
Novel Sequencing and Genomic Technologies Revolutionized Rice Genomic Study and Breeding


1
Rice Research Institute, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, China
2
Guangdong Key Laboratory of New Technology in Rice Breeding, Guangzhou 510640, China
3
Guangdong Rice Engineering Laboratory, Guangzhou 510640, China
*
Authors to whom correspondence should be addressed.
Agronomy 2022, 12(1), 218; https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy12010218
Submission received: 6 December 2021
/
Revised: 21 December 2021
/
Accepted: 1 January 2022
/
Published: 16 January 2022
(This article belongs to the Special Issue Breakthroughs and Prospects of Rice Breeding)
Abstract
:Rice is one of the most important food crops worldwide. Population growth and climate change posed great challenges for further rice production. In the past decade, we have witnessed an explosive development in novel sequencing and genomic technologies. These technologies have been widely applied in rice genomic study and improvement processes, and contributed greatly to increase the efficiency and accuracy of rice breeding. On the other hand, novel sequencing and genomic technologies also promote the shift of breeding schemes from conventional field selection processes to genomic assisted breeding. These technologies have revolutionized almost every aspect of rice study and breeding. Here, we systematically sorted out and reviewed the progress and advancements of sequencing and genomic technologies. We further discussed how these technologies were incorporated into rice breeding practices and helped accelerate the rice improvement process. Finally, we reflected on how to further utilize novel sequencing and genomic technologies in rice genetic improvement, as well as the future trends of advancement for these technologies. It can be expected that, as the sequencing and genomic technologies will develop much more quickly in the future, and be combined with novel bioinformatics tools, rice breeding will move forward into the genomic assisted era.
1. Introduction
Rice (Oryza sativa L) is one of the most important crops, feeding nearly half of the world population. As the world population is expected to reach 10 billion by 2050, there is an urgent need to enhance rice production by 50% in the coming decade [1]. Furthermore, the climate changes pose another great challenge to the stabilization of rice production. Rice’s adaptation to climate change and other imperatives will need to keep pace with the rate of change [2]. To overcome these challenges, it is an urgent task to breed more resilient, resource-use-efficient and ‘climate-smart’ varieties [3].
Tremendous efforts have been made worldwide in rice genetic study and molecular breeding to understand and improve the agronomic traits of rice, such as grain yield, environmental performance, biotic stresses resistance and nutrition quality. However, rice breeding is still restrained by many factors. These restraints relate mainly to two core questions, namely discovering the genetic variations and breeding tools for harnessing these variations. First of all, the access and characterization of genetic variations conferring the desired traits is the fundamental resource for rice improvement. However, the lack of high-throughput genotyping tools and highly efficient genetic analysis methods make these studies time- and cost-consuming. On the other hand, breeding methods and tools play key roles in utilizing the characterized genetic variations to create cultivars with the desired traits. Traditional rice improvement programs rely mainly on identifying and crossing plants with agronomically desirable phenotypes, and then selecting new varieties manually in the field. The efficiency and the accuracy of this selection are low.
In the past two decades, rice genomic and genetic studies have progressed greatly with the invention and development of novel sequencing technologies. Based on these new technologies, many novel genomic approaches and methods were invented or developed and have changed the landscape of rice study. For example, novel long-read sequencing technologies, scaffolding methods and newly developed assembly algorithms have made it much easier to obtain a complete genome of rice-specific resources. The availability of complete genomes facilitated the identification of genomic variations, especially the structural variations, which are very difficult to characterize without complete genome sequences. These technological advancements also facilitated the emergence of novel genomic concepts which fundamentally changed the framework of rice research. For example, the concept of pan-genome and the related methodology were developed based on the findings from modern genomic study, and were further enabled by new sequencing technologies and novel algorithms. The pan-genome concept has changed the genomic study framework from using single or a few reference genomes to using a pan-genome, which can catalog and present more genomic diversity within a species [4].
These novel sequencing and genomic technologies have been widely applied and revolutionized almost every aspect of rice basic and breeding researches: (1) Enabling the assembly of a number of high-quality rice reference genomes, which can provide the unified framework for further genomic and genetic research in rice. (2) Providing cost-effective and efficient tools for accessing the sequences of accessions within a rice gene bank. (3) Facilitating acquisition of genome sequences, and providing tools for the in-depth exploration of genetic resources, as well as the molecular mechanism underlying important traits. (4) Facilitating novel concepts and methodologies to study and harness genetic variations in rice germplasms. (5) Enabling novel techniques to utilize genetic variances in routine breeding processes and improve the efficiency and accuracy of rice breeding.
With a further reduction in costs and an increase in the quality of sequencing technologies, as well as a faster development of genomic and bioinformatics in the near future, we can expect a deeper incorporation and implementation of these sequencing and genomic technologies into rice genomic studies and breeding programs. Enabled by these technologies, genomics-assisted breeding in rice has come of age [5].
In this review, we systemically sort out and discuss the advancements of sequencing and genomic technologies. We further reviewed how these technologies have helped and shaped the landscape of rice genetic study and breeding efforts. We aim to provide a full picture and outlook on how these sequencing and genomic technologies can be incorporated into rice breeding, and the future shift of the rice breeding paradigm.
2. The Reference Genomes, the Fundamental Framework for Rice Genetic and Breeding Research
2.1. The First Rice Reference Genome and Its Refinement by Next-Generation Sequencing Technologies
A reference genome provides a uniform and fundamental framework for genomic and genetic study. A high-quality reference genome is therefore a prerequisite for genomics studies. Benefitting from having the smallest genome among the major cereals, rice is the first crop to have its whole genome sequenced. In 1997, the International Rice Genome Sequencing Project (IRGSP) started to sequence rice’s whole genome using a map-based clone-by-clone strategy [6]. Two draft genome sequences of the cultivated rice subspecies japonica Nipponbare and indica 93-11 were released in 2002 [7,8]. In 2005, the IRGSP reported the map-based and finished version of the Nipponbare sequence. In the same years, a genome sequence of an indica variety, 93-11, was also released by sequencing more bacterial artificial chromosomes (BACs) and P1-derived artificial chromosome (PACs) clones and adopting new assembly algorithms [9]. The completeness of the rice reference genome was the first step in a systematic and complete functional characterization of the rice genome [10].
As the Rice Genome Sequencing Project started, the next-generation sequencing (NGS) technologies had not been invented. However, after the emergence of NGS technologies, it helped researchers to further improve and refine the rice reference genome. In 2013, the Nipponbare rice genome was updated and validated using data from two short-read sequencing platforms, in which researchers found and corrected more than 4000 errors in the previous version of the Nipponbare genome [11].
A high-quality reference genome is defined not only by the continuity and correctness of the assembled sequences, but also by the annotation of the genome, which is the biological function of a specific part of the genome. However, despite the fast advance in the rice genome sequences available, the genome annotation has lagged far behind. Novel sequencing technologies and algorithms play important roles in refining the annotation of rice genomes. For example, an RNA-seq enabled by NGS technologies provides immense information about the gene expression at the whole-genome scale, which can help identify the transcription component of the genome. Using massive RNA-seq data, Sang et al. refined the gene model prediction of the Nipponbare genome and achieved a higher integrity and quality in genome annotation compared to the previous annotation system [12].
Successfully assembling a high-quality reference genome for rice was a milestone for rice research, since it not only provides a uniform framework for rice genomic studies, but also offers unprecedented information and insights into the genomics, evolution and biology of rice [13]. Functional gene cloning is the most dramatically boosted field regarding the availability of the rice reference genome and related resources. From 1990 to 1994, there were 20–30 papers per year describing studies on gene cloning in rice. From 1995 on, when the first rice BAC library was published, the number ranged from 40 to 80. After the full genome was available, the number jumped to more than 100 per year [14].
