1. Introduction
The production of healthy seed and plant material is a fundamental prerequisite for the establishment of ecologically stable and economically productive forest stands. As in the past, forest plant production is nevertheless threatened by harmful biotic factors, with new, invasive species playing an increasingly significant role as a result of climate change and globalization. From a forestry perspective, it is important that plant loss is reduced effectively and at a minimum of effort and cost. The fields of plant protection and plant breeding have a significant role to play in this regard, but novel strategies are necessary to meet the increasingly demanding requirements. The detection and identification of plant pathogens is one of the key prerequisites for the development of treatment concepts and the implementation of plant protection measures. Early detection in seed harvesting stands, seeds, and seedlings is of particular interest in this regard, and demands the development of efficient, practical diagnostic methods.
Methods based on the polymerase chain reaction (PCR) are generally characterized by a high level of sensitivity, specificity, and reliability. They are therefore already routinely used to identify pathogens in a forestry context. Examples include PCR-based methods for the detection of pathogens responsible for Dothistroma needle blight [
1,
2], various species of the genus
Fusarium [
3,
4],
Phytophthora [
5,
6], and
Rhabdocline [
7], as well as
Sphaeropsis sapinea (Fr.) Dyko & B. Sutton [
8],
Hymenoscyphus pseudoalbidus V. Queloz et al. [
9], and many other pathogens. Such methods are nevertheless time-consuming and require a level of investment in equipment that plant production companies are generally unable to afford. Loop-mediated isothermal amplification (LAMP) represents a sensitive and, above all, simple alternative to conventional PCR. The method was developed by Notomi et al. [
10] for the detection of hepatitis B viruses. The LAMP amplification involves the use of four to six primers that bind at six to eight regions of the target DNA. The synthesis of the new DNA strand occurs with the aid of a thermostable DNA polymerase characterized by a high level of strand displacement activity, thus enabling amplification to occur at constant temperature in a simple block heater or water bath. Another advantage is the simplicity of the results evaluation process. LAMP amplified products can be stained using fluorescent dyes and are therefore visible to the naked eye immediately after the reaction. The method is therefore particularly suitable for in situ analysis and laboratories that lack PCR equipment. A growing number of LAMP protocols for the detection of pathogens affecting herbaceous and woody plants have been established in recent years. To give an example, OptiGene Ltd. (Horsham, UK) offers an LAMP kit for the detection of
H. pseudoalbidus [
11]. Amongst others, LAMP protocols have also been published for
Fusarium graminearum Schwabe [
12],
Botrytis cinerea Pers. [
13],
Heterobasidion irregulare Garbelotto & Otrosina [
14],
Xylella fastidiosa Wells et al.,
Ceratocystis platani Engelbrecht & Harrington, and
Phytophthora ramorum Werres et al. [
15].
This paper presents the development of an LAMP-based quick test in combination with LAMP-optimized DNA extraction as a means of detecting Rhabdocline pseudotsugae Syd. in plant material from Douglas firs. The objective is to establish a robust detection method for practical forestry scenarios that not only enables plant production businesses to examine plant material quickly and easily, but also enhances quality management processes in the production and distribution of “source-identified” plant material over the long term.
2. Materials and Methods
2.1. Fungal Material, Plant Material
The establishment of the new LAMP method involved the use of a synthetic Rhabdocline fragment as a positive control. The templates for the synthetic fragment were a consensus sequence from the internal transcribed spacer (ITS) regions of the ribosomal DNA (rDNA) of R. pseudotsugae (the regions were available in the Genome Database maintained by the National Center for Biotechnology Information (NCBI)) as well as proprietary sequence data on ITS regions gathered within the context of previous studies. The Rhabdocline fragment was synthesized by a DNA service provider (Eurofins Genomics GmbH, Ebersberg, Germany).
Branch material with visible fruiting bodies of
R. pseudotsugae and branch material with no visible signs of infection were collected from a roughly 25-year-old stand of Douglas firs in Tharandter Wald in Saxony (50°58′ N, 13°28′ E) in 2018 and 2019 before being stored at −40 °C for further examination. Douglas fir seedlings from four German designated regions of origin for reproductive forest plant material were cultivated by project partner Biomasse Schraden e. V. (Großthiemig, Germany) in 2018 and also made available for the purpose of the study.
