3. Rationale for a GS Consortium
Developing a new cultivar requires assessing many lines to identify one that is acceptable for all targeted traits. There is an axiom that “breeding is a numbers game”. The validity of this axiom depends on if you are applying it to short or long terms breeding goals and whether you are assessing genetic gain, gain per season, gain per dollar spent, or the probability of attaining a new cultivar [
12]. If the goal is to develop a new cultivar, from a certain population, in a short time frame, then the axiom is quite valid. A primary rationale of the GSC is to effectively increase the size and impact of our programs without greatly increasing our investment.
Evaluating more lines from a population increases the probability of identifying a new variety and presents the opportunity to increase selection intensity. When traits and the genes controlling them are independent then the probability that a new cultivar (
Pnc) will exist in a population is
where
pi is the probability that a line in the population has an acceptable value for the
ith trait of t independent traits. The number of lines that must be evaluated (
N) to have a particular probability (
Pe) that one line suited to be a new cultivar will be in that evaluation is estimated as [
12]:
A breeder must either increase
Pnc,
N, or both if they want to increase
Pe. There are many strategies to increase
Pnc such as increasing the rate of genetic gain, selecting better parents, and using molecular breeding to select individuals prior to evaluations. All strategies should be considered. Regardless of
Pnc, the larger the evaluation (
N), the greater
Pe becomes, though with diminishing returns (
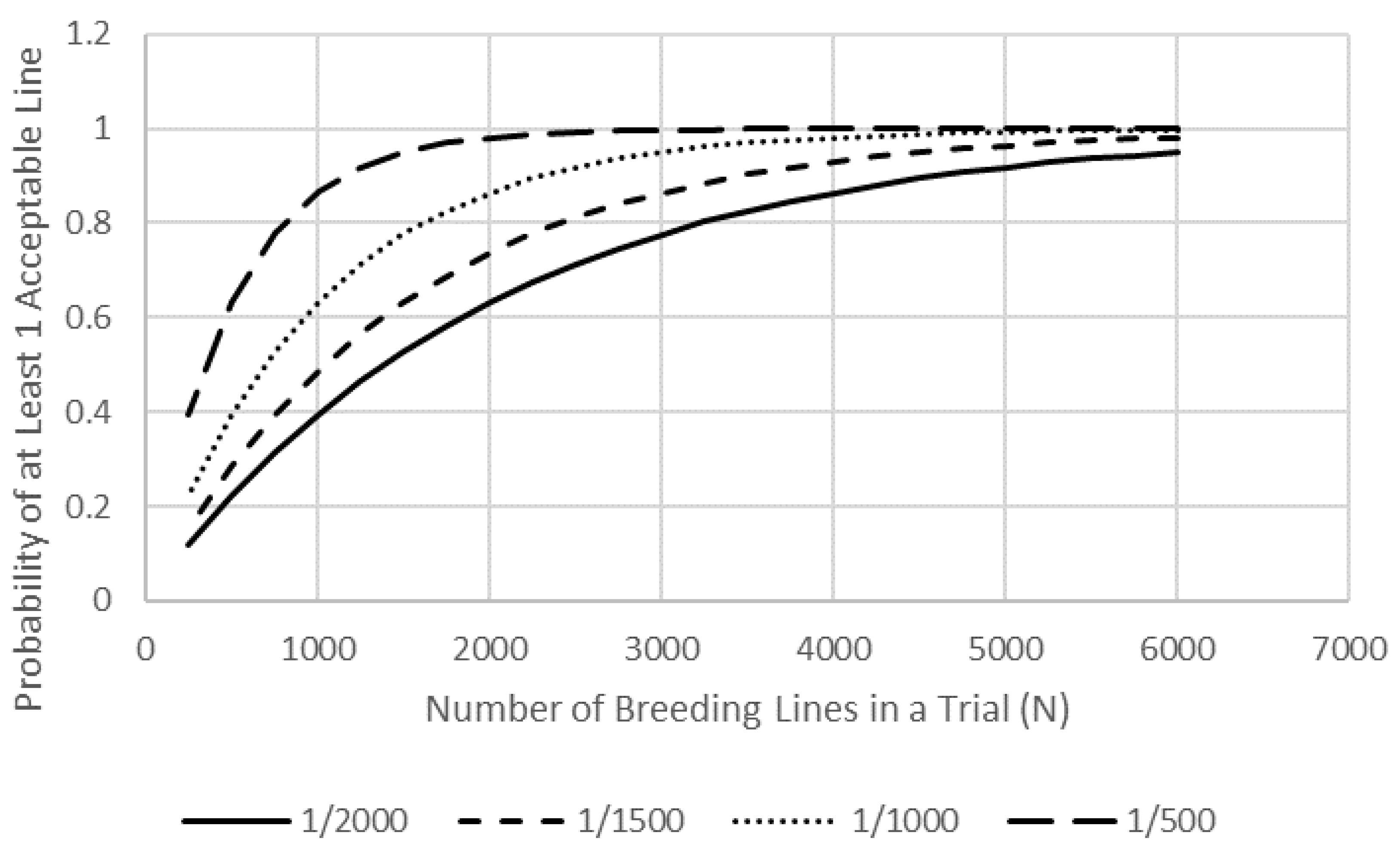
Figure 1). Each of the four MBPs in our GSC evaluates an average of 900 stage-1 lines per year. If we assume a
Pnc of 0.001, then each breeder has a
Pe of 0.59 by evaluating their 900 lines.
