.-D.V.D. New Approach on Quantification of Porosity of Thin Films via Electron-Excited X-ray Spectra. Anal. Chem. 2016, 88, 7083–7090. [Google Scholar] [CrossRef]
- Eggert, F. EDX-spectra simulation in electron probe microanalysis. Optimization of excitation conditions and detection limits. Microchim. Acta 2006, 155, 129–136. [Google Scholar] [CrossRef]
- Armigliato, A. Thin film X-ray microanalysis with the analytical electron microscope. J. Anal. At. Spectrom. 1999, 14, 413–418. [Google Scholar] [CrossRef]
- Christien, F.; Pierson, J.F.; Hassini, A.; Capon, F.; Le Gall, R.; Brousse, T. EPMA–EDS surface measurements of interdiffusion coefficients between miscible metals in thin films. Appl. Surf. Sci. 2010, 256, 1855–1860. [Google Scholar] [CrossRef]
- Pascual, R.; Cruz, L.R.R.; Ferreira, C.L.L.; Gomes, D.T.T. Thin film thickness measurement using the energy-dispersive spectroscopy technique in a scanning electron microscope. Thin Solid Films 1990, 185, 279–286. [Google Scholar] [CrossRef]
- Benoit, D.; Bresse, J.-F.; Van’t dack, L.; Werner, H.; Wernisch, J. (Eds.) Microbeam and Nanobeam Analysis; Springer Vienna: Vienna, Austria, 1996; ISBN 978-3-211-82874-8. [Google Scholar]
- Waldo, R.A.; Militello, M.C.; Gaarenstroom, S.W. Quantitative thin-film analysis with an energy-dispersive x-ray detector. Surf. Interface Anal. 1993, 20. [Google Scholar] [CrossRef]
- Gauvin, R. Quantitative X-ray microanalysis of heterogeneous materials using Monte Carlo simulations. Microchim. Acta 2006, 155. [Google Scholar] [CrossRef]
- Sokolov, S.A.; Kelm, E.A.; Milovanov, R.A.; Abdullaev, D.A.; Sidorov, L.N. Non-destructive determination of thickness of the dielectric layers using EDX. In Proceedings of the SPIE—The International Society for Optical Engineering, Vienna, Austria, 19–23 September 2016; Lukichev, V.F., Rudenko, K.V., Eds.; Volume 10224, p. 1022426. [Google Scholar]
- Stenberg, G.; Boman, M. Thickness measurement of light elemental films. Diam. Relat. Mater. 1996, 5, 1444–1449. [Google Scholar] [CrossRef]
- Willich, P.; Obertop, D. Composition and thickness of submicron metal coatings and multilayers on Si determined by EPMA. Surf. Interface Anal. 1988, 13, 20–24. [Google Scholar] [CrossRef]
- Hunger, H.-J.; Baumann, W.; Schulze, S. A new method for determining the thickness and composition of thin layers by electron probe microanalysis. Cryst. Res. Technol. 1985, 20, 1427–1433. [Google Scholar] [CrossRef]
- Ares, J.R.; Pascual, A.; Ferrer, I.J.; Sánchez, C. A methodology to reduce error sources in the determination of thin film chemical composition by EDAX. Thin Solid Films 2004, 450. [Google Scholar] [CrossRef]
- Sempf, K.; Herrmann, M.; Bauer, F. First results in thin film analysis based on a new EDS software to determine composition and/or thickness of thin layers on substrates. In Proceedings of the EMC 2008 14th European Microscopy Congress, Aachen, Germany, 1–5 September 2008; Springer: Berlin, Germany, 2008; pp. 751–752. [Google Scholar]
- Hodoroaba, V.-D.; Kim, K.J.; Unger, W.E.S. Energy dispersive electron probe microanalysis (ED-EPMA) of elemental composition and thickness of Fe-Ni alloy films. Surf. Interface Anal. 2012, 44. [Google Scholar] [CrossRef]
- Canli, S. Thickness Analysis of Thin Films by Energy Dispersive X-Ray Spectroscopy; Middle East Technical University: Ankara, Turkey, 2010. [Google Scholar]
- Moller, A.; Weinbruch, S.; Stadermann, F.J.; Ortner, H.M.; Neubeck, K.; Balogh, A.G.; Hahn, H. Accuracy of film thickness determination in electron-probe microanalysis. Mikrochim. Acta 1995, 119, 41–47. [Google Scholar] [CrossRef]
- Procop, M.; Radtke, M.; Krumrey, M.; Hasche, K.; Schädlich, S.; Frank, W. Electron probe microanalysis (EPMA) measurement of thin-film thickness in the nanometre range. Anal. Bioanal. Chem. 2002, 374, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.S.; Coleoni, E.A.; Trincavelli, J.C.; Kaschny, J.; Hubbler, R.; Soares, M.R.F.; Vasconcellos, M.A.Z. Metallic thin film thickness determination using electron probe microanalysis. X-Ray Spectrom. 2001, 30, 253–259. [Google Scholar] [CrossRef]
- Libo, Z.; Shengxiang, B.; Rong, W.; Shilan, L.; Lili, M.; Dechun, L.; Zhuang, L.; Bao, S.; Wang, R.; Li, S.; et al. Thin film thickness measurement using electron probe microanalyzer. In Proceedings of the 2009 International Conference on Applied Superconductivity and Electromagnetic Devices, Chengdu, China, 25–27 September 2009; pp. 142–144. [Google Scholar]
- Osada, Y. Electron probe microanalysis (EPMA) measurement of aluminum oxide film thickness in the nanometer range on aluminum sheets. X-Ray Spectrom. 2005, 34, 92–95. [Google Scholar] [CrossRef]
- Merlet, C. Thin Film Quantification by EPMA: Accuracy of Analytical Procedure. Microsc. Microanal. 2006, 12, 842–843. [Google Scholar] [CrossRef]
- August, H.; Wernisch, J. A Method for Determining the Mass Thickness of Thin Films Using Electron Probe Microanalysis. Scanning 1987, 9, 145–155. [Google Scholar] [CrossRef]
- Shang, Y.; Guo, Y.; Liu, Z.; Xu, L. An EPMA software for determination of thin metal film thickness and it’s application. Acta Metall. Sin. 1997, 33, 443–448. [Google Scholar]
- Watanabe, M.; Horita, Z.; Nemoto, M. Absorption correction and thickness determination using the zeta factor in quantitative X-ray microanalysis. Ultramicroscopy 1996, 65, 187–198. [Google Scholar] [CrossRef]
- Bishop, H.E.; Poole, D.M. A simple method of thin film analysis in the electron probe microanalyser. J. Phys. D Appl. Phys. 1973, 6, 1142–1158. [Google Scholar] [CrossRef]
- Rinaldi, R.; Llovet, X. Electron Probe Microanalysis: A Review of the Past, Present, and Future. Microsc. Microanal. 2015, 21, 1053–1069. [Google Scholar] [CrossRef]
- Colby, J.W. Quantitative Microprobe Analysis of Thin Insulating Films. Adv. X-ray Anal. 1967, 11, 287–305. [Google Scholar] [CrossRef]
- HUNGER, H.J. Thin Film Analysis by X-Ray Microanalysis Using Gaussian f (p z) Curves. Scanning 1988, 10, 65–72. [Google Scholar] [CrossRef]
- STRATAGem—SAMx. Available online: http://www.samx.com/index.html.en (accessed on 30 July 2020).