2.2. Novel Sequencing Technologies and Assembly Strategies Facilitated the Assembly of More High-Quality Rice Genomes
In the last decade, with the availability of a complete reference genome, the re-sequencing method based on NGS has emerged as a powerful tool to identify genomic variations across rice germplasms. The re-sequencing method maps NGS sequencing data to the reference genome, and then calls the variants according to the reference. However, most structural variations (SV) presented in rice germplasms cannot be detected by the re-sequencing method. Furthermore, increasing evidence demonstrate that the single reference genome is insufficient to fully capture the diversity within the rice gene pool, as large amounts of genomic variances found in one or a few individuals have not been found in genomes of other individuals. For example, the whole-genome assembly and comparative analyses of Zhenshan97 and Minghui63 uncovered surprising structural differences, especially with respect to inversions, translocations, presence/absence variations (PAV) and segmental duplications [15]. Therefore, it is critical to capture and understand genetic variations, especially SVs, in much more diverse rice gene banks, and higher-quality reference genomes representing diverse genetic variation are needed [16].
The invention and development of novel sequencing technologies, including next-generation sequencing technologies, and the third-generation sequencing technologies have accelerated the genome assembly of other accessions from rice gene banks.
Before the invention of third-generation sequencing technologies, researchers have tried to assemble rice genomes using NGS technologies. For example, Gao et al. used the re-sequencing strategy and got an update version of the genomes of PA64 and 93–11, which are parents of a pioneer super hybrid rice, LYP9 [17]. The emergence of third-generation sequencing technologies, which can obtain sequencing data with much longer reads, completely changed the landscape of rice genome assembly studies. The third-generation sequencing platforms can obtain and sequence much longer reads (>10 Kb) compared to NGS technologies (~150 bp). Much longer reads can solve the difficulties encountered in genome assembly, especially in the repetitive regions, which are as high as 45% in rice genomes. At the same time, many scaffolding technologies were developed to further help to scaffold genomes into chromosome-scale assemblies. For example, optical mapping can join assembled contigs to produce super-scaffolds by producing fingerprints of DNA sequences and construct a consensual optical map [18].
With the help of these novel technologies, the complex genomic structure of rice genomes can be easily solved, which will make the assembly of rice genomes a routine task. In 2016, Zhang et al. assembled the genome of two indica cultivars by BAC sequencing using a third-generation (PacBio) sequencing platform [15]. Then, in 2017, Du et al. reported a near-complete genome of the indica rice Shuhui498 through the integration of third-generation sequencing and mapping data, genetic map and fosmid sequence tags. The assembled genome covers more than 99% of the estimated genome size and served as a reference for the discovery of genes and structural variations in rice [19]. Since then, a number of rice genome assemblies were reported with unprecedented speed, and more than 100 de novo assembled rice genomes were released (Table 1) [20].
2.3. Pan-Genome, a Novel Genomic Concept and Reference Genome for Rice Genomic Studies
De novo genome assemblies of diverse rice accessions provide suitable references for probing genetic variations in diverse rice germplasms. However, routine analysis methods, such as a re-sequencing analysis, can only use a specific single genome for reference. As mentioned above, a single reference cannot present all genetic variations within a population or species, since a lot of variations are absent in a specific single genome. These problems raised the question of how to collect, present and characterize the highest number of genetic variations existing in a population or species. The pan-genome and the related methodology based on novel sequencing technologies are proposed as one of the feasible concepts and tools to solve these problems.
A pan-genome refers to a complete collection of the genomic sequences found in a biological clade, such as a species, or a sub-population, rather than a single individual [31]. The concept was first proposed by Tettelin et al., who found a lot of genes present and absent variations among different strains of bacteria by comparing bacterial genomes. They defined the pan-genome as the whole collection of genes found in all strains of bacteria [32]. Soon, the concept and the related methodology were extended to studies on humans, animals and plants [33,34,35]. The pan-genome provide a whole set of methodologies for estimating the genomic diversity of the dataset at hand and predicting the number of additional whole-genome sequences that would be necessary to fully characterize that diversity [36].
At present, three kinds of strategies were established for pan-genome construction [31]. The first one includes iterative construction or map-to-pan strategies, in which reads from NGS are mapped to a reference genome and then the unmapped reads are collected for de novo assembly. The first rice pan-genome was constructed using this strategy, in which the rice dispensable genome was constructed based on low-coverage (1–3×) sequencing data of 1483 cultivated rice accessions [37]. Thousands of protein-coding genes are successfully assembled, including most of the known agronomically important genes absent from the Nipponbare reference genome. The constructed pan-genome was helpful for understanding the rice dispensable genome and complementary to the reference genome for identifying candidate genes associated with phenotypic variation. Using a similar strategy, another rice pan-genome built on 3000 rice accessions was constructed [38,39].
The second strategy is based on identifying and collecting genomic differences among different accessions by whole-genome comparison. The prerequisite for this strategy is the availability of a number of de novo assembled genomes. With the completeness of de novo assembled genomes, this strategy greatly improves the quality of the pan-genome in terms of the completeness and continuity of genetic variations. In 2018, Zhao et al. applied this strategy and constructed a rice pan-genome by deep sequencing and de novo assembled 66 divergent rice accessions [40]. The ever-evolving sequencing technologies, particularly the third-generation long-read sequencing technologies, have made high-quality de novo assembly much more accessible, thus favoring the utilization of this strategy for pan-genome construction [41].
More recently, a third strategy, which utilized bi-directed variation graphs (VG), was developed to construct a graph-based pan-genome [42]. VG uses sequencing reads supported by linear genome references to produce a graph representation of the variable regions, avoiding assembly bias towards a reference genome [43,44]. Utilizing this approach, Qin et al. de novo assembled 31 high-quality genomes represented for genetic diversity in a rice population and constructed a graph-based pan-genome of rice. By leveraging this pan-genome, they were able to analyze the SV distribution and assessed the mechanisms of SV formation and their impacts on gene expression [30].
The constructed rice pan-genomes have deepened our understanding of rice genomic diversification, and offer much more informative platforms that enable researchers to obtain substantially more information than a single or a few reference genomes. They are also of great help for sequencing-based analyses such as RNA-seq or SV identification.
3. Sequencing and Genomic Technologies Enabled Novel Tools for Rice Functional Genomic Studies
Discovering genome components and further dissecting the regulatory mechanisms underlying rice agronomic traits, which is also termed as functional genomic study, is the base for rice molecular breeding. Functional genomics provides fundamental genetic variation resources for rice improvement. By utilizing high-quality reference genomes and the application of novel sequencing and genomic technologies, rice functional genomics have made tremendous progresses in the past decade. These progresses were mainly ascribed to the development of novel tools for the identification and characterization of the genetic variations controlling important agronomic traits in rice germplasm [45].
First of all, sequencing technologies facilitate the genotyping of the high density of the markers in a low-cost and high-throughput way. The availability of high-density markers further enabled the development of other genetic approaches for identifying quantitative trait locus (QTLs) or characterizing the underlying genomic variation effectively, such as bulk segregant analysis (BSA) and genome wide association study (GWAS) [46].