It is likely unwise and inefficient to greatly increase N within a single public breeding program given the cost of phenotyping and the diminished returns (Figure. 1). Our GSC though allows each breeder to use GS to predict the value of the 2700 stage-1 lines from the other three MBP resulting in an effective N of 3600 and a Pe of 0.97. The Pnc can be small when attempting to attain acceptable trait values for yield, agronomics, resistance to multiple pests, and quality such that N may need to be in the 2000–4000 range to attain a Pe > 0.9, an N that our GSC provides.
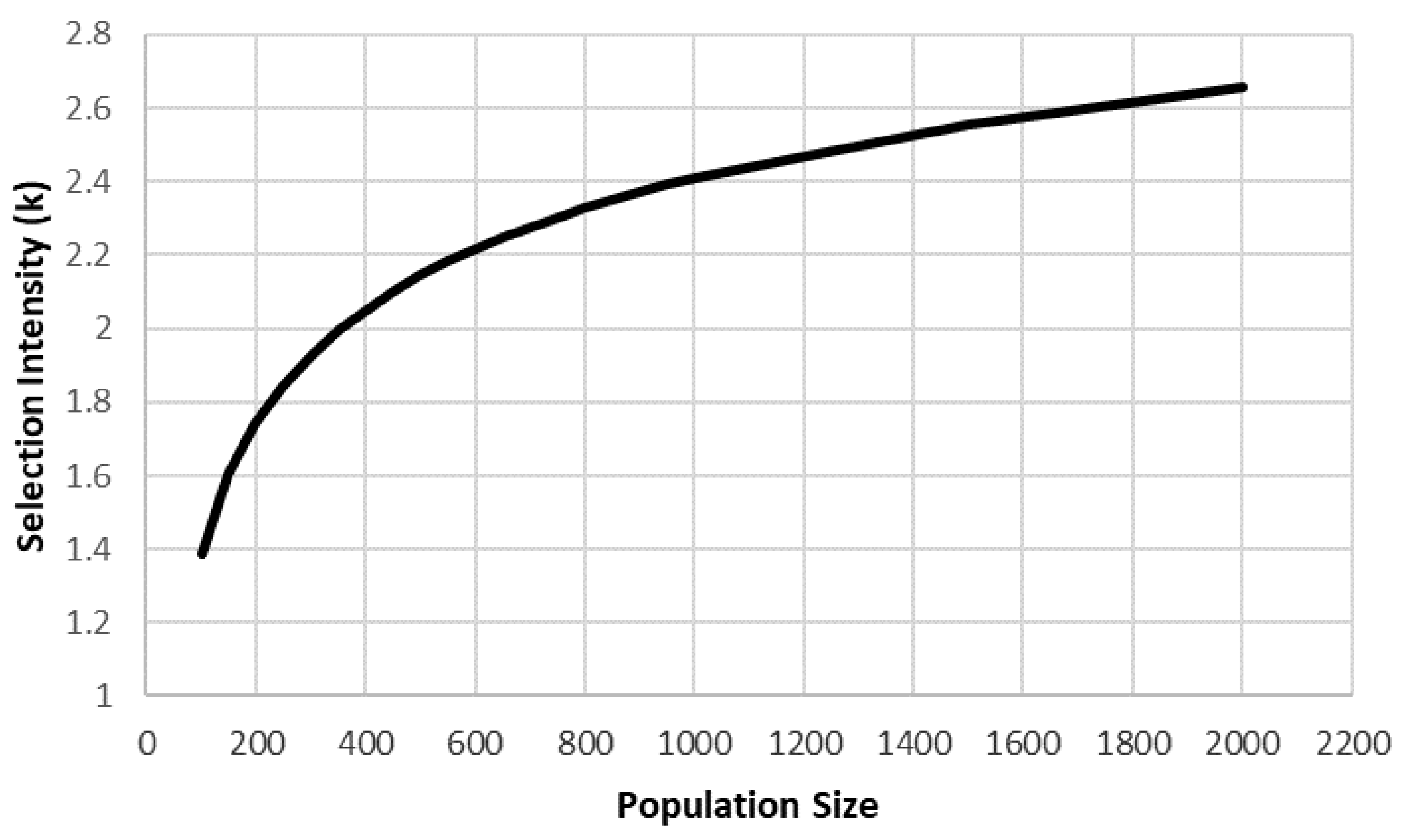
Selection intensity (
k) also increases as N increases, though also with diminishing returns (
Figure 2). Each MBP advances and average of 215 of their 900 lines to stage-2, providing a
k = 1.297. Selecting 215 of 3600 lines from the entire GSC produces a k of 1.980, a 53% increase compared no GSC. If a breeder selects 20 900 lines as parents, then
k = 2.233 while selecting 20 of the 3600 GSC lines produces
k = 2.849, a 28% increase compared to no GSC.
Increasing
N within public programs by investing funds to phenotype more lines is difficult due to limited funding and is likely inefficient in terms due to the diminishing returns in
Pnc and
k (
Figure 1 and
Figure 2). An alternative to increased phenotyping is to assess a large number of lines using GS as genotyping is often less expensive than phenotyping [
2]. Funding genotyping can also be problematic within a public program. The four MBP in our GSC were already genotyping all their new breeding lines prior to forming the GSC so genotyping was not a new expense required of the MBPs. Borrenpohl et al. [
13] presented evidence that GS could replace phenotyping of stage-1 lines such that the money saved by not conducting a stage-1 trial could be used to genotype lines. Thus, the genotyping required for a GSC is not necessarily an extra expense and could even save money.