- Kühn, J.; Hodoroaba, V.-D.; Linke, S.; Moritz, W.; Unger, W.E.S. Characterization of Pd-Ni-Co alloy thin films by ED-EPMA with application of the STRATAGEM software. Surf. Interface Anal. 2012, 44, 1456–1458. [Google Scholar] [CrossRef]
- Dumelié, N.; Benhayoune, H.; Balossier, G. TF_Quantif: A Quantification Procedure for Electron Probe Microanalysis of Thin Films on Heterogeneous Substrates; IOP Publishing: Bristol, UK, 2007; ISBN 9781600214554. [Google Scholar]
- Small World. Available online: http://www.small-world.net/efs.htm (accessed on 30 July 2020).
- Batanova, V.G.; Sobolev, A.V.; Kuzmin, D.V. Trace element analysis of olivine: High precision analytical method for JEOL JXA-8230 electron probe microanalyser. Chem. Geol. 2015, 419, 149–157. [Google Scholar] [CrossRef]
- ThinFilmID—Oxford Instruments. Available online: http://www.oxford-instruments.cn/OxfordInstruments/media/nanoanalysis/brochures and thumbs/ThinFilmID_Brochure.pdf (accessed on 30 July 2020).
- LayerProbe—Oxford Instruments. Available online: https://www.oxford-instruments.com/products/microanalysis/solutions/layerprobe (accessed on 30 July 2020).
- pyPENELOPE. Available online: http://pypenelope.sourceforge.net/ (accessed on 30 July 2020).
- Llovet, X.; Salvat, F. PENEPMA: A Monte Carlo Program for the Simulation of X-Ray Emission in Electron Probe Microanalysis. Microsc. Microanal. 2017, 23, 634–646. [Google Scholar] [CrossRef]
- Electron Beam Scattering Modeling. Available online: http://montecarlomodeling.mcgill.ca/ (accessed on 30 July 2020).
- Gauvin, R.; Lifshin, E.; Demers, H.; Horny, P.; Campbell, H. Win X-ray: A new Monte Carlo program that computes X-ray spectra obtained with a scanning electron microscope. Microsc. Microanal. 2006, 12, 49–64. [Google Scholar] [CrossRef]
- Gauvin, R.; Michaud, P. MC X-Ray, a new monte carlo program for quantitative X-ray microanalysis of real materials. Microsc. Microanal. 2009, 15, 488–489. [Google Scholar] [CrossRef]
- Llovet, X.; Merlet, C. Electron Probe Microanalysis of Thin Films and Multilayers Using the Computer Program XFILM. Microsc. Microanal. 2010, 16, 21–32. [Google Scholar] [CrossRef]
- CASINO. Available online: https://www.gel.usherbrooke.ca/casino/index.html (accessed on 30 July 2020).
- Demers, H.; Poirier-Demers, N.; Couture, A.R.; Joly, D.; Guilmain, M.; De Jonge, N.; Drouin, D. Three-dimensional electron microscopy simulation with the CASINO Monte Carlo software. Scanning 2011, 33, 135–146. [Google Scholar] [CrossRef]
- Hovington, P.; Drouin, D.; Gauvin, R. CASINO: A new Monte Carlo code in C language for electron beam interaction—Part I: Description of the program. Scanning 1997, 19, 1–14. [Google Scholar] [CrossRef]
- Drouin, D.; Couture, A.R.; Joly, D.; Tastet, X.; Aimez, V.; Gauvin, R. CASINO V2.42—A Fast and Easy-to-use Modeling Tool for Scanning Electron Microscopy and Microanalysis Users. Scanning 2007, 29, 92–101. [Google Scholar] [CrossRef] [PubMed]
- CalcZAF. Available online: http://www.probesoftware.com/Technical.html (accessed on 30 July 2020).
- Armstrong, J.T.; Donovan, J.; Carpenter, P. CALCZAF, TRYZAF and CITZAF: The Use of Multi-Correction-Algorithm Programs for Estimating Uncertainties and Improving Quantitative X-ray Analysis of Difficult Specimens. Microsc. Microanal. 2013, 19, 812–813. [Google Scholar] [CrossRef]
- Ritchie, N.W.M. NIST DTSA-II. Available online: http://www.cstl.nist.gov/div837/837.02/epq/dtsa2/ (accessed on 30 July 2020).
- Ritchie, N.W.m.M. Spectrum Simulation in DTSA-II. Microsc. Microanal. 2009, 15, 454–468. [Google Scholar] [CrossRef]
- Ritchie, N.W.M. Efficient Simulation of Secondary Fluorescence Via NIST DTSA-II Monte Carlo. Microsc. Microanal. 2017, 23, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Salvat, F.; Fernández-Varea, J.M.; Sempau, J. PENELOPE—2006: A Code System for Monte Carlo Simulation of Electron and Photon Transport; OECD/NEA Data Bank: Paris, France, 2006; ISBN 9264023011. [Google Scholar]
- Katz, L.; Penfold, A.S. Range-energy relations for electrons and the determination of beta-ray end-point energies by absorption. Rev. Mod. Phys. 1952, 24, 28–44. [Google Scholar] [CrossRef]
- Cockett, G.H.; Davis, C.D. Coating thickness measurement by electron probe microanalysis. Br. J. Appl. Phys. 1963, 14, 813–816. [Google Scholar] [CrossRef]
- Matthews, M.B.; Kearns, S.L.; Buse, B. The Accuracy of Al and Cu Film Thickness Determinations and the Implications for Electron Probe Microanalysis. Microsc. Microanal. 2018, 24, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.S.; Vasconcellos, M.A.Z.; Llovet, X.; Salvat, F. Thickness Determination of Ultra-Thin Films on Si Substrates by EPMA. Microchim. Acta 2004, 145, 13–17. [Google Scholar] [CrossRef]
- Chininis, J.; Whiteside, P.; Hunt, H.K. Metal-clad waveguide characterization for contact-based light transmission into tissue. In Photonic Therapeutics and Diagnostics XII; Choi, B., Kollias, N., Zeng, H., Kang, H.W., Wong, B.J.F., Ilgner, J.F., Tearney, G.J., Gregory, K.W., Marcu, L., Skala, M.C., et al., Eds.; SPIE: Bellingham, WA, USA, 2016; Volume 9689, p. 968915. [Google Scholar]
- Stanford, J.A.; Teslich, N.; Donald, S.; Saw, C.K.; Gollott, R.; Dinh, L.N. Measurement of PuO2 film thickness by electron probe microanalysis (EPMA) calibration curve method. J. Nucl. Mater. 2020, 530, 151968. [Google Scholar] [CrossRef]
- Bastin, G.F.F.; Heijligers, H.J.M.M. A systematic database of thin-film measurements by EPMA Part I—Aluminum films. X-Ray Spectrom. 2000, 29, 212–238. [Google Scholar] [CrossRef]
- Bastin, G.F.; Heijligers, H.J.M. A systematic database of thin-film measurements by EPMA Part II - Palladium films. X-Ray Spectrom. 2000, 29, 373–397. [Google Scholar] [CrossRef]
- Merlet, C. Analytical Multilayer Model Revisited for High Atomic Numbers at Low Voltage. Microsc. Microanal. 2014, 20, 736–737. [Google Scholar] [CrossRef]
- Merlet, C. Quantitative X-ray Microanalysis of Multi-layered Specimens: Capability and Accuracy. Microsc. Microanal. 2013, 19, 1240–1241. [Google Scholar] [CrossRef]
- Pazzaglia, A.; Maffini, A.; Dellasega, D.; Lamperti, A.; Passoni, M. Reference-free evaluation of thin films mass thickness and composition through energy dispersive X-ray spectroscopy. Mater. Charact. 2019, 153, 92–102. [Google Scholar] [CrossRef]
- Darznek, S.A.; Kuzin, A.Y.; Mityukhlyaev, V.B.; Stepovich, M.A.; Todua, P.A.; Filippov, M.N. Measurement of the Thickness Nonuniformity of Nanofilms Using an Electron Probe Method. Meas. Tech. 2016, 59, 822–825. [Google Scholar] [CrossRef]
Figure 1.