Table 1 summarizes the origin (region and forest stand), year of ripeness, and germination capacity of the seeds used:
The specificity of the LAMP reaction was tested using isolates of endophytic fungi supplied by project partner Institut für Pflanzenkultur e. K. (IFP; Schnega, Germany). The isolates were cultivated from surface-disinfected Douglas fir needles. The plant material was collected from the aforementioned Douglas fir stand in Tharandter Wald in spring 2018. DNA was extracted from the fungal isolates using an innuPREP Plant DNA Kit, Protocol 1 (Analytik Jena AG, Jena, Germany) and stored at −25 °C.
Table 2 summarizes the samples used. DNA taken from an environmental sample colonized with arbuscular mycorrhizal fungi (AMF) from a greenhouse trial at project partner IFP was also made available (see
Table 2). The fungal species present in the AMF sample were determined using targeted amplified product sequencing of the ITS region of the rDNA with the aid of nanopore sequencing (Oxford Nanopore Technologies Ltd., Oxford, UK) followed by taxonomical classification of the sequences using the Kaiju program [
16].
2.2. LAMP Primer Design
A set of LAMP primers consists of a basic set comprising an inner primer pair (FIP/BIP) and an outer primer pair (F3/B3). Reaction rate can be improved with the aid of additional loop primers (LF/LB). In the study at hand, suitable LAMP primers were selected using the PrimerExplorer V5 (Eiken Chemical Co., Ltd.; Tokyo, Japan). It was possible to use the Rhabdocline-specific consensus sequence of the ITS region to identify a large number of potential primer binding sites. Five basic sets with binding sites between the 5.8S and 28S rRNA genes were selected alongside three basic sets that bind in the ITS1 and ITS2 region. Wherever possible, additional loop primers (LF/LB) were determined and a TTTT spacer variation in the FIP and BIP primer tested for each basic set. The specificity of the binding sites was tested using BLAST analysis.
2.3. Establishment of the LAMP Assay
A total of 32 LAMP primer combinations were compared in terms of their reaction rate and yield, as well as the reproducibility and specificity of the amplification. Tests were carried out using the WarmStart LAMP Kit, WarmStart Colorimetric LAMP 2x Master Mix and Bst 3.0 DNA polymerase from New England Biolabs GmbH (Frankfurt am Main) as well as the GspSSD Isothermal Mastermix and the GspSSD 2.0 Isothermal Mastermix from OptiGene Ltd. (supplied by Amplex Diagnostics GmbH, Gießen, Germany). All work carried out in connection with the establishment of the LAMP assay was conducted using a qTower3 real-time PCR cycler (Analytik Jena AG).
Project partner Biomasse Schraden e. V. provided 50 Douglas fir seedlings per region of seed origin for the examination of the sensitivity of the LAMP reaction. DNA was extracted using the DNeasy Plant Mini Kit (Qiagen GmbH, Hilden, Germany) in accordance with the manufacturer’s instructions. All samples were first tested for infection with
R. pseudotsugae with the aid of nested PCR. This involved an initial PCR using universal primers ITS1F/ITS4 [
17,
18] followed by the nesting of the PCR products with
R. pseudotsugae-specific primer pair RPP1/RPP4 [
7] in a second PCR. A detailed description of the PCR parameters is provided in Morgenstern et al. [
19]. Fragment length of the final PCR product was measured using a Fragment Analyzer™ (Agilent Technologies Deutschland GmbH, Waldbronn, Germany). The results were evaluated using ProSize 3.0 software (Agilent Technologies Deutschland GmbH).
The successful establishment of the LAMP assay was followed by the retesting of the DNA samples from the same seedlings for infection with R. pseudotsugae. The LAMP reaction mixture contained 15.0 µL of GspSSD Isothermal Master Mix (OptiGene Ltd.), 4.0 µL of RNase-free water (Qiagen GmbH), 5.0 µL of LAMP Primer Mix (Eurofins Genomics GmbH; FIP/BIP: 4.0 µM each; F3/B3: 1.0 µM each; LF/LB: 2.0 µM each), and 1.0 µL of the DNA template. The LAMP mixture was incubated for 1.5 h at 65 °C in a qTower3 real-time PCR cycler (Analytik Jena AG).