The GSC concept allows a breeder to leverage their current investment to effectively increase the size of their selection population without much extra investment. This can increase Pnc, k, and gain per unit of investment. A GSC converts separate breeding programs into one large cooperative program allowing each breeder to assess the local and broad value of all lines from all breeders in. This extensive cooperation among public programs is now feasible due to the low cost of genotyping and the proven success of GS.
There are other benefits to a GSC. GS could be used to predict the value of all lines from all breeders, in all phenotyping environments, and over all environments, without actually phenotyping all lines in all environments. This is especially beneficial when applied to lines in the early stages of evaluation of each MBP where the population size is large and there is maximum genetic variation. A breeder can also get the predicted value of their lines for traits they did not phenotype when another MBP phenotypes for that trait.
A GSC offers benefits for genotyping. One condition for a GSC to operate is that all MBP must genotype their lines with the same marker system. Samples for genotyping can be consolidated within a GSC and result in volume discount and a lower genotyping cost per sample. The use of a common genotyping platform across all members also facilitates analyses of the diversity within and across the MBPs. The GSC also presents unique opportunities for training graduate students and learning from each MBP.
4. Requirements for a Successful GSC
The general operating plan for a GSC is to genotype all new breeding lines from each MBP prior to field testing, and to distribute the lines among the MBPs for testing, predictions, and selection. The GS models would be trained using phenotypic data from past and current field trials associated with the product development phase of each MBP. While it may be desirable to integrate all aspects of the MBPs, this article focuses on integrating the product development phase involving the evaluation of new lines.
