Genome-Wide Association Study Identifies Candidate Genes Associated with Feet and Leg Conformation Traits in Chinese Holstein Cattle

 ,
, 
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Body Measurements and DNA Samples Collection
2.3. Phenotypic and Genetic Parameters
2.4. Genotyping and Quality Control
2.5. Population Structure Analysis
2.6. Genome-Wide Association Studies
2.7. Gene Identification
2.8. Function and Pathway Enrichment and Network Analysis
3. Results
3.1. Descriptive Statistics and Heritability Estimation of Feet and Legs Traits
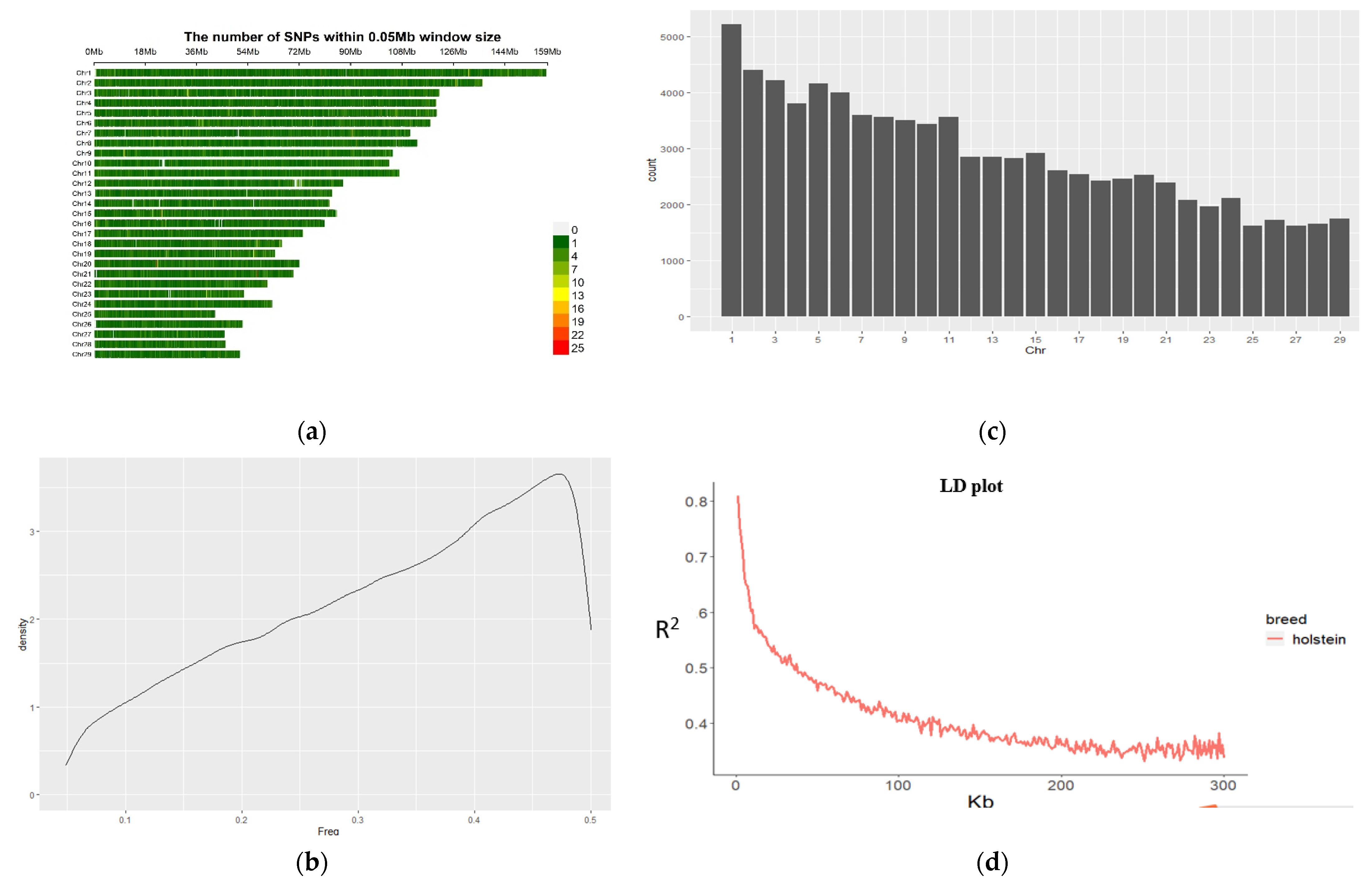
3.2. Information of SNPs
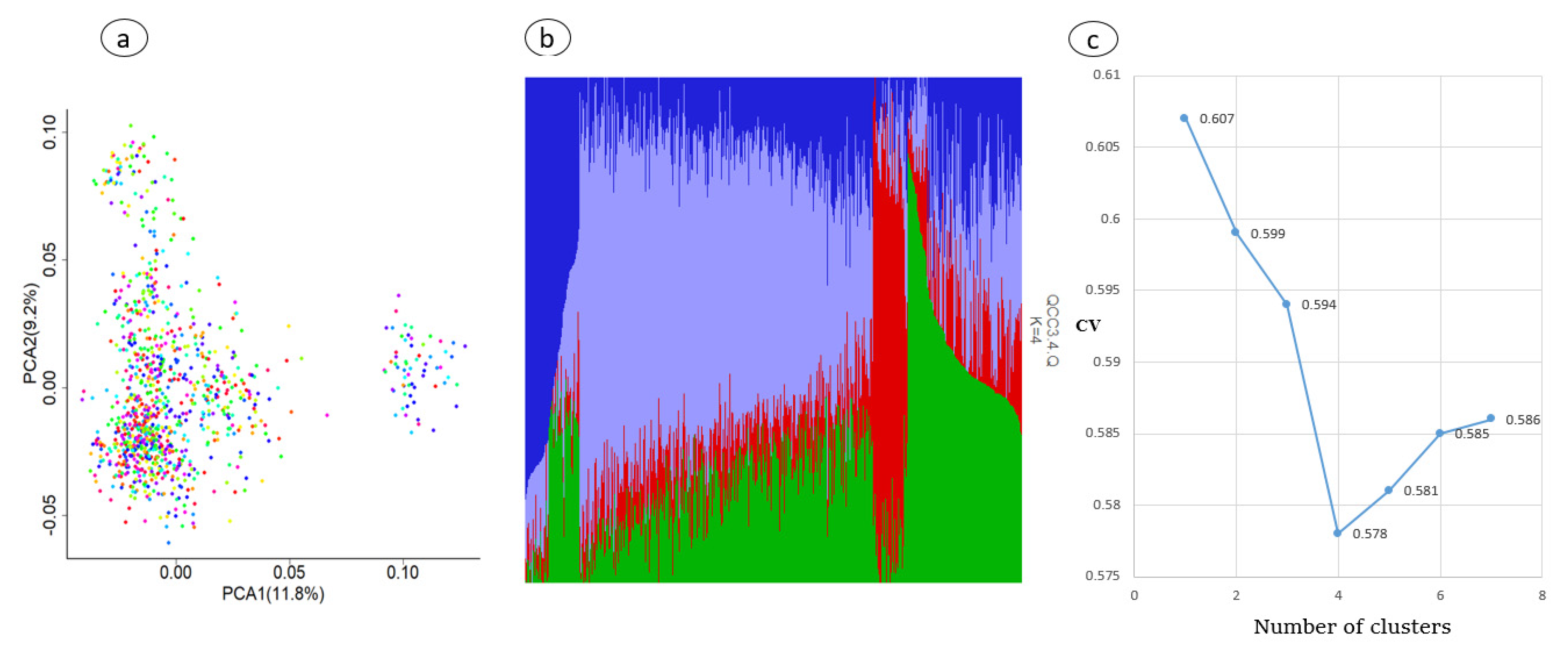
3.3. Population Stratification
3.4. GWAS Results
3.5. Identified Candidate Genes
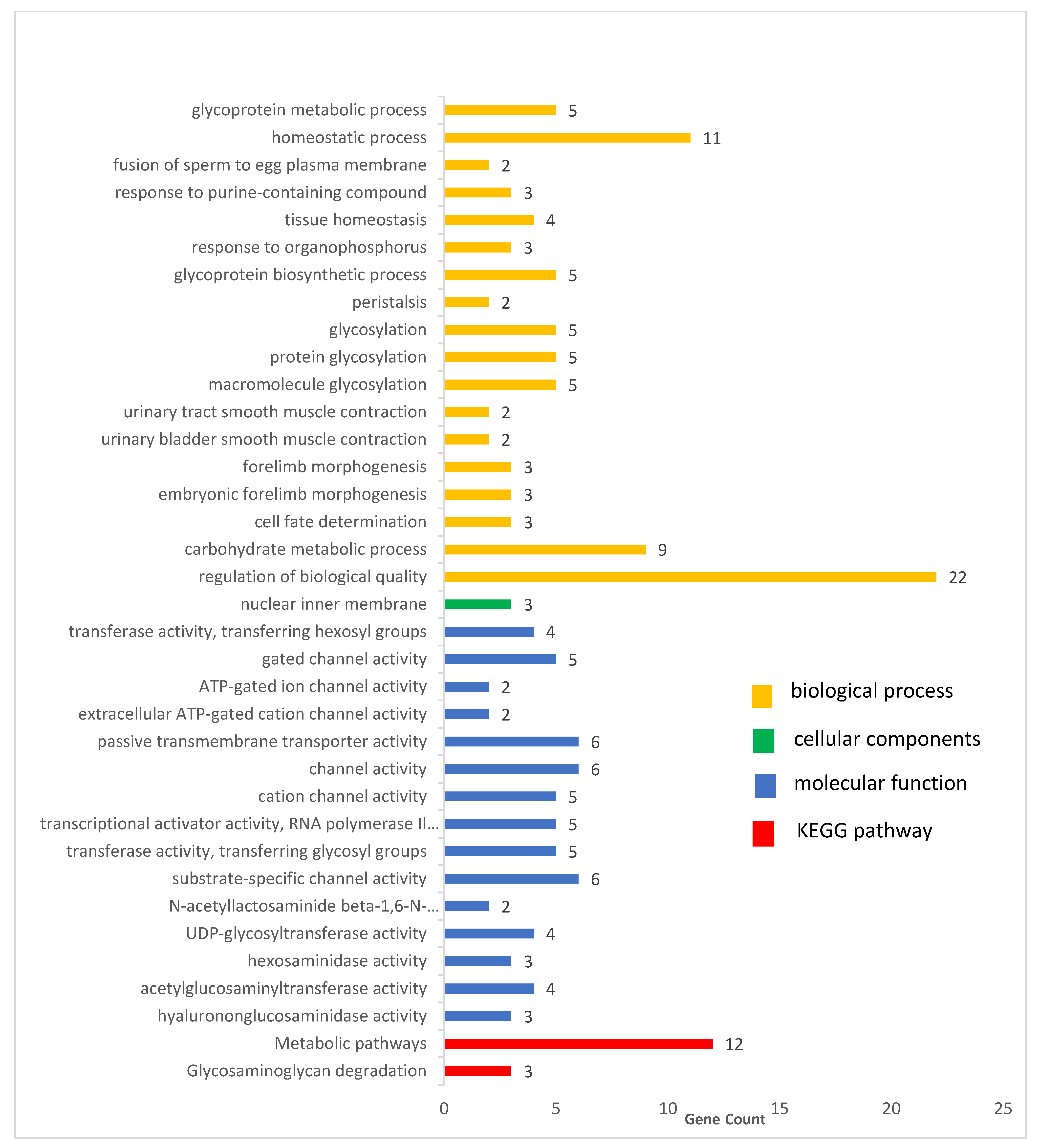
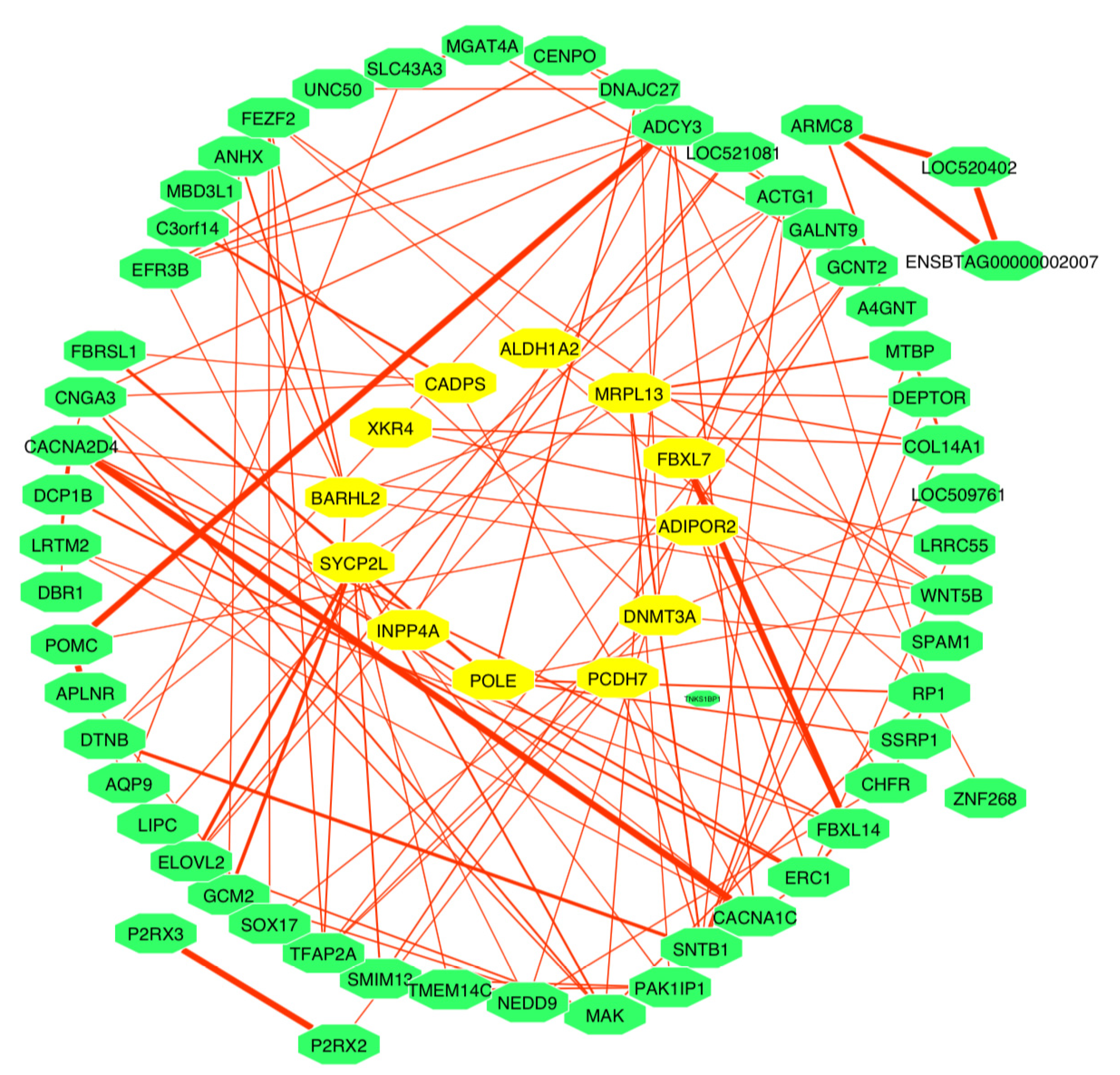
3.6. Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miglior, F.; Fleming, A.; Malchiodi, F.; Brito, L.F.; Martin, P.; Baes, C.F. A 100-Year Review: Identification and genetic selection of economically important traits in dairy cattle. J. Dairy Sci. 2017, 100, 10251–10271. [Google Scholar] [CrossRef]
- Pérez-Cabal, M.A.; García, C.; González-Recio, O.; Alenda, R. Genetic and phenotypic relationships among locomotion type traits, profit, production, longevity, and fertility in Spanish dairy cows. J. Dairy Sci. 2006, 89, 1776–1783. [Google Scholar] [CrossRef]
- Wu, J.J.; Wathes, D.C.; Brickell, J.S.; Yang, L.G.; Cheng, Z.; Zhao, H.Q.; Xu, Y.J.; Zhang, S.J. Reproductive performance and survival of Chinese Holstein dairy cows in central China. Anim. Prod. Sci. 2012, 52, 11–19. [Google Scholar] [CrossRef]
- Liu, S.; Yi, J.; Yang, L. Genetic parameters estimates for locomotion score, body condition score and final type score of Holstein cattle in southern China. J. Appl. Anim. Res. 2013, 41, 240–243. [Google Scholar] [CrossRef] [Green Version]