The cross-section multilayer sample (brass/Cu/bronze/Pd/Au/Ni) observed with the optical microscope (top) and electron microscope (bottom).
Figure 1.
The cross-section multilayer sample (brass/Cu/bronze/Pd/Au/Ni) observed with the optical microscope (top) and electron microscope (bottom).

Figure 2.
An example of the FIB cross-sectioning workflow.

Figure 3.
A comparison between film thickness determination methods: traditional cross-sectioning (left column) and angle lapping (right column).
Figure 3.
A comparison between film thickness determination methods: traditional cross-sectioning (left column) and angle lapping (right column).

Figure 4.
The experimental setup, working functionality, and the equation used to calculate the film thickness with a Calo tester.
Figure 4.
The experimental setup, working functionality, and the equation used to calculate the film thickness with a Calo tester.

Figure 5.
SEM images of the main steps regarding TEM lamella preparation: (a) FIB digging; (b) border cut; (c) nanomanipulator welding; (d–f) lamella displacement; (g) support welding; (h) nanomanipulator detachment; (i) thinning; (j) lamella analysis with STEM.
Figure 5.
SEM images of the main steps regarding TEM lamella preparation: (a) FIB digging; (b) border cut; (c) nanomanipulator welding; (d–f) lamella displacement; (g) support welding; (h) nanomanipulator detachment; (i) thinning; (j) lamella analysis with STEM.

Figure 6.
The volumes of interaction of SE, BSE, and EDS.

Figure 7.