There are many factors that will affect the success of a GSC. Some factors are related to management and administrative (see items I–M below). These are not trivial, but they will not be discussed in this article. Items A–H are related to principals of breeding and GS. We will discuss each within the context of our recently formed GSC.
Germplasm among the member programs must be related
Shared breeding goals among the members
Germplasm from each member must offers value to the other members
GS must be effective for target traits and populations
Development of optimal breeding and testing schemes to enhance predictions
A common, affordable marker platform
A common database for storing phenotypic and genotypic data
Ability to accurately phenotype for target traits
Communication among members
Skills in GS analyses
Coordinator for organizing samples, data files, and for executing analyses within and across programs
Ability of members to fund genotyping
Ability to share germplasm
A. The elite lines in the cultivar development portion of each MBP must be related to one another to obtain meaningful GS predictions [
14,
15,
16]. The degree of relatedness also affects what size training population (TP) is required [
5]. Bassi et al. [
17] estimated that to attain a prediction accuracy of 0.5 required a TP size of at least 50 when selection candidates are full sibs of individuals in the TP, at least 100 for half-sibs, and at least 1000 for more distant relationships. In simulations of maize biparental populations, Hickey et al. [
18] reported that a TP between 400 and 1000 individuals was required to achieve prediction accuracies above 0.6 when half-sib F
2 families were used as the TP to predict phenotypes in a target F
2 population. Utilizing a TP from unrelated biparental populations produced poor prediction accuracy unless >4000 phenotypes were used).
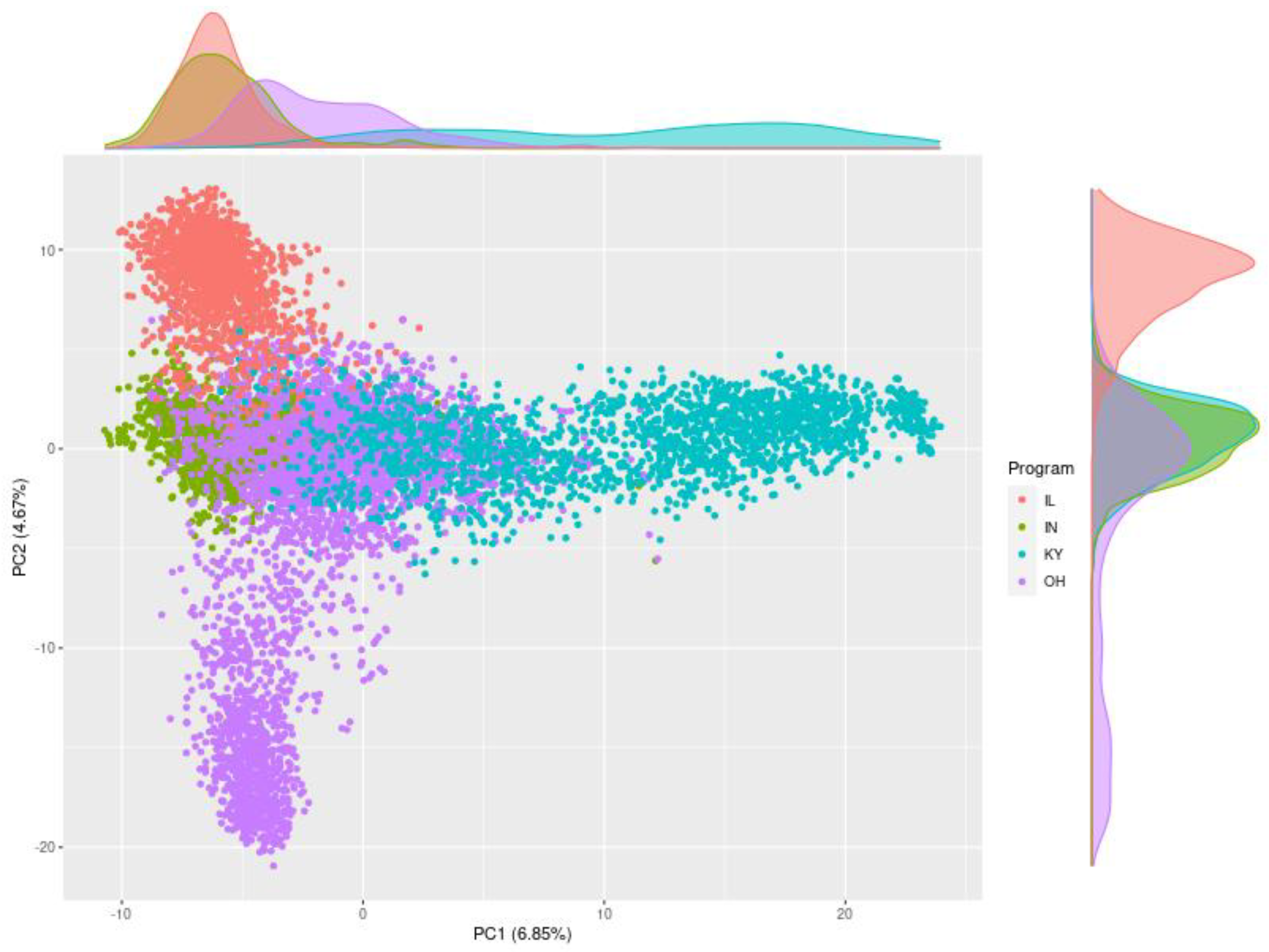
We used GBS to genotyped 8943 recent and historical germplasm accessions from the four MBPs of our GSC. The principal components graph (
Figure 3) indicates considerable diversity and some structure among the lines of the four MBPs. There is also considerable relatedness as 5745 (64%) of the lines are within 1.25 standard deviations from the origin in
Figure 3: this includes 35% of the lines from IL, 98% of the lines from IN, 43% of the lines from KY, and 77% of the lines from OH. The analysis of Fst values also suggest that the germplasm from the MBPs is fairly related especially lines from IL and OH, IN and OH, and KY and OH. (