- Heringstad, B.; Egger-Danner, C.; Charfeddine, N.; Pryce, J.E.; Stock, K.F.; Kofler, J.; Sogstad, A.M.; Holzhauer, M.; Fiedler, A.; Müller, K.; et al. Invited review: Genetics and claw health: Opportunities to enhance claw health by genetic selection. J. Dairy Sci. 2018, 101, 4801–4821. [Google Scholar] [CrossRef] [Green Version]
- Chapinal, N.; Koeck, A.; Sewalem, A.; Kelton, D.F.; Mason, S.; Cramer, G.; Miglior, F. Genetic parameters for hoof lesions and their relationship with feet and leg traits in Canadian Holstein cows. J. Dairy Sci. 2013, 96, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Z.; Wang, S.; Li, H. Retraction: Progress of genome wide association study in domestic animals. J. Anim. Sci. Biotechnol. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Ning, C.; Liu, J.F.; Zhang, Q.; Jiang, L. Short communication: Replication of genome-wide association studies for milk production traits in Chinese Holstein by an efficient rotated linear mixed model. J. Dairy Sci. 2019, 102, 2378–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Liu, J.; Sun, D.; Ma, P.; Ding, X.; Yu, Y.; Zhang, Q. Genome wide association studies for milk production traits in Chinese Holstein population. PLoS ONE 2010, 5, e13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Li, J.; Gao, X.; Song, W.; Chen, C.; Yao, D.; Ma, J.; Xu, L.; Ma, Y. Genome-wide association study of milk components in Chinese Holstein cows using single nucleotide polymorphism. Livest. Sci. 2020, 233. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, J.; Chen, C.J.; Zhang, J.; Wen, W.; Tian, J.; Zhang, Z.; Gu, Y. GWAS-based identification of new loci for milk yield, fat, and protein in holstein cattle. Animals 2020, 10, 2048. [Google Scholar] [CrossRef]