2.4. Optimization of DNA Extraction
In the first step, the influence of Douglas fir DNA on the LAMP reaction was examined. This involved the mixing of a specific amount of R. pseudotsugae DNA with various amounts of Douglas fir DNA and the evaluation of the rate and yield of the LAMP reaction. A seedling from the Dillenburg stand that had been tested with R. pseudotsugae-specific primers RPP1/RPP4 and found to be free of infection was selected as the Douglas fir sample. A sample with a specific concentration of R. pseudotsugae was obtained by dissecting fruiting bodies from Douglas fir needles collected in Tharandter Wald and extracting DNA using the innuPREP Plant DNA Kit, Protocol 1 (Analytik Jena AG). The concentration of the fungal DNA was increased by means of standard PCR using primer pair ITS1F/ITS4, with the amplified ITS fragment subsequently diluted to 0.2 ng/μL.
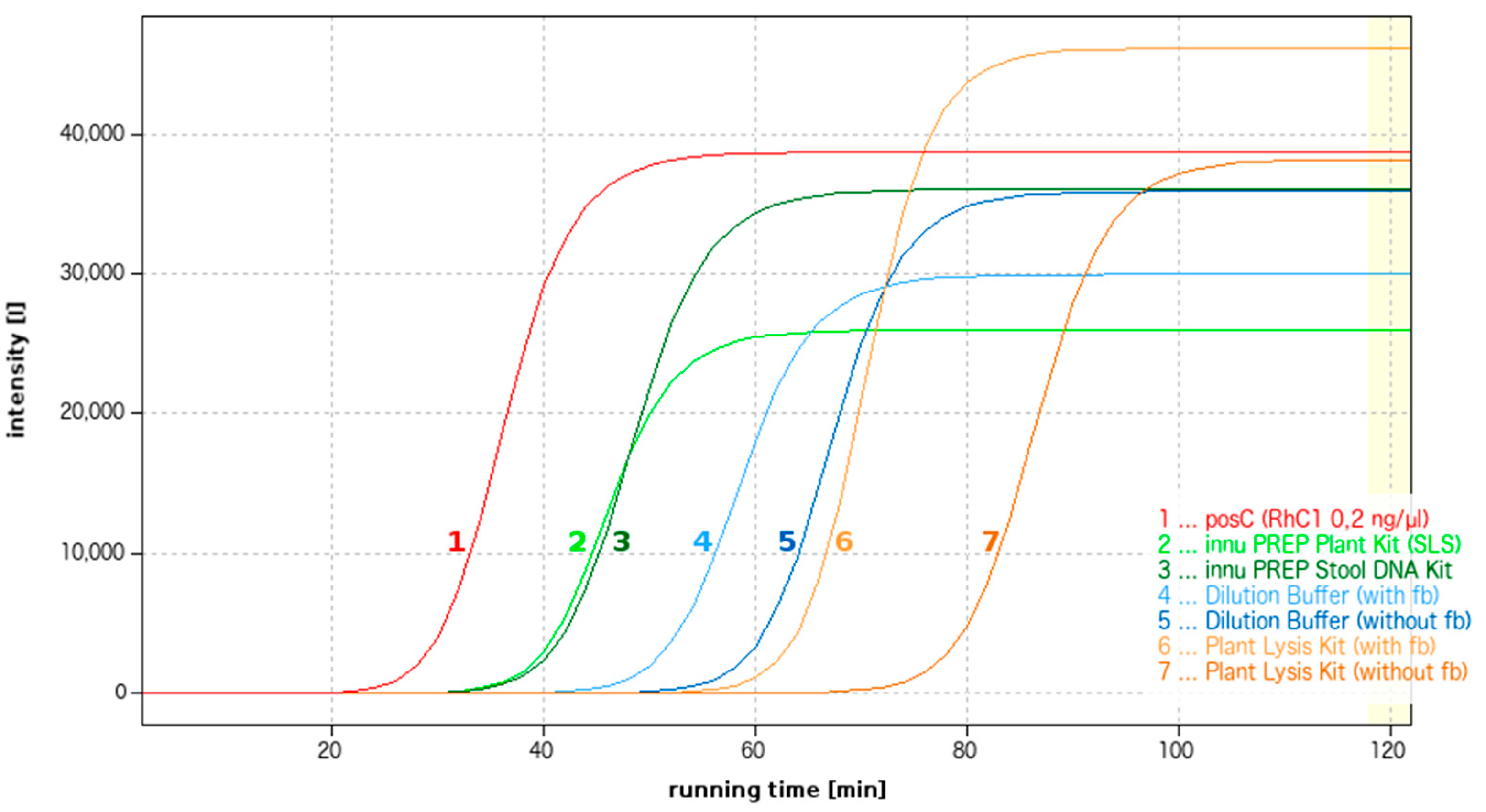
DNA extraction methods suitable for use in combination with LAMP amplification were researched from a variety of aspects. On the one hand, the methods tested included conventional DNA kits recommended by a variety of manufacturers for either the extraction of fungal DNA (DNeasy Plant Mini Kit (Qiagen GmbH); NucleoSpin® Plant II (Macherey-Nagel GmbH & Co. KG, Düren); innuPREP Plant DNA Kit (Analytik Jena AG)) or the extraction of DNA from difficult environmental samples (E.Z.N.A.® Soil DNA Kit (Omega Bio-tek, Inc., distributed by VWR International GmbH, Darmstadt, Germany); innuPREP TCM DNA Extraction Kit; innuPREP Stool DNA Kit (Analytik Jena AG)). All tests were successfully used to extract DNA from Douglas fir needles featuring fresh fruiting bodies of R. pseudotsugae. The primary objective was to extract fungal DNA that was as pure as possible and free from inhibitors.