Table 2). Producing a large TP is a benefit of the GSC. And thehe 5745 centralized accessions can serve as a large and suitable TP for predictions within and between MBPs and optimized TPs can be derived from this set [
18].
The general relatedness among the four MBPs is not surprising as these MBP have participated in cooperative trials of each other’s germplasm and exchanged parents for decades. A similar relationship could arise among programs that acquire much of their germplasm and parents from a common source, such as the Consultive Group for International Agriculture Research (CGIAR) institutes. This is a common practice among National breeding programs in the developing world.
B. Each member of the GSC should target the same primary traits in their product profile. The four MBP in our GSC share the primary goals of improving grain yield, test weight, and resistance to Fusarium Head Blight (FHB) cause by the fungus Fusarium graminearum. In addition, the four MBP have similar target values for maturity and plant height.
C. It is vital that the germplasm of each MBP of the GSC offers value to the other MBPs. It is important to analyze the value of germplasm among the MBPs and the genotype x environment interaction (GEI) pattern among lines and MBP environments to assess the degree of specific adaptation of each MBP’s germplasm. We assessed the GEI pattern and value of the germplasm from each MBP to the other MBPs in an analysis of data from cooperative trials.
The trait averages show that lines from one MBP perform reasonably well when tested in the other MBP’s environments (
Table 3) indicating that the germplasm of each MBP offers value to the others. This is particularly true for yield where the highest average yielding lines in IN, KY, and OH were from a different MBP. The average GEI values also indicate that there is not a pronounced MBP-specific adaptation of one MBP’s line to their program’s environments.
D. The accuracy of GS must be sufficient to warrant its use for the target traits of the GSC. Published results of static TPs show that GS can be effective in soft red winter wheat for our primary traits. The prediction accuracy for grain yield has ranged from 0.20 to 0.65, from 0.37 to 0.62 for FHB resistance, and from 0.30 to 0.66 for test weight (
Table 4).