- Li, C.; Sun, D.; Zhang, S.; Wang, S.; Wu, X.; Zhang, Q.; Liu, L.; Li, Y.; Qiao, L. Genome wide association study identifies 20 novel promising genes associated with milk fatty acid traits in Chinese Holstein. PLoS ONE 2014, 9, 103951. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Buitenhuis, A.J.; Lund, M.S.; Li, C.; Sun, D.; Zhang, Q.; Poulsen, N.A.; Su, G. inese and Danish Holstein populJoint genome-wide association study for milk fatty acid traits in Chations. J. Dairy Sci. 2015, 98, 8152–8163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Li, C.; Cai, W.; Liu, S.; Yin, H.; Shi, S.; Zhang, Q.; Zhang, S. Genome-wide association study for milk protein composition traits in a chiniese holstein population using a single-step approach. Front. Genet. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Wang, Z.; Zhang, Q.; Yue, S.; Yin, B.; Jiang, Y.; Shi, K. Identification of whole-genome significant single nucleotide polymorphisms in candidate genes associated with body conformation traits in Chinese Holstein cattle. Anim. Genet. 2020, 51, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Fang, M.; Liu, L.; Wang, S.; Liu, J.; Ding, X.; Zhang, S.; Zhang, Q.; Zhang, Y.; Qiao, L.; et al. Genome wide association studies for body conformation traits in the Chinese Holstein cattle population. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chu, Q.; Guo, G.; Dong, G.; Li, X.; Zhang, Q.; Zhang, S.; Zhang, Z.; Wang, Y. Genome-wide association studies identified multiple genetic loci for body size at four growth stages in Chinese Holstein cattle. PLoS ONE 2017, 12, e0175971. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, P.; Fu, W.; Dong, T.; Qi, C.; Liu, L.; Guo, G.; Li, C.; Cui, X.; Zhang, S.; et al. Genome-wide association study for pigmentation traits in Chinese Holstein population. Anim. Genet. 2014, 45, 740–744. [Google Scholar] [CrossRef]
- Liu, A.; Wang, Y.; Sahana, G.; Zhang, Q.; Liu, L.; Lund, M.S.; Su, G. Genome-wide Association Studies for Female Fertility Traits in Chinese and Nordic Holsteins. Sci. Rep. 2017, 7, 8487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Wan, Z.; Zhang, J.; Xu, L.; Han, B.; Sun, D. Genome-wide association studies for the concentration of albumin in colostrum and serum in chinese holstein. Animals 2020, 10, 2211. [Google Scholar] [CrossRef]