On the other hand, the work carried out targeted the identification of methods that facilitate quick DNA extraction at a minimum of technical effort. DNA extraction was tested with the aid of FTA Indicating Micro Cards (Whatman, part of GE Healthcare; distributed by VWR International GmbH), the Dilution Buffer from the Phire Plant Direct PCR Kit (Fisher Scientific GmbH, Schwerte, Germany), and the Plant Material Lysis Kit (OptiGene Ltd.). The needle material used came from infected Douglas firs with visible fruiting bodies as well as needles without visible fruiting bodies.
4. Discussion
As described above, the ongoing TreeLAMP project has led to the successful establishment of a quick, sensitive LAMP assay for the detection of
R. pseudotsugae, the pathogen that causes Rhabdocline needle cast. Working with
R. pseudotsugae is accompanied by special challenges. On the one hand, the fungus is not yet known to feature any distinctive mycelia, with the visual examination of specimens for fruiting bodies therefore the only available means of macroscopic identification. On the other, the successful cultivation of this obligate biotrophic needle parasite has not yet been reported. The continuous availability of fungal material of high purity is nevertheless a fundamental prerequisite for the establishment of a quick test specific to
R. pseudotsugae. The study at hand initially used fruiting bodies dissected from infected needles. With the harvesting of fresh fruiting bodies essentially only possible in May, and with fruiting bodies only found on a limited number of Douglas firs in both years of harvesting, the LAMP assay went on to be established using a synthetic Rhabdocline fragment as a positive control. Neither nested PCR using primer pair RPP1/RPP4 [
7] nor the newly-established LAMP assay identified any differences between the synthetic fragment and samples taken from fruiting bodies of
R. pseudotsugae.
The detection limit of the LAMP assay was found to stand at 0.02 pg/µL (reaction time: 1.5 h) in the case of pure R. pseudotsugae samples as well as mixed fungal/Douglas fir samples. A comparison between PCR with subsequent fragment length measurement and LAMP amplification found LAMP to detect a 21% higher infection rate in seedlings. It is nevertheless to be noted that the ITS product used for nested PCR was also used for the LAMP reaction. On the one hand, this was due to the fact that the DNeasy Plant Mini Kit (Qiagen GmbH) was still being used to extract DNA from the seedlings and only identified as unsuitable during the subsequent optimization of DNA extraction for the LAMP assay. On the other, extraction rendered the respective plant material unusable, thus preventing the repeated extraction of DNA from the same plant material. The newly-established LAMP assay can nevertheless be assumed to be highly sensitive, as the use of an identical DNA template for both methods ensured the availability of a comparable amount of R. pseudotsugae DNA for the respective reaction.