3.1. High-Throughput Genotyping Tools Developed Based on Sequencing Technologies
Three factors, i.e., population size, phenotyping and number of markers, determine the success of functional gene mapping and cloning. Two decades ago, these tasks relied mainly on a conventional map-based cloning strategy using low-throughput molecular markers. The throughput and cost of acquiring and genotyping molecular markers was one of the main constraints for map-based cloning in the pre-NGS era [47]. The re-sequencing approach enabled by NGS technologies can genotype a massive number of DNA polymorphisms in a whole-genome scale. At the same time, the NGS technologies have allowed the parallelization of the sequencing process, thereby reducing the sequencing cost 1000-fold since its invention [48]. The cost for library construction and whole-genome sequencing is below $30 for each rice accession at present, which makes the sequencing-based genotyping approach a routine process used in rice [49].
These advancements facilitated high-density linkage mapping and the efficient accomplishment of genetic studies in a population scale [47]. For example, Gao et al. identified 43 yield-related QTLs, including 20 newly identified QTLs, by re-sequencing a recombinant inbred line (RIL) population, whose approved sequencing-based approaches will greatly facilitate QTL cloning [17].
High-density molecular markers also booted the emergence of many novel genetic tools and approaches for a fast identification of causal genes conferring desirable agronomic traits in rice. Feng et al. reported a genetic tool for directly targeting the specific causal genes by using a single-gene resolution linkage map leveraging NGS technologies and a large F2 population [50]. By implementing this tool, they not only successfully mapped to their specific causal genes of QTLs, but also constructed a complex genetic interaction network containing 30 QTL–QTL interactions [50].
3.2. Bulk Segregant Analysis
Bulk segregant analysis (BSA) was firstly reported by Michelmore et al., who identified random amplified polymorphic DNA (RAPD) markers tightly linked to genes for resistance to lettuce downy mildew [51]. Theoretically, BSA is amenable to any type of markers. Zhang et al. reported detecting QTLs related to heat tolerance in rice by BSA using SSR markers [52]. The invention of NGS technologies has made BSA much more rapid and cost-effective. By sequencing bulks of offspring with contracting phenotypes in a segregating population along with their parental lines, BSA can quickly and cost-effectively identify the loci controlling the phenotype. This approach was successfully demonstrated for a faster identification of QTLs for blast resistance in rice [53]. Soon, the method was widely and successfully utilized in identifying QTLs of many important traits in rice [54,55,56].
The combination of the BSA approach and sequencing technologies further enabled the invention of many alternative or novel methods for accelerating traditional positional cloning for qualitative traits. Quantitative trait gene sequencing (QTG-Seq) was devised by Zhang et al. to accelerate QTL fine-mapping by combing QTL partitioning and BSA sequencing. QTG-Seq combines QTL partitioning to convert a quantitative trait into a near-qualitative trait, sequencing bulked segregant pools from a large segregating population and using a robust new algorithm for identifying candidate genes. QTG-Seq can fine-map the genomic interval into a ~150 kb, whereas the mapping resolution of a traditional QTL mapping is ~2 Mb [57]. Another method, MutMap, combines mutant generation, the BSA method and sequencing technologies to accelerate functional gene identification in rice [58]. QTL-Seq is an extension of the BSA and MutMap methodology for mapping major effect QTLs using the NGS platform. In QTL-Seq, Each bulk DNA will be sequenced and aligned independently to reference the genome to calculate the SNP index. The SNP index of the lowest bulk is subtracted from the SNP index of the highest bulk, which results in the formation of a peak in the plot of the SNP index giving the position of the QTL [53]. Using this method, Arikit et al. were able to rapidly identify QTLs controlling rice cook grain elongation, as well as the candidate genes underlying the QTL [59].
3.3. Genome-Wide Association Study (GWAS)
GWAS was another genetic approach quickly developed according to the advancement of sequencing and genomic technologies. The concept of GWAS was first developed in humans and was subsequently introduced into plant genetics [60]. GWAS enabled researchers to quickly identify a limited interval of region controlling the phenotype, which is normally 200–400 kb, depending on the linkage disequilibrium of the population. Similar results may take 3–5 years to get in traditional bi-parental QTL mapping. High-density molecular markers are prerequisites for GWAS, since this method needs to consider the relationship between the phenotype and the markers at the whole-genome level. In this way, the high density of the molecular markers provided by NGS technologies facilitated the wide implementation of GWAS in gene mapping and cloning in rice.
Rice has a few advantages in conducting GWAS: (1) It has abundant germplasm resources with a much higher genetic diversity than that of other plants. An abundant diversity is the fundamental factor determining the success of the identification of functional genes by GWAS. (2) Rice is a self-pollinated and inbred species, which make GWAS more efficient. With just a one-time genotyping of a population, the panel of inbred lines can be kept immortal in seed banks and can be phenotyped for different traits in different environments in both present and future studies [61].
The GWAS platform was well developed in rice a decade ago. The first GWAS study in rice using NGS technologies was reported in 2010. The SNPs of 517 rice landraces were identified by sequencing and used for GWAS for 14 agronomic traits [62]. GWAS was then further refined and developed to investigate the genetic basis of natural variation in biological traits in rice.
GWAS has helped accelerate QTL identification and the functional cloning of the genes underlying the QTLs. OsSPL13 was the first functional gene identified in rice by GWAS. OsSPL13 was firstly identified as a candidate gene for grain shape by combining the results from GWAS and expression profiling. A further gene functional validation was conducted by genetic complementarity [63]. Since then, more and more functional genes were identified by GWAS. For example, by utilizing GWAS, a natural allele of a C2H2-type transcription factor was identified to confer non-race-specific resistance to blast, and natural variations in OsGSK2 were found to determine rice mesocotyl’s length variation [64,65].
A wealth of resources, including a number of diverse rice accessions, a dense genomic variation map and a set of high-quality genotype and phenotype data are prerequisites for an effective GWAS implementation. At present, two major international diverse rice panels were established to facilitate large-scale GWAS study. The first one is a 2 K panel, which contains 2000 international diverse accessions collected by the International Rice Research Institute [66]. The other panel is a 3 K panel, which is parallel to the 2 K panel collected by the International Rice Research Institute and the Chinese Academy of Agricultural Science [39]. Both panels contain whole-genome SNP information and are suitable for GWAS study. Previous, we selected a subset of the 2 K panel containing more than 500 accessions according to the diversity and phylogenic analysis. Based on this subset, we conducted a GWAS on more than 30 phenotypes and succeeded in identifying many novel QTLs and subsequently the candidate functional genes for important agronomical traits of rice [67,68]. Our results proved this diverse panel is a valuable resource for identifying QTLs by GWAS.
Despite many successfully cases, GWAS in rice also faces a number of challenges, including missing genotypic data in some genome regions because of the coverage of the NGS data, as well as the problems involved in solving G × E interactions [69]. The development of new statistical analysis methods and the corresponding experimental designs can help to solve these problems. As for the missing data problems, they can be partially solved by genotypic data imputation methods, which can impute the missing genotypic data through statistical methods, due to linkage disequilibrium among the polymorphisms of local regions in rice [70]. The advancement in algorithms is another aspect to fully exploit the potential of GWAS. Compared to the traditional statistical modeling used in GWAS, Bayesin methods and machine learning methods are showing an increasing performance when implemented in GWAS [71]. A few pioneering studies have been conducted to utilize these new methods for GWAS and gain mechanistic insights into the genotype–phenotype associations [72,73].