There are several GS predictions of particular interest within a GSC: (1) breeder A predicting the value of their own lines in their own environments, (2) breeder A predicting the value of lines from the other MBP in breeder A’s environments, and (3) breeder A predicting the value of their own lines in other MBP environments The values in
Table 4 are encouraging but what is more relevant is the accuracy of a GS model that uses the accumulating phenotypic data from the annual successive stage trials of a breeding trials as the TP, referred to as stage-gate trials. Several studies have shown that GS provides useful prediction with TP whose phenotypes come from unbalanced designs that commonly occur within breeding programs [
5,
13,
25,
30,
31,
32,
33,
34,
35]. We assessed GS accuracy using the yield, test weight, and FHB resistance data from the breeding trials of each MBP (
Table 5). The cross-validation estimates of GS accuracy within a program (diagonal elements in
Table 5) were similar to the GS accuracy as obtained using the static TPs (
Table 4). These results show GS can be effective within a program using stage-gate data.
The accuracy of GS though was quite low when using the stage-gate data from one MBP to predict the phenotypes of lines in another MBP, (off diagonal elements,
Table 5). The low GS accuracy could arise from (1) low genetic relatedness among the lines in the programs, (2) each MBP may represent a unique environment which promotes large GEI, or (3) too few lines are tested over all environments to effectively connect the data among programs (e.g., low connectivity). Our analysis of diversity (
Figure 3) and GEI (
Table 3) suggest that the diversity and GEI in the data are unlikely to be major issues, leaving a lack of data connectivity over between the stage-gate trials of the MBP as the likely problem. Prior to the GSC, our early stage-gate trials (stages 1 and 2) had zero connectivity and in a typical year about 1% of all lines in the GSC were tested by more than one MBP.
The MBP of our GSC conduct cooperative trials where all lines in a trial are tested by all MBPs providing 100% connectivity. We data from cooperative trials to assess GS accuracy among MBP using these connected data sets. Data from the 5-State trials was used to assess GS accuracy for yield and test weight. Data from the P+NUWWSN FHB trials were used to assess GS accuracy for FHB resistance. We used BLUES estimated over trials conducted by three MBPs to predict the phenotype of the lines in trials of the fourth MBP: this is called a 3->1 prediction. We used BLUEs from one MBP trials to predict the phenotype of lines over the trials of the other three MBP (1->3 predictions). Finally, we used BLUEs from one MBP’s trials to predict the phenotypes of one other MBP’s trials (1->1 predictions).
The accuracy of GS between MBPs improved when using the highly connected data from the cooperative trials relative to the analysis using stage-gate data. Previously the average 1->1 accuracy for yield was 0.058 with a range of −0.1 to 0.18 (
Table 5). The average 1->1 accuracy for yield in the 5-State analysis was 0.22 with a range of 0.04 to 0.43 (
Table 6). This is similar to the inter-program accuracy reported by Saranelli et al. [
22] by using data from the Sungrains cooperative wheat trial. Using BLUEs from data compiled over three MPB’s trials greatly improved prediction accuracies shown by the values of the 3->1 and the 1->3 predictions (
Table 6). These results show the (1) increasing the connectivity between testing sites and MBP by co-testing more lines can increase the accuracy of GS and (2) that compiling phenotypic data over multiple MBPs can increase GS accuracy, a practice facilitated by a GSC.
E. There is a conundrum to be addressed. The primary benefit of a GSC is to effectively increase the size of each MBP by facilitating access to all the breeding lines of the other MBP. This benefit is only realized with accurate GS predictions between MBPs which can only be attained with connectivity of the phenotyping of the MBPs. This suggests that each MBP needs to increase their phenotyping if they continue to phenotype all of their own lines as well as lines from other MBP to attain connectivity. This is a major challenge given that an individual MBP cannot greatly increase the number of lines they phenotype. Sparse testing and the use of marker x environment interaction (MEI) in a GS model can be used to create an effective evaluation scheme that will produce connectivity and useful inter-program predictions without extensive replication of lines over all programs. Training population optimization algorithms [
36,
37,
38] could potentially be used to identify the optimum set of lines to evaluate across the MBPs to achieve sufficient GS accuracy with fixed phenotyping resources.