Published data on the sensitivity of LAMP reactions varies greatly. To give an example, the LAMP kit for
H. pseudoalbidus from OptiGene Ltd. (Horsham, UK) is described as achieving a sensitivity of 7 pg of DNA based on a reaction time of 15.5 min [
11]. The LAMP assay for
F. graminearum detects up to 2 pg of the fungus in 30 min [
12], while the assays for
X. fastidiosa,
C. platani, and
P. ramorum are characterized by a detection limit of 0.02 pg/µL based on a reaction time of 30 min [
15]. By way of contrast, it is possible to reliably detect 65 pg of
B. cinerea after a reaction time of ≤20 min [
13], with 20 pg of
H. irregulare detectable in under 40 min [
14]. Despite the large number of reasons to which the varying sensitivity of LAMP assays may be attributable, it is the pathogens involved and the DNA regions upon which LAMP primer selection is based that are most likely to have an influence. The use of a synthetic fragment as a positive control for
R. pseudotsugae is unlikely to have played a role, as all sensitivity tests performed within the context of the work at hand were also carried out in combination with DNA extracted from fruiting bodies of the fungus as well as mixed DNA samples.
There is nevertheless a fundamental difference between the newly-established LAMP assay for
R. pseudotsugae and other assays in terms of reaction rate. In the work at hand, 1.5 h are required in order to detect 0.02 pg/µl of
R. pseudotsugae DNA. Target DNA concentration is known to have an influence on reaction rate, but amplification was found to take 60 min even at a concentration of 20 pg/µL. Other LAMP assays can be evaluated after a reaction time of between 15 min [
11] and 40 min [
14]. In the case of the new
R. pseudotsugae assay, it is possible that the relatively high GC content in the ITS2 region (59%)—the region in which the binding sites for primer set 5–6 are located—affects the optimum hybridization of the LAMP primers at the start of the reaction in particular, thus resulting in a delay in the start of the exponential amplification of the LAMP products. A reaction time of 1.5 h is nevertheless acceptable given the high level of sensitivity achieved by the newly-established LAMP assay.
As reaction time increases, so does the risk of non-specific amplification such as that observed in polymerases from New England Biolabs GmbH, in particular in the study at hand. To give an example, this may be caused by a decrease in intact nucleotides and intact polymerases as reaction time increases, which can lead to mishybridization. Non-specific amplification and false positive results may naturally also be attributable to non-specific primer binding. Cross-reactions with closely related species are also regularly described in connection with a variety of LAMP assays. Examples include the ability of the LAMP assay for
F. graminearum to detect other species of the genus
Fusarium from a reaction time of 50 min and upwards [
12], as well as the cross-reactions with
C. fimbriata and
P. lateralis described in Aglietti et al. [
15].
The specificity of the newly-established LAMP primers for R. pseudotsugae was tested with the aid of the AMF sample as well as 15 fungal isolates from Douglas fir needles. The AMF sample facilitated the relatively simple, successful testing of the LAMP primers on a range of different fungal sequences. The fungal isolates, on the other hand, were found to enable specific tests for fungi, including the Douglas fir endophyte R. parkeri and N. gaeumannii, the pathogen that causes Swiss needle cast. Cross-reactions with other pathogenic species of the genus Rhabdocline were not tested for in the study at hand due to the fact that only R. pseudotsugae has been found to exist in Europe to date.
The LAMP amplifications initially observed in the case of R. parkeri and P. macrostoma are to be regarded not as an indication that the LAMP primers lack specificity, but rather as evidence of a high level of sensitivity. The LAMP reaction tests all samples according to the principle of double determination. A positive signal was measured in the case of one of four R. parkeri isolates and one of two samples but not reproduced in the repeated test. The positive result in the case of R. parkeri must therefore be attributable to either a non-specific reaction or a sample handling error. In the case of P. macrostoma, on the other hand, the positive result is suspected to have been caused by the contamination of the culture with R. pseudotsugae, as the LAMP reaction only led to a negative test for P. macrostoma after the repetition of the DNA extraction.
In summary, the newly-established LAMP assay is characterized by a high level of sensitivity, ease of handling, and minimal investment in equipment. In the case of in situ applications, the LAMP assay is nevertheless only practical in combination with a sample preparation and DNA extraction process that is equally quick and simple to perform. A variety of suitable extraction options were tested, with considerable differences observed between the various methods and manufacturers involved. As expected, the sensitivity of the LAMP reaction when used with direct analysis protocols is somewhat limited when compared with that of a conventional DNA kit. Extraction with the aid of a DNA kit is therefore preferable, provided the technical prerequisites are fulfilled. The innuPREP Stool DNA Kit from Analytik Jena can be recommended for the detection of R. pseudotsugae in mixed samples. In the case of testing under more minimalistic conditions, the Plant Material Lysis Kit from OptiGene Ltd. is recommended over other options due to its ease of handling.






{kind=link}
{kind=link}
{kind=link}