4. Breeding Schemes Revolutionized by Sequencing Technologies and Genomic Approaches
Reference genomes and novel functional genomic tools accelerated the identification of the genetic components controlling rice traits, which serve as the source for rice improvement. How to efficiently and accurately utilize these genetic resources in rice improvement is another important aspect in rice breeding. A traditional rice breeding process depends on crossing cultivars using germplasms with desired traits and then selecting plants mainly by manual inspection in the field. The selection and subsequent homogenization of the traits are often time- and cost-consuming, and it normally takes 5–10 years to bring out a new variety [74]. What is more, if the trait is a complex trait with many low heritability genes, the uncertainty of the selection increases significantly. Therefore, labor intensity, time consumption, low efficiency, environment dependence, etc., are major barriers that impede conventional rice breeding.
Sequencing and genomic technologies advancement provided many tools and methods which can incorporate into and accelerate the conventional breeding process. These tools and methods also revolutionized the landscape and schemes of rice breeding, and led to a new concept, ‘genomics-assisted breeding’ [75].
4.1. Sequencing Technologies Accelerate Rice Marker-Assisted Selection
In the past decade, as high-throughput DNA sequencing decreases in cost, the focus of plant breeding has gradually switched from phenotype-based to genotype-based selection [76]. The high-density markers provided by NGS are now widely applied to improve the breeding efficiency. On the one hand, as mentioned above, the utilization of high-density markers can accelerate the identification of QTLs and superior haplotypes. These identified relationships between phenotypes and genotypes facilitate phenotype selection based on genotype selection, which is also termed as marker-assisted selection (MAS). MAS is used extensively in breeding programs to track the desired traits using linked molecular markers. Facilitated by NGS technologies, millions of SNP markers can be detected with low cost and high throughput, and used to monitor genomic loci that are linked to markers in breeding pedigrees in order to assemble combinations of loci.
‘Haplotype assembly’ was another novel concept proposed and developed based on MAS and NGS technologies. Recent re-sequencing of germplasm collections in a few crops has facilitated the identification of a small number of strong marker-trait associations, as well as haplotypes for the target traits, which facilitate ‘haplotype assembly’. ‘Haplotype assembly’ means stacking or combining superior haplotypes of desired traits to develop ideal varieties. This concept was proposed as a new approach for developing improved crops through assembling superior haplotypes of the targeted traits [74]. ‘Haplotype assembly’ had been further suggested by Abbai et al., who reported superior haplotypes upon phenotyping the subset of the 3000 rice panel [77].
4.2. Genomic Selection
The high-density molecular markers provided by NGS have also prompted the development of ‘genomic selection’ (GS). GS is based on integrating germplasm accessions, genomic resources, molecular technology and breeding tools [78]. Unlike MAS, GS used genome-wide DNA markers to predict the genetic merit of breeding individuals for complex traits.
In 1994, Rex Bernardo achieved an early milestone for the development of GS by replacing pedigree-based kinship with a marker-based kinship estimated using Restriction Fragment Length Polymorphism (RFLP) markers spanning the entire maize genome [79]. Meuwissen et al. introduced the second milestone of GS, in which effects of all markers in a parental generation were estimated and added together to predict the performance of future progeny [80,81]. In Meuwissen’s proposed framework, breeding values were predicted using genome-wide marker information. However, the technology to generate genome-wide markers did not exist at that time. High-throughput DNA sequencing technology fundamentally enables the power of GS by providing whole-genome high-density markers and many other related genomic information at an affordable cost and a large scale [82]. Genotyping-by-sequencing and commercially available SNP arrays are the first two established and low-cost genotyping platforms allowing for the rapid genotyping of large number of markers across a genome, which is the foundation platform for GS.
GS does not need markers specifically associated with a trait because breeding lines are selected for crossing and advancing generations based on genomic-estimated breeding values calculated from genome-wide marker data. GS has a higher accuracy of prediction of elite genetic materials in the initial generations and permits shorter breeding cycles [83]. The key advantage of GS over MAS is that it considers all markers irrespective of their role in the phenotype, which can consider all major and minor effects on the traits [84].
In 2015, Spindel et al. reported a GS application in rice breeding in which they performed a GWAS in conjunction with a five-fold GS cross-validation on a population of 363 elite breeding lines from the IRRI-irrigated rice breeding program [85]. In their reports, the prediction accuracies of GS ranged from 0.31 and 0.34 for grain yield and plant height to 0.63 for flowering time [85]. In addition to inbreed lines, GS has also been applied in hybrid breeding in rice. Cui et al. used genomic best linear unbiased prediction to perform a hybrid performance prediction using an existing rice population of 1495 hybrids. This study showed that the prediction abilities on ten agronomic traits ranged from 0.35 to 0.92 [86].
The platforms of GS breeding in rice have been established based on the research findings of genomics, breeding chips and high-throughput sequencing technologies. Specifically, the Green Super Rice project, first proposed in 2005, and which aims to develop new rice varieties with various green traits, has developed high-throughput breeding chips of rice based on Illumina’s Infinium technology, which constitute a GS technical system in rice [78]. The prediction accuracy of GS can be further improved by taking the significant loci detected by GWAS and the functional genes verified by molecular biological experiments into consideration as fixed effects in the GS model [87]. Furthermore, GS models can be optimized by including other types of omic information, such as transcriptomic data [88].
4.3. Genome Editing
Genome editing is a novel technique, proposed to be the most efficient breeding strategy today [89]. Genome editing allows the targeted modification of almost any crop genome sequence to generate novel variation and accelerate breeding efforts, which is totally different from traditional breeding strategies based on crossing [90]. Genome editing is highly dependent on accurate sequence information for a precise determination of the target position. This sequence information can be easily acquired by novel sequencing technologies. The other great challenge for genome editing is to understand the extensive and complicated genetic networks controlling agronomic traits and their interaction with environmental factors. The development of sequencing and genomic technologies together with the establishment of a large number of ‘omics’ databases will facilitate the identification of feasible targets for genome editing in rice [91].
With the availability of precise genome editing tools and precise information on the rice genome, as well as the knowledge about rice domestication, a novel concept, termed as de novo domestication, was developed as a revolutionary breeding method in rice. The theory behind this concept is that the initial morphological changes that occurred during crop domestication are mainly controlled by a few large-effect loci. Therefore, genetic modifications of a few major genes from ‘undomesticated’ species could bring about a rapid evolution [92]. The success of de novo domestication relies on fully understanding the domestication process and the genes controlling these domestication traits. With the advancement of sequencing and genomic technologies, the genomic study of Oryza species can provide further insights into rice domestication, and also help to identify critical genes controlling rice domestication traits [93,94]. With all the information available, Yu et al. reported a successful example of de novo domesticating of an allotetraploid rice. They rapidly improved six agronomically important traits by editing homologs of the genes controlling these traits in diploid rice [95]. De novo domestication can be developed and used to create new staple rice varieties to strengthen global food security.
5. Summary of Sequencing and Genomic Technologies Incorporating and Accelerating Rice Breeding Processes
In summary, with the deep incorporation of novel sequencing and genomic technologies into rice study and breeding practices, the scheme and framework of rice breeding have changed profoundly. At present, rice germplasms and genetic resources are first characterized by sequencing technologies, and then genomic approaches and bioinformatics tools are applied to acquire masses of genomic data of these diverse resources. These data are further investigated to gain deep insights about the molecular mechanisms of important rice traits. Furthermore, these genomic data also provide large-scale tools and molecular information to enable genomic-assisted breeding and genome editing. With this knowledge about the molecular mechanisms of rice traits, combined with molecular information and tools, the breeding and improvement will be much faster than the traditional breeding scheme (Figure 1).