Sparse testing is where alleles, not lines, are replicated over environments and facilitates testing more lines with the same phenotyping resources [
39]. The performance of lines in environment where they were not tested can be predicted using GS models that includes a MEI term. Endelman et al. [
39] concluded that sparse testing at early stages with GS was superior to testing all lines in all environments as it enabled sampling a broader array of lines and environments. They also concluded that GS would be cost effect if the TP size was large and consisted of related individuals, a scenario that exists within a well-designed GSC. Others have noted that that GS can be effective for selection in early-stage trials [
13,
40] and that GS can alter the distribution of breeding resources [
33,
37,
39].
To increase connectivity and balance in our phenotyping, our GSC will disperse the stage-1 and stage-2 lines from each MBP among all four MBPs. Prior to the GSC, none of our stage-1 stage-2 were tested by another MBP, and less than 1% of all our breeding lines.
Table 7 shows how we dispersed stage stage-1 and stage-2 lines over the four programs of the GSC for the 2020–2021 season. A total of 35% of the stage-1 and stage-2 lines are now being tested outside of their MBP of origin, and 12% are being tested by multiple MBP providing connectivity among the four MBP.
We are currently investigating different sparse-testing schemes with a goal of attaining sufficient connectivity while not significantly increasing the amount of phenotyping any one MBP must conduct. An example is shown in
Table 8 assuming Breeder A normally tests 1000 lines in a stage-1 trial. Without a GSC, Breeder A will only test their own 1000 lines. In a GSC, breeder A could test 400 of their own lines and 200 lines from each of the other three MBPs Breeder A would send 600 of their stage-1 lines to the other MBP for testing. We are proposing to disperse the lines across the MBPs by families so pedigrees and alleles are replicated over MBPs. Connectivity could increase at advanced stage of testing. We envision that in stage-2 trials that some lines will be evaluated by multiple, MBPs. Our most advanced lines will still be evaluated by all MBPs in a cooperative trial that provides complete connectivity. Data from the stage-1, stage-2, and cooperative trials would be combined to make predictions.
Assessing GEI is the major rationale for uniform testing that allows estimation of both broad (stable, main effect) and local (main effect + GEI) adaptation of lines. GEI must be dealt with in sparse-testing [
41]. There are two scenarios for using GS models that incorporate GEI: (1) CV1 = predicting the value of lines that have not been phenotyped in any environment and (2) CV2 = predicting the value of lines that have been tested in some environments but not others (i.e., sparse testing) [
42,
43,
44]. The CV2 scenario is particularly applicable to our GSC where the vast majority of our lines are tested by just one MBP in their local environments in stage-1 trials. Crossa et al. [
44] indicated that GEI could be used to estimate the value of lines in environments where they were not tested by using information from relatives that were tested in those environments. Burgueño et al. [
42] extended this to GS models using marker data and the CV1 and CV2 testing schemes. They reported that prediction accuracy was significantly greater in the CV2 sparse testing scenario than for the CV1 scenario. This emphasizes the importance of testing related lines with shared marker alleles across environments. Sparse testing allows for estimation of main effects (broad adaption) and site-specific trait values. Lopez-Cruz et al. [
45] incorporated MEI into the prediction models for wheat yield. They found that the use of MEI increased GS accuracy by up to 30% compared to models without MEI, and that GS accuracy was greater for CV2 than for CV1. Others have reported increased GS accuracy by incorporating GEI (or MEI) into prediction models for wheat [
5,
46,
47,
48] in sparse-testing trials and CV2. Studies in rye [
49], maize, [
50], rice [
51], rubber [
52], and coffee [
53] also show benefits to incorporating GEI (or MEI) into GS predictions.
F. Genotyping Platforms. Applied GS requires low-cost genotyping and a GSC requires all MBP to genotype their lines with the same marker platform. High-throughput multiplexed SNP genotyping is a widely used genotyping method. Genotyping-by-sequencing (GBS) is a low-cost SNP genotyping platform that has been widely used in plant breeding [
54,
55,
56]. With GBS target SNPs need not be identified a priori which reduces assay development cost and ascertainment bias while enabling application across a wide range of diverse populations. The disadvantages of GBS include (1) SNP calls with a greater proportion of randomly missing data for some genotypes, which must be imputed [
57,
58], (2) generation of markers that do not match across datasets, and (3) the bioinformatics workload for GBS can be intensive in terms of data storage, CPU usage and labor as the number of samples reach 10,000 s after many years and new samples are added each season requiring new SNP calls.