(a) An Au film between two Cu Layers. The picture is reported as it was acquired from the instrument; (b) the same picture of (a), but post-processed to enhance the separation between the layers; (c) a plot profile of the pictures (a,b) and the extrapolated thickness of the Au film.
Figure 7.
(a) An Au film between two Cu Layers. The picture is reported as it was acquired from the instrument; (b) the same picture of (a), but post-processed to enhance the separation between the layers; (c) a plot profile of the pictures (a,b) and the extrapolated thickness of the Au film.

Figure 8.
X-ray attenuation length as a function of the energy of the photons for copper and gold.

Figure 9.
A log-log plot of the emission of Au film on Cu as function of the thickness using a 50 kV X-ray beam. The 10–90% thickness range is marked.
Figure 9.
A log-log plot of the emission of Au film on Cu as function of the thickness using a 50 kV X-ray beam. The 10–90% thickness range is marked.

Figure 10.
The penetration depth of electrons with an energy of 25 keV in copper and gold.

Figure 11.
A log-log plot of the emission of Pd film on Cu as a function of the thickness using a 5 kV and 10 kV electron beam. The 10–90% thickness range is marked.
Figure 11.
A log-log plot of the emission of Pd film on Cu as a function of the thickness using a 5 kV and 10 kV electron beam. The 10–90% thickness range is marked.

Figure 12.
Range of thicknesses that a metal sample must have to be properly analyzed for each technique.
Figure 12.
Range of thicknesses that a metal sample must have to be properly analyzed for each technique.

Figure 13.
The time needed for the measurement and preparation of a sample for each technique.