6. Future Perspectives
Human explosion and adverse climatic changes are posing extreme challenges for global food security. Breeding new rice varieties with stress resilience and high yielding is an urgent demand to solve these problems. As reviewed above, many new concepts, techniques and methods enabled by novel sequencing technologies and genomic approaches have greatly improved the efficiency and effectiveness of rice breeding. These technologies have actually reshaped the framework of rice genomic study and breeding practices. In the near future, we can expect much faster advances in the field of sequencing and genomic technologies. Sequencing and genomic approaches will likely be incorporated more deeply into rice improvement programs. The development of novel tools development and a revolution in breeding concepts will be the next trends for rice research and breeding.
6.1. Novel Bioinformatics Tools and Platforms Play Key Roles in Translating Genomic Data into Future Tools for Rice Breeding
The application of genomics for trait dissection is well established; however, translating the findings from genomic studies into breeding programs is challenging [96]. In this process, bioinformatics tools and platforms play central roles, since they provide powerful methods, theoretical approaches, standards and software tools to analyze the ever-increasing genomic data. Databases host a combination of various genomic information alongside analysis tools providing a useful platform for genomic information utilization in breeding. In rice breeding studies, a large number of functional genes, QTLs and genomic information have been identified. For an efficient usage, these data and information need to be integrated into a suitable platform for further exploration and utilization [97]. However, most of these databases lack supporting tools for breeders, which is an important aspect for future database construction. Software solutions to integrate breeding and research resources are another highly demanded field. With the rise in multi-omic technologies, the future development of bioinformatics will focus more on enabling the integration of multi-omics data to allow a system-level understanding of the agronomic traits in rice. This will pave the way for multi-omic breeding schemes which can help to develop elite rice cultivars much more rapidly.
In term of bioinformatic algorithm development, Artificial Intelligent (AI) is the most expeditiously rising technology to deal with big biological data mining [98]. Machine learning (ML) is an emerging and promising application of AI. Advanced ML algorithms have revealed a great potential for making highly precise and efficient pipelines for data analysis to enhance the breeding performance and ultimately crop productivity. ML tools can help rice breeders to explore biological patterns by analyzing a large amount of genomic data. ML has also been applied in genomic selection to process large genotyping data sets for genomic prediction [99]. In the future, ML models are expected to be largely applied in the different -omics disciplines, enhancing their integration toward the resolution of key biological questions in breeding.
6.2. From Conventional Breeding Practices to Genomic-Assisted Breeding
Current advances in genome sequencing techniques and biological “Big Data” have profoundly transformed rice breeding. In the coming year, huge amounts of genomic data will become much more accessible. Rice breeding based on genomic information, which is termed as genomic-assisted breeding (GAB), will be more practical and widely applied. GAB has been successfully implemented in rice mainly by MAS, which rely on one or a few markers linked to the phenotype. Novel sequencing technologies have enabled the access to high-density molecular markers, which will promote a paradigm shift from individual markers to the combination of markers (haplotype), and further to the assessment of markers at a whole-genome scale. These changes will greatly accelerate the progress of rice breeding, since more markers and related genomic information will improve the efficiency and accuracy of trait improvement. By this trend, more novel concepts and methods which can fully utilize genomic information in breeding practices are needed. Furthermore, valuable genetic variations from rice’s wild relatives or landraces will be characterized and harnessed under novel concepts and approaches, such as pan-genomic, de novo genome assembly and de novo domestication. Therefore, rice breeding will be benefited by utilizing valuable genetic variations from rice’s wild relatives, which will help to overcome the bottleneck of genetic diversity in modern rice breeding.
In summary, a comprehensive application of new sequencing and genomic technologies, as well as recently developed multi-omic and data mining technologies, have become a feasible path for enhancing the precision, efficiency and effectiveness of rice breeding programs to develop climate-resilient, high-yielding and nutritious varieties. Genomic-assisted breeding is coming of age for rice breeding with these advancements in technologies.
Author Contributions
H.G.: writing the original draft and critically revising the manuscript; S.L. and J.Z.: writing, reviewing, revising and finally approving the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Key Areas Research Projects of Guangdong Province (2020B0202090003), Guangdong Provincial International Cooperation Project of Science & Technology (2021A0505030048), the “YouGu” Plan of Rice Research Institute of Guangdong Academy of Agricultural Sciences (2021YG001), the Evaluation and Operation Funds of Guangdong Key Laboratories (2020B1212060047), the Innovation Team Project of Guangdong Modern Agricultural Industrial System (2021KJ106).
Conflicts of Interest
The authors declare no conflict of interest.
References
- OECD; FAO. OECD-FAO Agricultural Outlook 2020–2029; OECD: Paris, France, 2020. [Google Scholar]
- Lee, J.-S.; Chebotarov, D.; Platten, J.D.; McNally, K.; Kohli, A. Advanced Strategic Research to Promote the Use of Rice Genetic Resources. Agronomy 2020, 10, 1629. [Google Scholar] [CrossRef]
- Meynard, D.; Vernet, A.; Meunier, A.C.; Mieulet, D.; Bes, M.; Portefaix, M.; Breitler, J.-C.; Périn, C.; Guiderdoni, E. Thirty years of genome engineering in rice: From gene addition to gene editing. Annu. Plant Rev. 2020, 3, 1–76. [Google Scholar]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Bohra, A.; Yu, J.; Graner, A.; Zhang, Q.; Sorrells, M.E. Designing future crops: Genomics-assisted breeding comes of age. Trends Plant Sci. 2021, 26, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Burr, B. International Rice Genome Sequencing Project: The effort to completely sequence the rice genome. Curr. Opin. Plant Biol. 2000, 3, 138–142. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.-H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, J.; Lin, W.; Li, S.; Li, H.; Zhou, J.; Ni, P.; Dong, W.; Hu, S.; Zeng, C. The genomes of Oryza sativa: A history of duplications. PLoS Biol. 2005, 3, e38. [Google Scholar] [CrossRef] [Green Version]