- Cai, Z.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Prioritizing candidate genes post-GWAS using multiple sources of data for mastitis resistance in dairy cattle. BMC Genom. 2018, 19, 656. [Google Scholar] [CrossRef]
- Wang, X.; Ma, P.; Liu, J.; Zhang, Q.; Zhang, Y.; Ding, X.; Jiang, L.; Wang, Y.; Zhang, Y.; Sun, D.; et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015, 16, 1–9. [Google Scholar] [CrossRef]
- Cole, J.B.; Wiggans, G.R.; Ma, L.; Sonstegard, T.S.; Lawlor, T.J.; Crooker, B.A.; Van Tassell, C.P.; Yang, J.; Wang, S.; Matukumalli, L.K.; et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genom. 2011, 12, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abo-Ismail, M.K.; Brito, L.F.; Miller, S.P.; Sargolzaei, M.; Grossi, D.A.; Moore, S.S.; Plastow, G.; Stothard, P.; Nayeri, S.; Schenkel, F.S. Genome-wide association studies and genomic prediction of breeding values for calving performance and body conformation traits in Holstein cattle. Genet. Sel. Evol. 2017, 49, 82. [Google Scholar] [CrossRef] [Green Version]
- Vargas, G.; Neves, H.H.R.; Camargo, G.M.F.; Cardoso, V.; Munari, D.P.; Carvalheiro, R. Genome-wide association study and functional analysis of feet and leg conformation traits in Nellore cattle. J. Anim. Sci. 2018, 96, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.; Milkevych, V.; Gao, H.; Christensen, O.F.; Jensen, J. DMU-a package for analyzing multivariate mixed models in quantitative genetics and genomics. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014; pp. 18–22. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, 1005957. [Google Scholar] [CrossRef]
- Wang, Q.; Tian, F.; Pan, Y.; Buckler, E.S.; Zhang, Z. A SUPER powerful method for genome wide association study. PLoS ONE 2014, 9, e107684. [Google Scholar] [CrossRef]