An emerging alternative to GBS protocols are pooled, multiplexed targeted sequencing assays that target specific SNPs [
59,
60] Commercial implementations of pooled, multiplexed sequencing technology include Illumina’s AmpliSeq (Illumina, San Diego, CA, USA), Integrated DNA Technologies’ rhAmpSeq (Integrated DNA Technologies, Coralville, IA, USA), and Diversity Array Technology’s DArTag (Diversity Array Technology, Bruce, Australia). These platforms require a significant up-front design cost but can produce more repeatable SNPs, can accurately identify heterozygotes and require less bioinformatics than GBS.
In collaboration with the USDA Eastern Regional Small Grains Genotyping Lab at North Carolina State University (
https://www.ars.usda.gov/southeast-area/raleigh-nc/plant-science-research/docs/small-grains-genotyping-laboratory/main/, accessed on 2 August 2021), we are in the process of designing a pooled, multiplexed sequencing genotypic assay that will provide data on ~2500 highly polymorphic markers at a cost of ~
$5–
$9 per sample. The final assay will also include ~100 well-characterized markers for critical agronomic and disease-resistance traits. This will facilitate GS and marker-assisted selection in the same genotyping platform.
The targeting sequencing assays provide fewer markers that GBS. Several studies in wheat have shown that 1000–2000 markers can provide the same GS accuracy as marker sets that are 10 times larger [
13,
56,
61]. The low cost, repeatability, identification of heterozygotes, ability to combine GS and MAS in one genotyping operation, and the lightened bioinformatics favor the use of targeting sequencing assays in a GSC.
G. Building a GSC requires careful collection, curation, and standardization of phenotypic and genotypic data from all MBP so all data is accessible to all MBP.
Recently, multiple research groups have been working on developing centralized, open-source relational databases for breeding. The Breeding Application Programming Interface (BrAPI) has been developed to implement standardized tools for interacting with breeding databases [
62]. BrAPI standards have been implemented in both commercially supported breeding databases such as the Integrated Breeding Platform (
https://integratedbreeding.net, accessed on 2 August 2021) and in open source projects such as Breedbase (
https://breedbase.org, accessed on 2 August 2021) and Breeding4Results (
https://b4r.irri.org, accessed on 2 August 2021). Currently there several large Breedbase instances supporting public breeding efforts for cassava (
Manihot esculenta;
https://cassavabase.org, accessed on 2 August 2021), bananas (
Musa spp.;
https://musabase.org, accessed on 2 August 2021), yams (
Dioscorea spp.;
https://yambase.org, accessed on 2 August 2021), sweet potato (
Ipomoea batatas;
https://sweetpotatobase.org, accessed on 2 August 2021), and Solanaceous crops (
Solanaceae spp.;
https://solgenomics.net, accessed on 2 August 2021). Genotyping data management software such as the Genomics Open-source Breeding Informatics Initiative Genomics Data Manager (GOBii GDM;
https://gobiiproject.atlassian.net/wiki/spaces/GD/overview, accessed on 2 August 2021) and GenomicsDB (
https://www.genomicsdb.org/, accessed on 2 August 2021) features efficient storage and querying of genotype data.
Any breeding database utilized by multiple breeding programs should include support for the generation and management of trial information and phenotypic data at the plot level. It should enable the management of information on accessions, locations, and genotypic data. In addition, any database solution should support the use of trait ontology systems such as the crop ontology (CO) database [
63] to ensure standardization of trait measurements across MBP, as well as integration with tablet data collection programs such as the Field Book Android app [
64]. Our experience has shown that transitioning to a common breeding data management system is not trivial, and resources and technical support for this transition are vital.
H. Accurate phenotypes. The foundation of all successful breeding, including GS and marker-assisted selection, is accurate phenotyping. Each member of the GSC should be able to provide reliable phenotypic data for the key traits of the GSC. The data should be filtered for outliers and assessed for validity by the MBP that generates the data. Each MBP should also be phenotyping in some environments that are relevant to other MBP.







{kind=link}
{kind=link}
{kind=link}