- International Rice Genome Sequencing Project; Sasaki, T. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Sang, J.; Zou, D.; Wang, Z.; Wang, F.; Zhang, Y.; Xia, L.; Li, Z.; Ma, L.; Li, M.; Xu, B.; et al. IC4R-2.0: Rice genome reannotation using massive RNA-seq data. Genom. Proteom. Bioinf. 2020, 18, 161–172. [Google Scholar] [CrossRef]
- Song, S.; Tian, D.; Zhang, Z.; Hu, S.; Yu, J. Rice genomics: Over the past two decades and into the future. Genom. Proteom. Bioinf. 2018, 16, 397–404. [Google Scholar] [CrossRef]
- Jackson, S.A. Rice: The first crop genome. Rice 2016, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, L.-L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.-M.; Xie, W. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Chebotarov, D.; Kudrna, D.; Llaca, V.; Lee, S.; Rajasekar, S.; Mohammed, N.; Al-Bader, N.; Sobel-Sorenson, C.; Parakkal, P. A platinum standard pan-genome resource that represents the population structure of Asian rice. Sci. Data 2020, 7, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.-Y.; Zhao, S.-C.; He, W.-M.; Guo, L.-B.; Peng, Y.-L.; Wang, J.-J.; Guo, X.-S.; Zhang, X.-M.; Rao, Y.-C.; Zhang, C. Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc. Natl Acad. Sci. USA 2013, 110, 14492–14497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelton, J.M.; Coleman, M.C.; Herndon, N.; Lu, N.; Lam, E.T.; Anantharaman, T.; Sheth, P.; Brown, S.J. Tools and pipelines for BioNano data: Molecule assembly pipeline and FASTA super scaffolding tool. BMC Genom. 2015, 16, 734. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef]
- Jia, L.; Xie, L.; Lao, S.; Zhu, Q.-H.; Fan, L. Rice bioinformatics in the genomic era: Status and perspectives. Crop J. 2021, 9, 609–621. [Google Scholar] [CrossRef]
- Sakai, H.; Kanamori, H.; Arai-Kichise, Y.; Shibata-Hatta, M.; Ebana, K.; Oono, Y.; Kurita, K.; Fujisawa, H.; Katagiri, S.; Mukai, Y. Construction of pseudomolecule sequences of the aus rice cultivar Kasalath for comparative genomics of Asian cultivated rice. DNA Res. 2014, 21, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Nie, S.-J.; Liu, Y.-Q.; Wang, C.-C.; Gao, S.-W.; Xu, T.-T.; Liu, Q.; Chang, H.-L.; Chen, Y.-B.; Yan, P.-C.; Peng, W. Assembly of an early-matured japonica (Geng) rice genome, Suijing18, based on PacBio and Illumina sequencing. Sci. Data 2017, 4, 170195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, J.C.; Yu, Y.; Copetti, D.; Zwickl, D.J.; Zhang, L.; Zhang, C.; Chougule, K.; Gao, D.; Iwata, A.; Goicoechea, J.L. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018, 50, 285–296. [Google Scholar] [CrossRef]
- Jain, R.; Jenkins, J.; Shu, S.; Chern, M.; Martin, J.A.; Copetti, D.; Duong, P.Q.; Pham, N.T.; Kudrna, D.A.; Talag, J. Genome sequence of the model rice variety KitaakeX. BMC Genom. 2019, 20, 905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, L.; Zhang, X.; Zhang, Q.; Jia, Y.; Wang, G.; Li, S.; Tian, D.; Li, W.-H.; Yang, S. Large-scale identification and functional analysis of NLR genes in blast resistance in the Tetep rice genome sequence. Proc. Natl Acad. Sci. USA 2019, 116, 18479–18487. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Lye, Z.N.; Groen, S.C.; Dai, X.; Rughani, P.; Zaaijer, S.; Harrington, E.D.; Juul, S.; Purugganan, M.D. Nanopore sequencing-based genome assembly and evolutionary genomics of circum-basmati rice. Genome Biol. 2020, 21, 21. [Google Scholar] [CrossRef]
- Tanaka, T.; Nishijima, R.; Teramoto, S.; Kitomi, Y.; Hayashi, T.; Uga, Y.; Kawakatsu, T. De novo genome assembly of the indica rice variety ir64 using linked-read sequencing and nanopore sequencing. G3-Genes Genom. Genet. 2020, 10, 1495–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panibe, J.P.; Wang, L.; Li, J.; Li, M.-Y.; Lee, Y.-C.; Wang, C.-S.; Ku, M.S.; Lu, M.-Y.J.; Li, W.-H. Chromosomal-level genome assembly of the semi-dwarf rice Taichung Native 1, an initiator of Green Revolution. Genomics 2021, 113, 2656–2674. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Deng, C.; Zhao, S.; Zhang, P.; Feng, J.; Huang, W.; Kang, S.; Qian, Q.; Xiong, G. High-quality genome assembly of Huazhan and Tianfeng, the parents of an elite rice hybrid Tian-you-hua-zhan. Sci. China Life Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e16. [Google Scholar] [CrossRef] [PubMed]
- Golicz, A.A.; Bayer, P.E.; Bhalla, P.L.; Batley, J.; Edwards, D. Pangenomics comes of age: From bacteria to plant and animal applications. Trends Genet. 2020, 36, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Sherman, R.M.; Forman, J.; Antonescu, V.; Puiu, D.; Daya, M.; Rafaels, N.; Boorgula, M.P.; Chavan, S.; Vergara, C.; Ortega, V.E. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 2019, 51, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, R.; Fu, W.; Li, Y.; Wang, X.; Li, M.; Du, D.; Tang, Q.; Cai, Y.; Long, Y. Building a sequence map of the pig pan-genome from multiple de novo assemblies and Hi-C data. Sci. China Life Sci. 2019, 63, 750–763. [Google Scholar] [CrossRef]
- Zhao, J.; Bayer, P.E.; Ruperao, P.; Saxena, R.K.; Khan, A.W.; Golicz, A.A.; Nguyen, H.T.; Batley, J.; Edwards, D.; Varshney, R.K. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 2020, 18, 1946–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, G.; Zhao, H.; Wang, G.; Lian, X.; Xie, W. Exploring the rice dispensable genome using a metagenome-like assembly strategy. Genome Biol. 2015, 16, 187. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Hu, Z.; Zheng, T.; Lu, K.; Zhao, Y.; Wang, W.; Shi, J.; Wang, C.; Lu, J.; Zhang, D. RPAN: Rice pan-genome browser for ∼3000 rice genomes. Nucleic Acids Res. 2017, 45, 597–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F. Genomic variation in 3010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and exploiting pan-genomics for crop improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Jordan, D.R.; Mace, E.S. A graph-based pan-genome guides biological discovery. Mol. Plant 2020, 13, 1247–1249. [Google Scholar] [CrossRef]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Danilevicz, M.F.; Fernandez, C.G.T.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, J.; Chen, L.; Huang, X.; Cheng, Z.; Han, B.; Zhang, Q.; Wu, C. Rice functional genomics research: Past decade and future. Mol. Plant 2018, 11, 359–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Gao, Z.; Qian, Q. Application of resequencing to rice genomics, functional genomics and evolutionary analysis. Rice 2014, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, D.; Bohra, A.; Thudi, M.; Varshney, R.K. Fine mapping and gene cloning in the post-NGS era: Advances and prospects. Theor. Appl. Genet. 2020, 133, 1791–1810. [Google Scholar] [CrossRef] [Green Version]