- Duggal, P.; Gillanders, E.M.; Holmes, T.N.; Bailey-Wilson, J.E. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genom. 2008, 9, 516. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, K.; Lee, S.H.; Chung, K.Y.; Park, J.E.; Jang, G.W.; Park, M.R.; Kim, N.Y.; Kim, T.H.; Chai, H.H.; Park, W.C.; et al. A gene-set enrichment and protein–protein interaction network-based gwas with regulatory snps identifies candidate genes and pathways associated with carcass traits in hanwoo cattle. Genes 2020, 11, 316. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, 183. [Google Scholar] [CrossRef] [Green Version]
- Carbon, S.; Dietze, H.; Lewis, S.E.; Mungall, C.J.; Munoz-Torres, M.C.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Fey, P.; Thomas, P.D.; et al. Expansion of the gene ontology knowledgebase and resources: The gene ontology consortium. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Olasege, B.S.; Zhang, S.; Zhao, Q.; Liu, D.; Sun, H.; Wang, Q.; Ma, P.; Pan, Y. Genetic parameter estimates for body conformation traits using composite index, principal component, and factor analysis. J. Dairy Sci. 2019, 102, 5219–5229. [Google Scholar] [CrossRef] [PubMed]
- Bolormaa, S.; Pryce, J.E.; Reverter, A.; Zhang, Y.; Barendse, W.; Kemper, K.; Tier, B.; Savin, K.; Hayes, B.J.; Goddard, M.E. A Multi-Trait, Meta-analysis for Detecting Pleiotropic Polymorphisms for Stature, Fatness and Reproduction in Beef Cattle. PLoS Genet. 2014, 10, e1004198. [Google Scholar] [CrossRef] [Green Version]
- Vargas, C.A.; Elzo, M.A.; Chase, C.C.; Olson, T.A. Genetic parameters and relationships between hip height and weight in Brahman cattle. J. Anim. Sci. 2000, 78, 3045–3052. [Google Scholar] [CrossRef]