{kind=link}
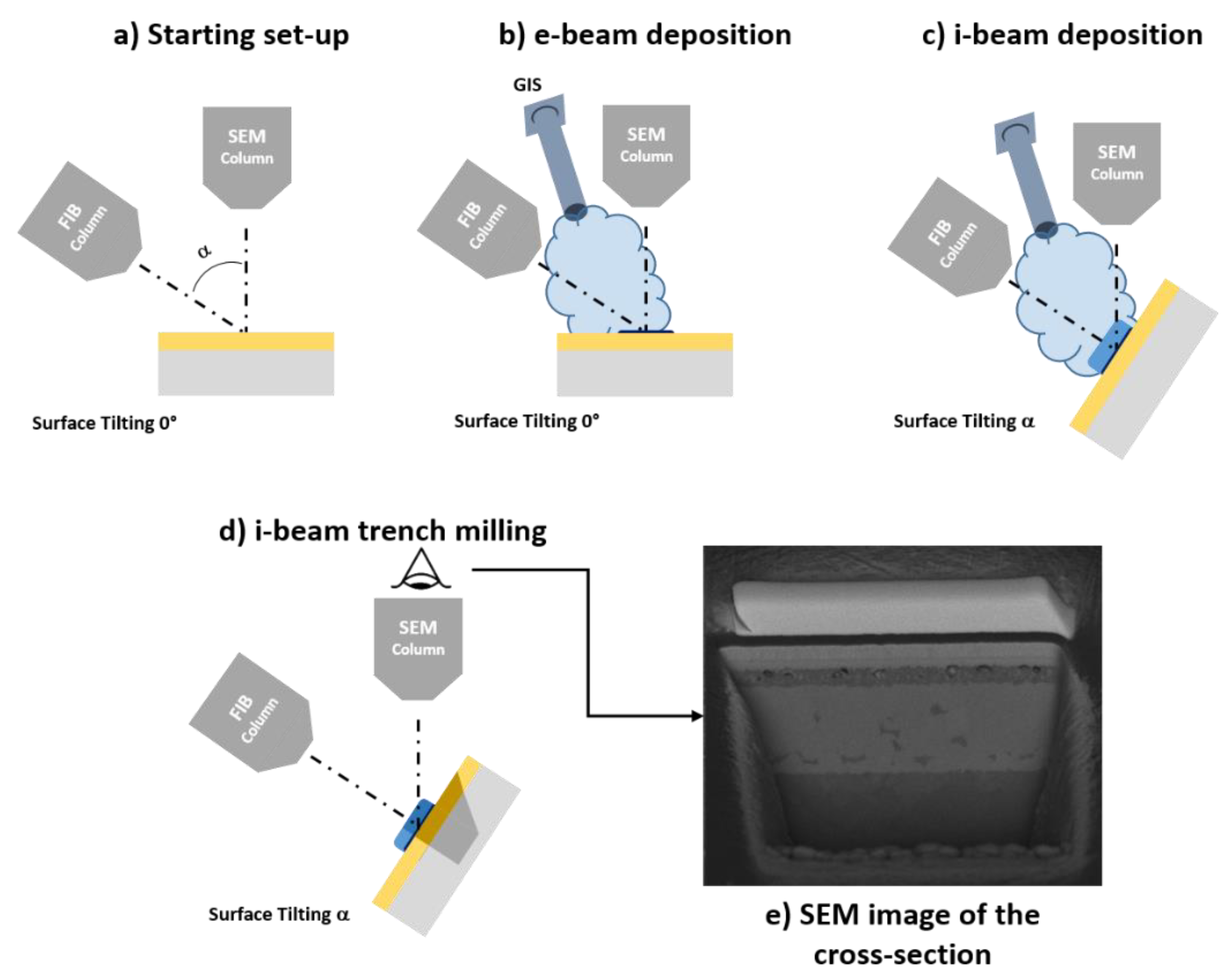{kind=link}
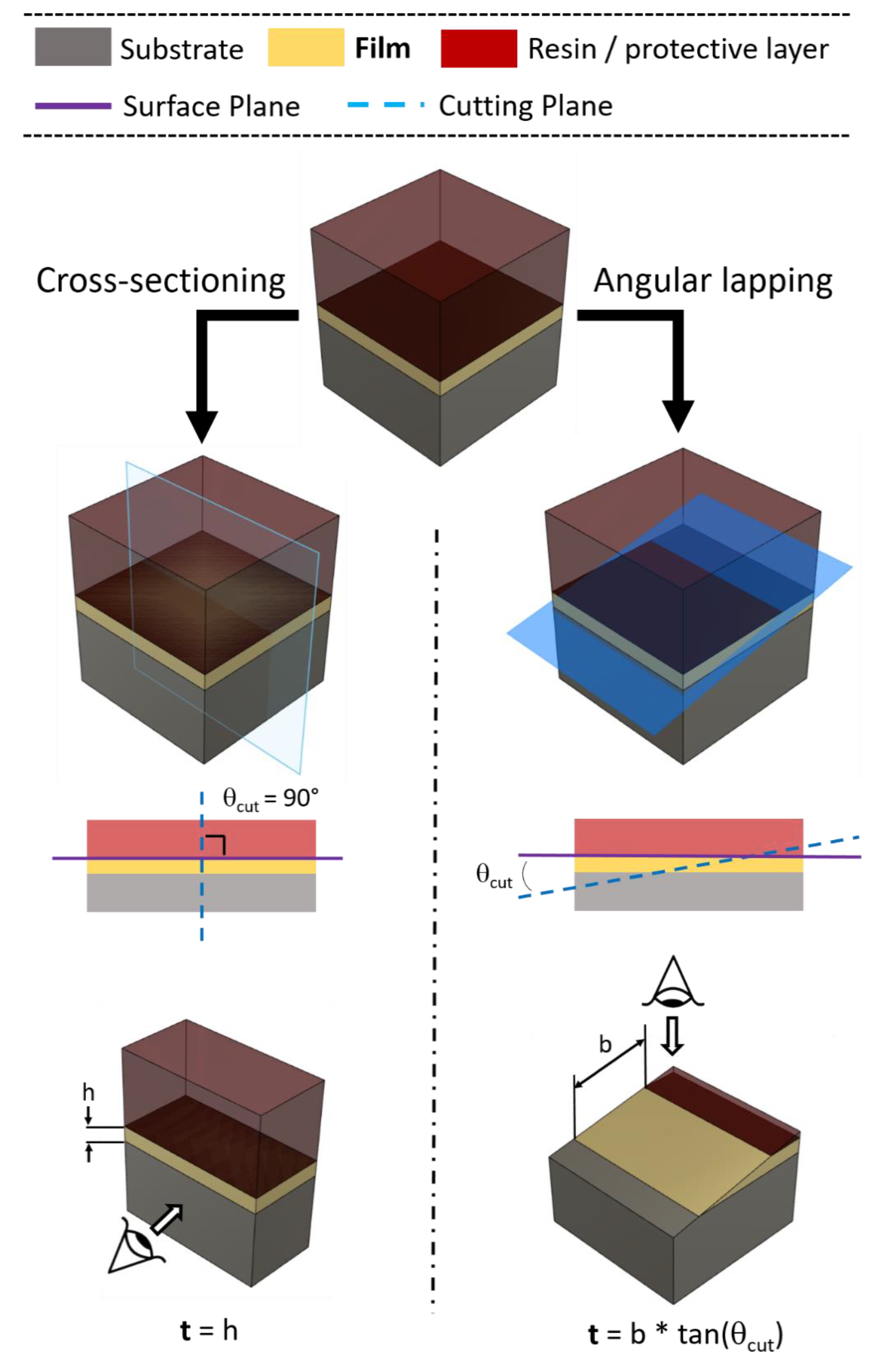{kind=link}
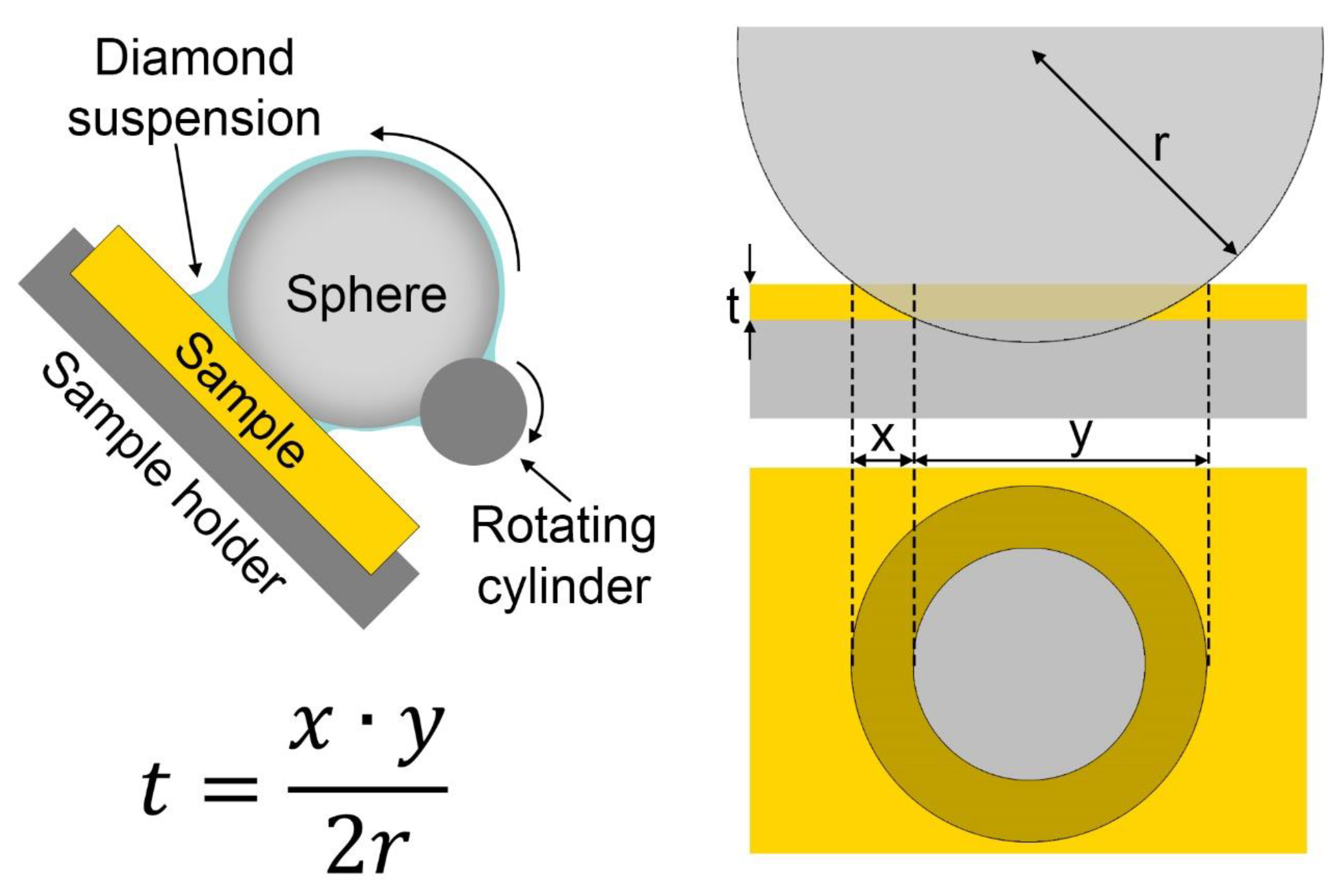{kind=link}
{kind=link}
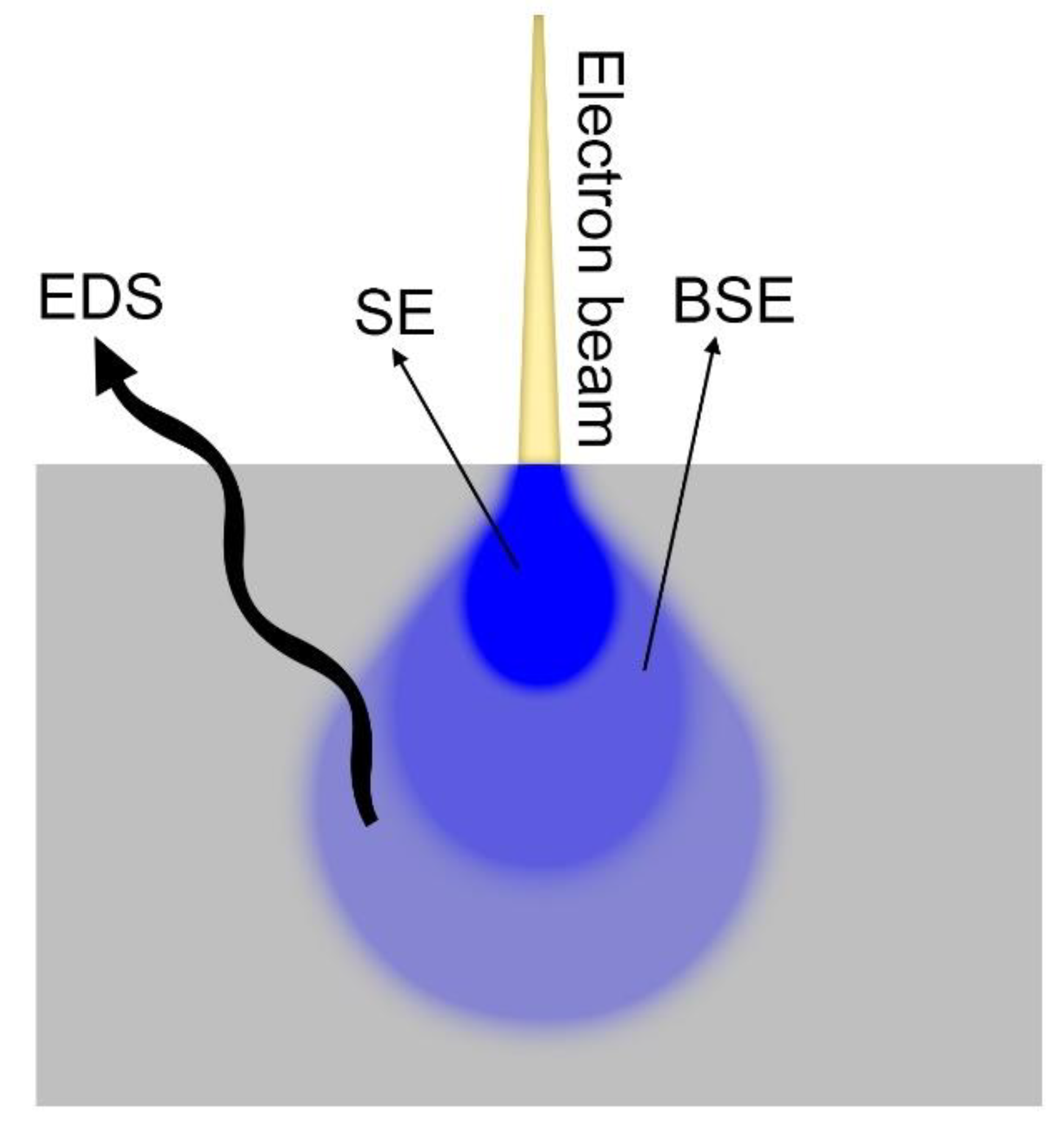{kind=link}
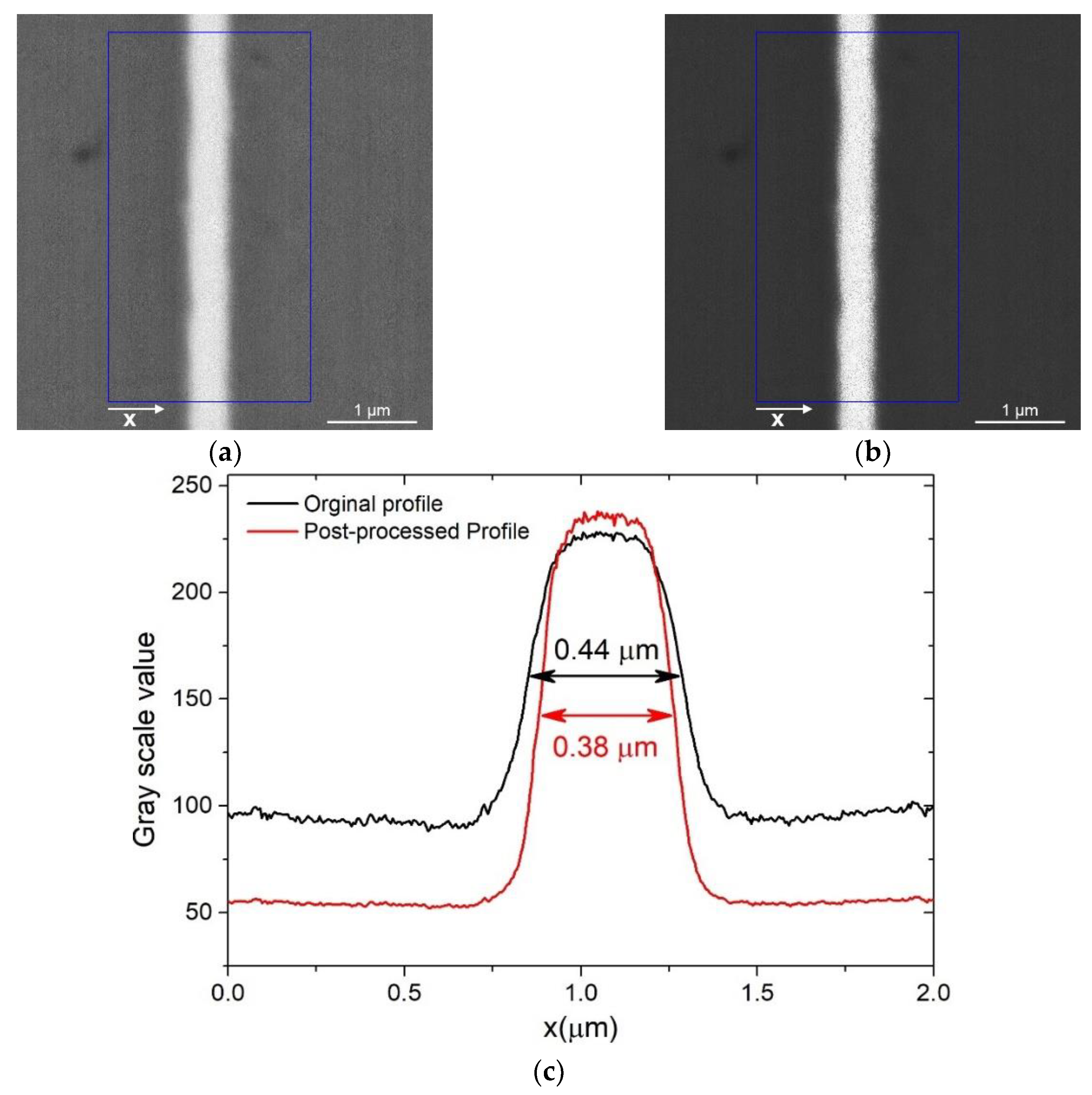{kind=link}
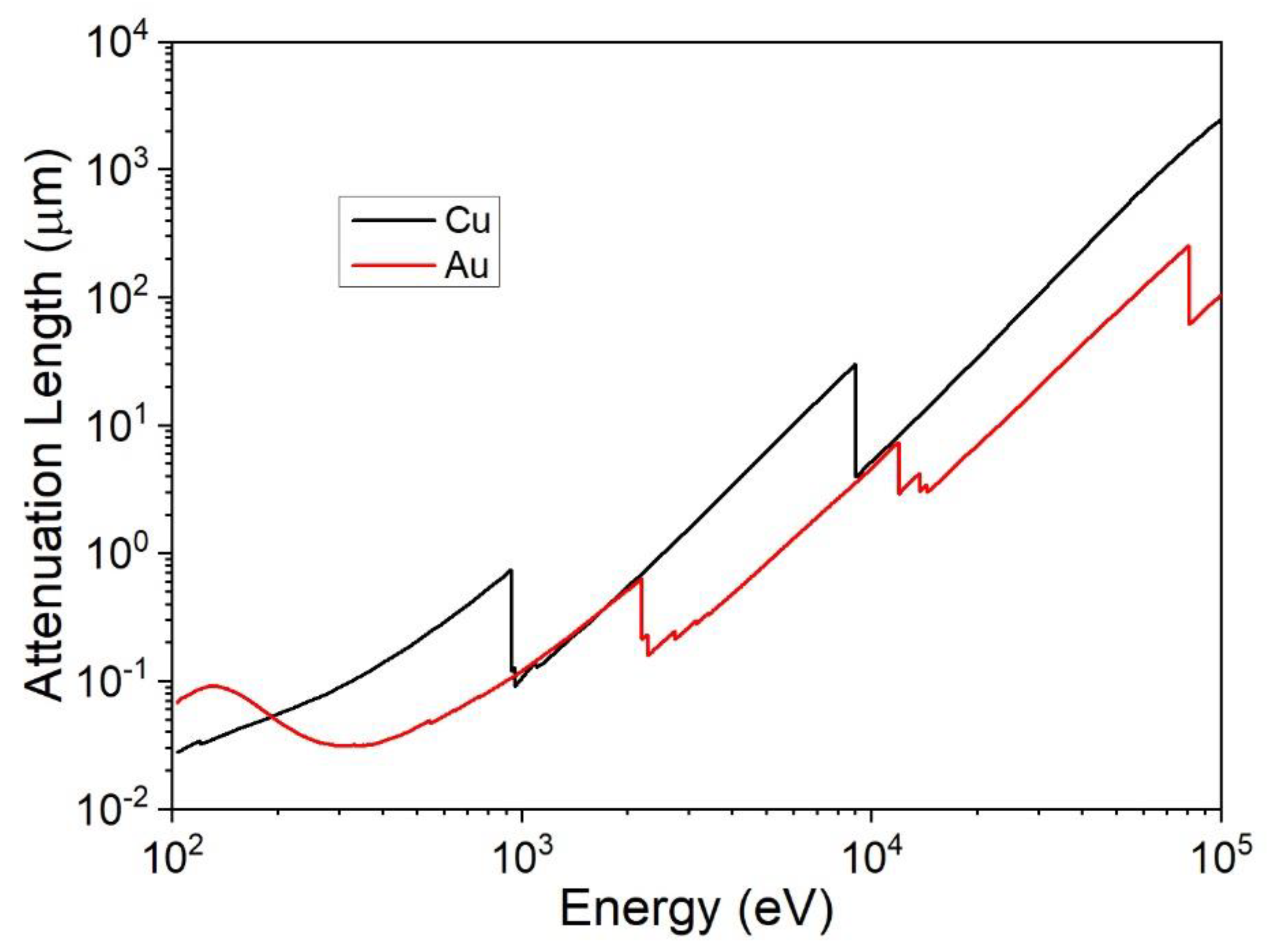{kind=link}
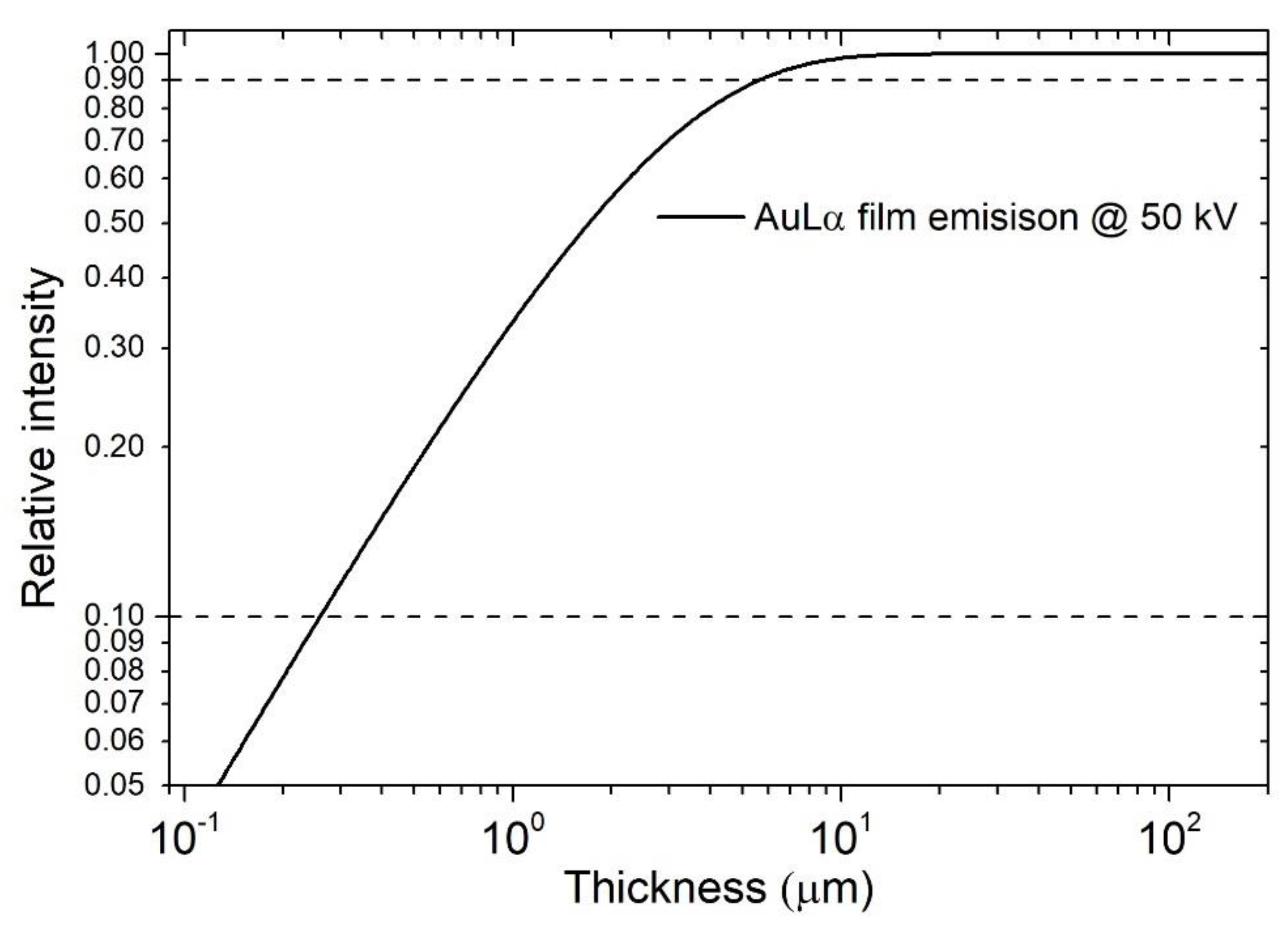{kind=link}
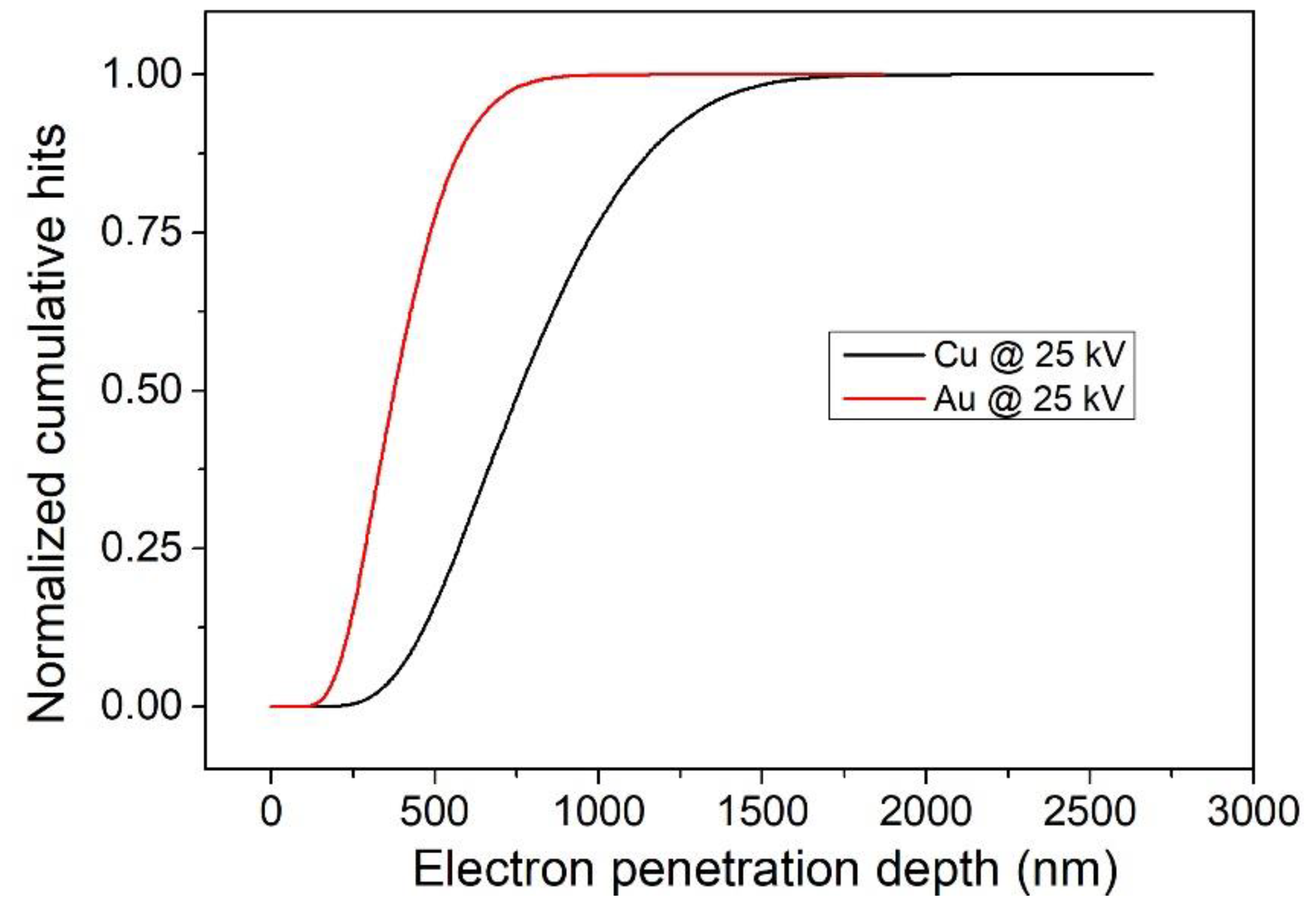{kind=link}
{kind=link}
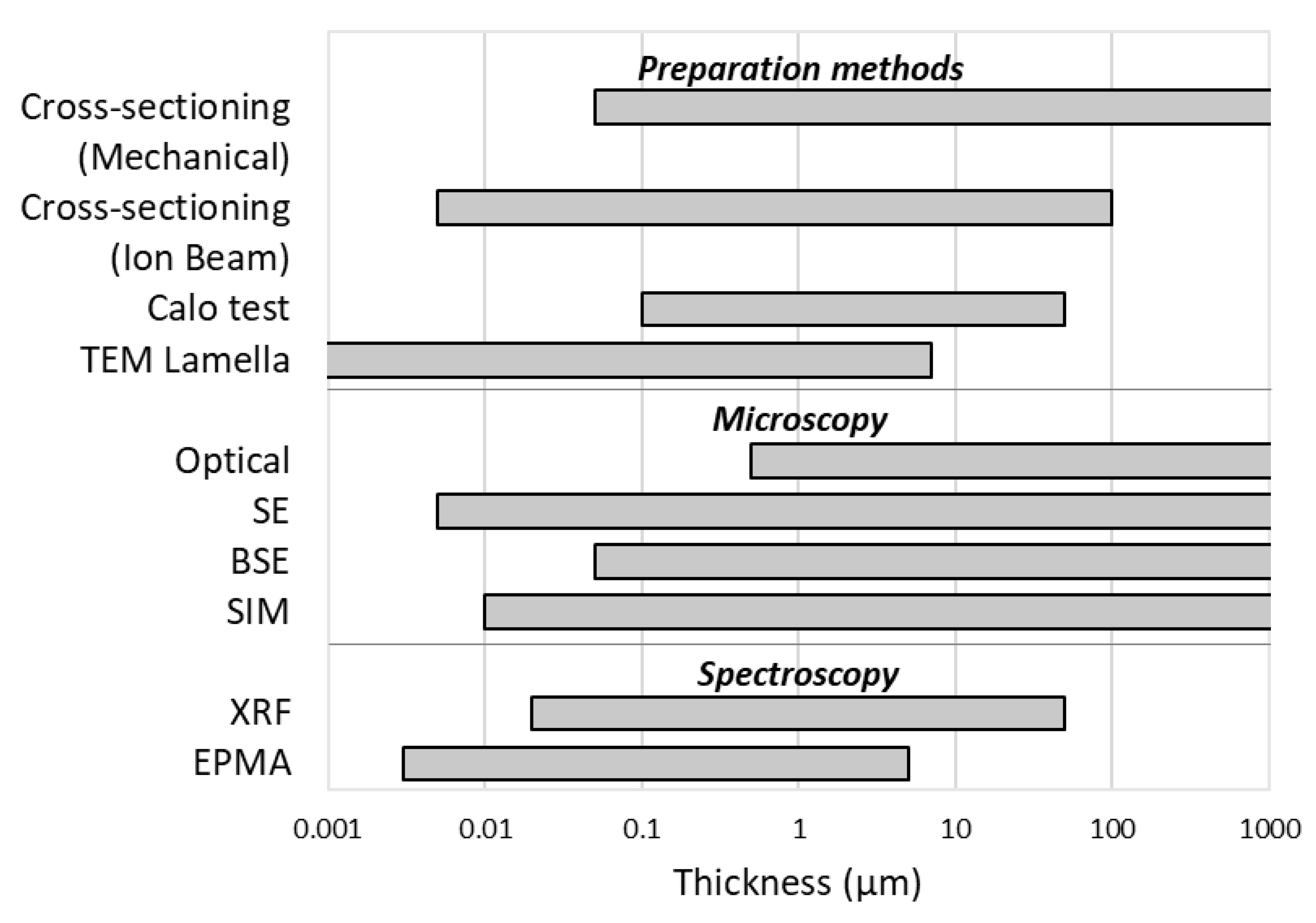{kind=link}
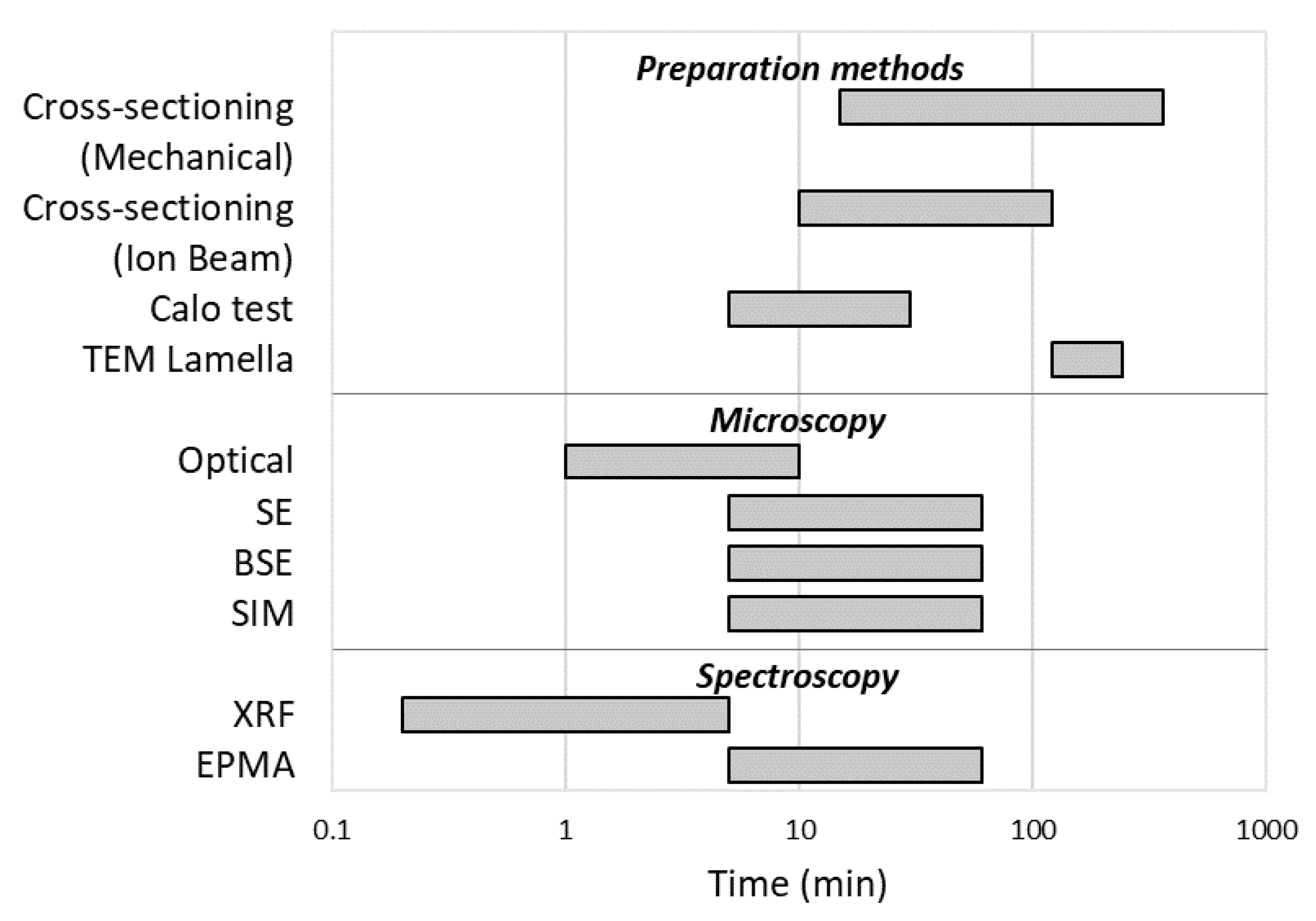{kind=link}
Table 1.
Penetration depths of SE, BSE, and EDS in Al and Au with a 20 kV beam.
| 20 kV Beam Penetration Depth | SE | BSE | X-Ray |
|---|---|---|---|
| Al | 50 nm | 700 nm | 2000 nm |
| Au | 5 nm | 70 nm | 200 nm |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Giurlani, W.; Berretti, E.; Innocenti, M.; Lavacchi, A. Measuring the Thickness of Metal Films: A Selection Guide to the Most Suitable Technique. Mater. Proc. 2020, 2, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/CIWC2020-06823
AMA Style
Giurlani W, Berretti E, Innocenti M, Lavacchi A. Measuring the Thickness of Metal Films: A Selection Guide to the Most Suitable Technique. Materials Proceedings. 2020; 2(1):12. https://0-doi-org.brum.beds.ac.uk/10.3390/CIWC2020-06823
Chicago/Turabian StyleGiurlani, Walter, Enrico Berretti, Massimo Innocenti, and Alessandro Lavacchi. 2020. "Measuring the Thickness of Metal Films: A Selection Guide to the Most Suitable Technique" Materials Proceedings 2, no. 1: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/CIWC2020-06823