- Park, S.T.; Kim, J. Trends in next-generation sequencing and a new era for whole genome sequencing. Int. Neurourol. J. 2016, 20, S76. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tang, J.; Han, B.; Huang, X. Advances in genome-wide association studies of complex traits in rice. Theor. Appl. Genet. 2020, 133, 1415–1425. [Google Scholar] [CrossRef]
- Feng, L.; Ma, A.; Song, B.; Yu, S.; Qi, X. Mapping causal genes and genetic interactions for agronomic traits using a large F2 population in rice. G3-Genes Genom. Genet. 2021, 11, jkab318. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.-L.; Chen, L.-Y.; Xiao, G.-Y.; Xiao, Y.-H.; Chen, X.-B.; Zhang, S.-T. Bulked segregant analysis to detect QTL related to heat tolerance in rice (Oryza sativa L.) using SSR markers. Agric. Sci. China 2009, 8, 482–487. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Wambugu, P.; Ndjiondjop, M.N.; Furtado, A.; Henry, R. Sequencing of bulks of segregants allows dissection of genetic control of amylose content in rice. Plant Biotechnol. J. 2018, 16, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Tang, S.; Zhan, Q.; Hou, Q.; Zhao, Y.; Zhao, Q.; Feng, Q.; Zhou, C.; Lyu, D.; Cui, L. Dissecting a heterotic gene through GradedPool-Seq mapping informs a rice-improvement strategy. Nat. Commun. 2019, 10, 2982. [Google Scholar] [CrossRef] [PubMed]
- Nubankoh, P.; Wanchana, S.; Saensuk, C.; Ruanjaichon, V.; Cheabu, S.; Vanavichit, A.; Toojinda, T.; Malumpong, C.; Arikit, S. QTL-seq reveals genomic regions associated with spikelet fertility in response to a high temperature in rice (Oryza sativa L.). Plant Cell Rep. 2020, 39, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, X.; Pan, Q.; Li, P.; Liu, Y.; Lu, X.; Zhong, W.; Li, M.; Han, L.; Li, J. QTG-Seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing of bulked segregant samples. Mol. Plant 2019, 12, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Arikit, S.; Wanchana, S.; Khanthong, S.; Saensuk, C.; Thianthavon, T.; Vanavichit, A.; Toojinda, T. QTL-seq identifies cooked grain elongation QTLs near soluble starch synthase and starch branching enzymes in rice (Oryza sativa L.). Sci. Rep. 2019, 9, 8328. [Google Scholar] [CrossRef] [Green Version]
- Consortium, W.T.C.C. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661. [Google Scholar]
- Wang, H.; Xu, X.; Vieira, F.G.; Xiao, Y.; Li, Z.; Wang, J.; Nielsen, R.; Chu, C. The power of inbreeding: NGS-based GWAS of rice reveals convergent evolution during rice domestication. Mol. Plant 2016, 9, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; Li, M. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961. [Google Scholar] [CrossRef]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef]
- Sun, S.; Wang, T.; Wang, L.; Li, X.; Jia, Y.; Liu, C.; Huang, X.; Xie, W.; Wang, X. Natural selection of a GSK3 determines rice mesocotyl domestication by coordinating strigolactone and brassinosteroid signaling. Nat. Commun. 2018, 9, 2523. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, Z.; Chern, M.; Yin, J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 2017, 170, 114–126.e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCouch, S.R.; Wright, M.H.; Tung, C.-W.; Maron, L.G.; McNally, K.L.; Fitzgerald, M.; Singh, N.; DeClerck, G.; Agosto-Perez, F.; Korniliev, P. Open access resources for genome-wide association mapping in rice. Nat. Commun. 2016, 7, 10532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Zhao, J.; Zhang, S.; Chen, L.; Yang, T.; Dong, J.; Fu, H.; Ma, Y.; Zhou, L.; Wang, J. Genome-Wide Association Mapping and Gene Expression Analysis Reveal the Negative Role of OsMYB21 in Regulating Bacterial Blight Resistance in Rice. Rice 2021, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, W.; Zhang, S.; Yang, T.; Liu, Q.; Dong, J.; Fu, H.; Mao, X.; Liu, B.J.R. Genome-wide association study and candidate gene analysis of rice cadmium accumulation in grain in a diverse rice collection. Rice 2018, 11, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Huang, X. Genome-wide association studies in rice: How to solve the low power problems? Mol. Plant 2019, 12, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.R.; Agosto-Pérez, F.J.; Chebotarov, D.; Shi, Y.; Marchini, J.; Fitzgerald, M.; McNally, K.L.; Alexandrov, N.; McCouch, S.R. An imputation platform to enhance integration of rice genetic resources. Nat. Commun. 2018, 9, 3519. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Dong, B.; Zou, Q. Revisiting genome-wide association studies from statistical modelling to machine learning. Brief. Bioinform. 2021, 22, bbaa263. [Google Scholar] [CrossRef]
- Kavvas, E.S.; Yang, L.; Monk, J.M.; Heckmann, D.; Palsson, B.O. A biochemically-interpretable machine learning classifier for microbial GWAS. Nat. Commun. 2020, 11, 2580. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Zeng, L.; Schunkert, H.; Söding, J. Bayesian multiple logistic regression for case-control GWAS. PLoS Genet. 2018, 14, e1007856. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.W.; Uauy, C.; Wulff, B.B.; Zhou, J.; Krasileva, K.; Clark, M.D.J.N. Genomic innovation for crop improvement. Nature 2017, 543, 346–354. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genomics-assisted breeding for crop improvement. Trends Plant Sci. 2005, 10, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Zhao, X.; Ponce, K.; Ye, G.; Leung, H. QTL mapping for agronomic traits using multi-parent advanced generation inter-cross (MAGIC) populations derived from diverse elite indica rice lines. Field Crops Res. 2016, 189, 19–42. [Google Scholar] [CrossRef] [Green Version]
- Abbai, R.; Singh, V.K.; Nachimuthu, V.V.; Sinha, P.; Selvaraj, R.; Vipparla, A.K.; Singh, A.K.; Singh, U.M.; Varshney, R.K.; Kumar, A. Haplotype analysis of key genes governing grain yield and quality traits across 3K RG panel reveals scope for the development of tailor-made rice with enhanced genetic gains. Plant Biotechnol. J. 2019, 17, 1612–1622. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Ali, J.; Zhang, C.; Li, Z.; Zhang, Q. Genomic breeding of green super rice varieties and their deployment in Asia and Africa. Theor. Appl. Genet. 2020, 133, 1427–1442. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, R. Prediction of maize single-cross performance using RFLPs and information from related hybrids. Crop Sci. 1994, 34, 20–25. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetic 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- McGowan, M.; Wang, J.; Dong, H.; Liu, X.; Jia, Y.; Wang, X.; Iwata, H.; Li, Y.; Lipka, A.E.; Zhang, Z. Ideas in Genomic Selection with the Potential to Transform Plant Molecular Breeding: A Review. Plant Breed. Rev. 2020, 45, 273–319. [Google Scholar]