- Price, A.L.; Zaitlen, N.A.; Reich, D.; Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldar, T.; Ghosh, S. Effect of Population Stratification on False Positive Rates of Population-Based Association Analyses of Quantitative Traits. Ann. Hum. Genet. 2012, 76, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Bittles, A.H. Population stratification and genetic association studies in South Asia. J. Mol. Genet. Med. 2005, 1, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharmaa, A.; Lee, J.S.; Dang, C.G.; Sudrajad, P.; Kim, H.C.; Yeon, S.H.; Kang, H.S.; Lee, S.H. Stories and challenges of genome wide association studies in livestock—A review. Asian-Australas. J. Anim. Sci. 2015, 28, 1371–1379. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef]
- Devlin, B.; Roeder, K. Genomic control for association studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef]
- Wang, D.; Sun, Y.; Stang, P.; Berlin, J.A.; Wilcox, M.A.; Li, Q. Comparison of methods for correcting population stratification in a genome-wide association study of rheumatoid arthritis: Principal-component analysis versus multidimensional scaling. BMC Proc. 2009, 3, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hödebeck, M.; Scherer, C.; Wagner, A.; Hecker, M.; Korff, T. TonEBP/NFAT5 regulates ACTBL2 expression in biomechanically activated vascular smooth muscle cells. Front. Physiol. 2014, 5, 467. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.W.; Edwards, J.R.; Naylor, A.J.; McGettrick, H.M. Adiponectin signalling in bone homeostasis, with age and in disease. Bone Res. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Yao, C.; Pang, D.; Lu, C.; Xu, A.; Huang, P.; Yu, H. Data mining and validation of ampk pathway as a novel candidate role affecting intramuscular fat content in pigs. Animals 2019, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Makina, S.O.; Muchadeyi, F.C.; Van Marle-Köster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 2015, 47, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shengwei, H.; Cunyuan, L.; Ming, L.; Xiaoyue, L.; Wei, N.; Yueren, X.; Rui, Y.; Bin, W.; Mengdan, Z.; Huixiang, L.; et al. Whole-genome resequencing reveals loci associated with thoracic vertebrae number in sheep. Front. Genet. 2019, 10, 674. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, J.; Kofuji, S.; Itoh, R.; Momiyama, T.; Takayama, K.; Murakami, H.; Chida, S.; Tsuya, Y.; Takasuga, S.; Eguchi, S.; et al. The PtdIns(3,4)P 2 phosphatase INPP4A is a suppressor of excitotoxic neuronal death. Nature 2010, 465, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Edea, Z.; Dadi, H.; Kim, S.W.; Park, J.H.; Shin, G.H.; Dessie, T.; Kim, K.S. Linkage disequilibrium and genomic scan to detect selective loci in cattle populations adapted to different ecological conditions in Ethiopia. J. Anim. Breed. Genet. 2014, 131, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Bongiorni, S.; Gruber, C.E.M.; Bueno, S.; Chillemi, G.; Ferrè, F.; Failla, S.; Moioli, B.; Valentini, A. Transcriptomic investigation of meat tenderness in two Italian cattle breeds. Anim. Genet. 2016, 47, 273–287. [Google Scholar] [CrossRef]
- Kamei, Y.; Suganami, T.; Ehara, T.; Kanai, S.; Hayashi, K.; Yamamoto, Y.; Miura, S.; Ezaki, O.; Okano, M.; Ogawa, Y. Increased expression of DNA methyltransferase 3a in obese adipose tissue: Studies with transgenic mice. Obesity 2010, 18, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Usman, T.; Wang, Y.; Wang, Z.; Xu, X.; Wu, M.; Zhang, Y.; Zhang, X.; Li, Q.; Liu, L.; et al. Polymorphisms in epigenetic and meat quality related genes in fourteen cattle breeds and association with beef quality and carcass traits. Asian-Australas. J. Anim. Sci. 2015, 28, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Londoño-Gil, M.; Rincón Flórez, J.C.; Lopez-Herrera, A.; Gonzalez-Herrera, L.G. Genome-Wide Association Study for Growth Traits in Blanco Orejinero (Bon) Cattle from Colombia. Livest. Sci. 2021, 243, 4366. [Google Scholar] [CrossRef]