- He, T.; Li, C. Harness the power of genomic selection and the potential of germplasm in crop breeding for global food security in the era with rapid climate change. Crop J. 2020, 8, 688–700. [Google Scholar] [CrossRef]
- Varshney, R.K.; Sinha, P.; Singh, V.K.; Kumar, A.; Zhang, Q.; Bennetzen, J.L. 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 2020, 56, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Gosal, S.S.; Wani, S.H. Accelerated Plant Breeding, Volume 1: Cereal Crops; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redona, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar]
- Cui, Y.; Li, R.; Li, G.; Zhang, F.; Zhu, T.; Zhang, Q.; Ali, J.; Li, Z.; Xu, S. Hybrid breeding of rice via genomic selection. Plant Biotechnol. J. 2020, 18, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Spindel, J.; Begum, H.; Akdemir, D.; Collard, B.; Redoña, E.; Jannink, J.; McCouch, S. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 2016, 116, 395–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing genetic gain through genomic selection: From livestock to plants. Plant Commun. 2020, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Joshi, R.K.; Zhao, K. Genome editing in rice: Recent advances, challenges, and future implications. Front. Plant Sci. 2018, 9, 1361. [Google Scholar] [CrossRef]
- Scheben, A.; Wolter, F.; Batley, J.; Puchta, H.; Edwards, D. Towards CRISPR/Cas crops–bringing together genomics and genome editing. New Phytol. 2017, 216, 682–698. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.; Zhang, J.; Botella, J.R.; Ma, C.; Kong, F.; Liu, B.; Zhu, J.-K. Perspectives on the application of genome-editing technologies in crop breeding. Mol. Plant 2019, 12, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Li, W.; Tan, L.; Tian, F. Harnessing knowledge from maize and rice domestication for new crop breeding. Mol. Plant 2020, 14, 9–26. [Google Scholar] [CrossRef]
- Huang, X.; Kurata, N.; Wang, Z.-X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; Guo, Y. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Huang, X.; Tian, Z.; Wing, R.A.; Han, B. The genomics of Oryza species provides insights into rice domestication and heterosis. Annu. Rev. Plant Biol. 2019, 70, 639–665. [Google Scholar] [CrossRef]
- Yu, H.; Lin, T.; Meng, X.; Du, H.; Zhang, J.; Liu, G.; Chen, M.; Jing, Y.; Kou, L.; Li, X. A route to de novo domestication of wild allotetraploid rice. Cell 2021, 184, 1156–1170.e1114. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.I.; Hu, H.; Gill, M.; Batley, J.; Edwards, D. Crop breeding for a changing climate: Integrating phenomics and genomics with bioinformatics. Theor. Appl. Genet. 2021, 134, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wang, K.; Chen, Z.; Cao, Y.; Gao, Q.; Li, Y.; Li, X.; Lu, H.; Du, H.; Lu, M. MBKbase for rice: An integrated omics knowledgebase for molecular breeding in rice. Nucleic Acids Res. 2020, 48, D1085–D1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzaq, A.; Kaur, P.; Akhter, N.; Wani, S.H.; Saleem, F. Next-generation breeding strategies for climate-ready crops. Front. Plant Sci. 2021, 12, 620420. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Nikoloski, Z. Machine learning approaches for crop improvement: Leveraging phenotypic and genotypic big data. J. Plant Physiol. 2021, 257, 153354. [Google Scholar] [CrossRef] [PubMed]
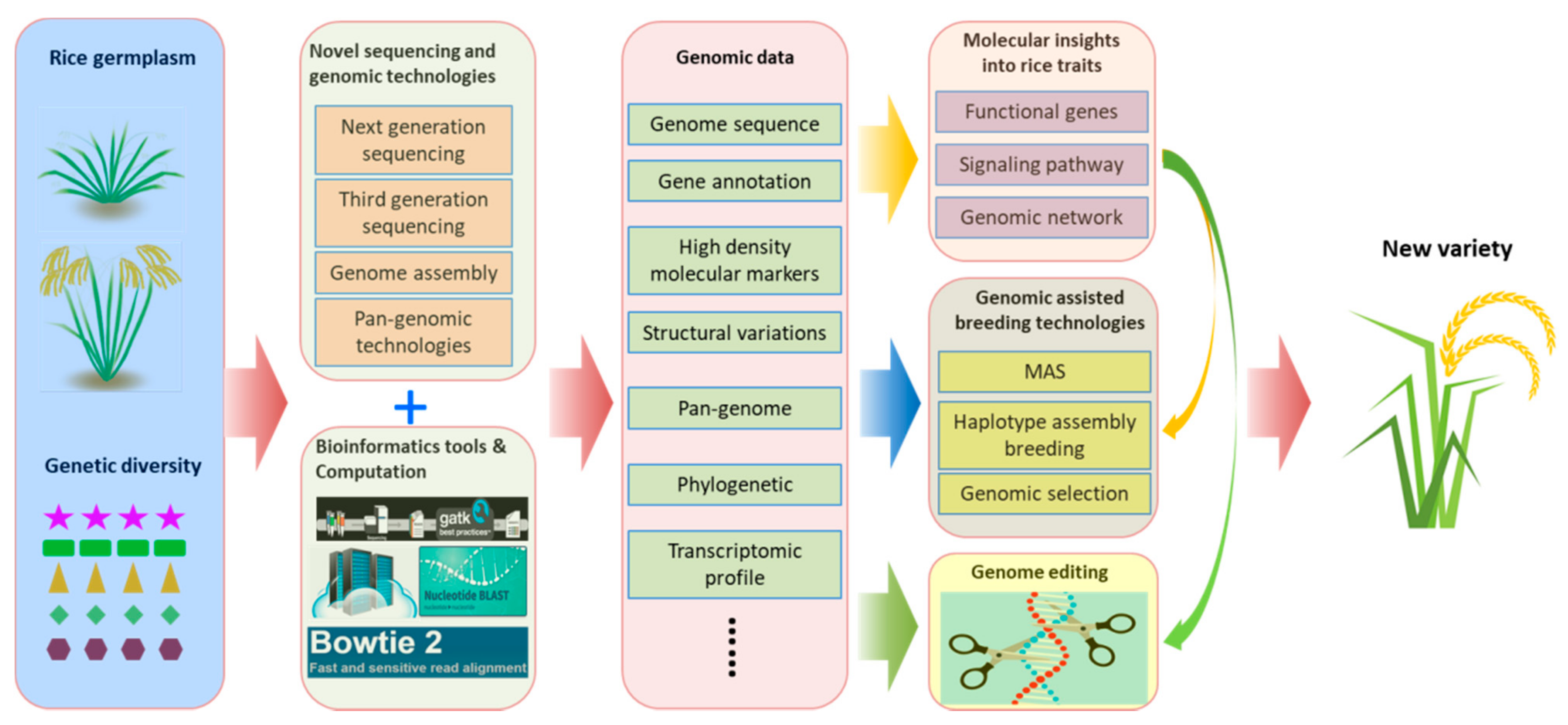
Figure 1.
Schematic diagram of novel sequencing and genomic technologies implemented into and accelerating rice research and breeding practices. Rice germplasms or diverse genetic resources are first characterized by novel sequencing and genomic technologies combined with bioinformatics tools. The genomic data acquired from sequencing and genomic technologies, including genome sequences, gene annotation, etc., will be further processed to gain biological insights about rice traits. These insights and knowledge will be utilized to accelerate and enable genomic-assisted breeding progress, as well as rice improvement by genomic editing to bring out new varieties.
Figure 1.
Schematic diagram of novel sequencing and genomic technologies implemented into and accelerating rice research and breeding practices. Rice germplasms or diverse genetic resources are first characterized by novel sequencing and genomic technologies combined with bioinformatics tools. The genomic data acquired from sequencing and genomic technologies, including genome sequences, gene annotation, etc., will be further processed to gain biological insights about rice traits. These insights and knowledge will be utilized to accelerate and enable genomic-assisted breeding progress, as well as rice improvement by genomic editing to bring out new varieties.


{kind=link}
Table 1.
High quality reference genomes of Asian rice.
| Species | Accession Name | Year of Publication | Reference |
|---|---|---|---|
| Japonica | Nipponbare | 2005 | [10] |
| Indica | 93-11 | 2005 | [9] |
| Aus | Kasalath | 2014 | [21] |
| Indica | MH63 | 2016 | [15] |
| Indica | ZS97 | 2016 | [15] |
| japonica | Suijing18 | 2017 | [22] |
| indica | R498 | 2017 | [19] |
| Aus | N22 | 2018 | [23] |
| Indica | IR8 | 2018 | [23] |
| Japonica | KitaakeX | 2019 | [24] |
| Indica | Tetep | 2019 | [25] |
| Aromatic | Basmatic 334 | 2020 | [26] |
| Aromatic | Dom Sufid | 2020 | [26] |
| All subpopulations | 12 accessions | 2020 | [16] |
| Indica | IR64 | 2020 | [27] |
| Indica | Taichung Native 1 | 2021 | [28] |
| Indica | Huazhan | 2021 | [29] |
| Indica | Tianfeng | 2021 | [29] |
| All subpopulations | 31 accessions | 2021 | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gu, H.; Liang, S.; Zhao, J. Novel Sequencing and Genomic Technologies Revolutionized Rice Genomic Study and Breeding. Agronomy 2022, 12, 218. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy12010218
AMA Style
Gu H, Liang S, Zhao J. Novel Sequencing and Genomic Technologies Revolutionized Rice Genomic Study and Breeding. Agronomy. 2022; 12(1):218. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy12010218
Chicago/Turabian StyleGu, Haiyong, Shihu Liang, and Junliang Zhao. 2022. "Novel Sequencing and Genomic Technologies Revolutionized Rice Genomic Study and Breeding" Agronomy 12, no. 1: 218. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy12010218
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.