- Laghouaouta, H.; Sosa-Madrid, B.S.; Zubiri-Gaitán, A.; Hernández, P.; Blasco, A. Novel genomic regions associated with intramuscular fatty acid composition in rabbits. Animals 2020, 10, 2090. [Google Scholar] [CrossRef]
- Do, D.N.; Bissonnette, N.; Lacasse, P.; Miglior, F.; Sargolzaei, M.; Zhao, X.; Ibeagha-Awemu, E.M. Genome-wide association analysis and pathways enrichment for lactation persistency in Canadian Holstein cattle. J. Dairy Sci. 2017, 100, 1955–1970. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.; Xia, J.; Xu, L.; Wang, X.; Zhu, B.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; Gao, H. A genome-wide association study suggests several novel candidate genes for carcass traits in Chinese Simmental beef cattle. Anim. Genet. 2018, 49, 312–316. [Google Scholar] [CrossRef]
- Wu, P.; Wang, K.; Yang, Q.; Zhou, J.; Chen, D.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A.; et al. Identifying SNPs and candidate genes for three litter traits using single-step GWAS across six parities in landrace and large white pigs. Physiol. Genom. 2018, 50, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- An, B.; Xu, L.; Xia, J.; Wang, X.; Miao, J.; Chang, T.; Song, M.; Ni, J.; Xu, L.; Zhang, L.; et al. Multiple association analysis of loci and candidate genes that regulate body size at three growth stages in Simmental beef cattle. BMC Genet. 2020, 21, 32. [Google Scholar] [CrossRef] [Green Version]
- Tizioto, P.C.; Coutinho, L.L.; Decker, J.E.; Schnabel, R.D.; Rosa, K.O.; Oliveira, P.S.N.; Souza, M.M.; Mourão, G.B.; Tullio, R.R.; Chaves, A.S.; et al. Global liver gene expression differences in Nelore steers with divergent residual feed intake phenotypes. BMC Genom. 2015, 16, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahana, G.; Kadlecová, V.; Hornshøj, H.; Nielsen, B.; Christensen, O.F. A genome-wide association scan in pig identifi es novel regions associated with feed effi ciency trait. J. Anim. Sci. 2013, 91, 1041–1050. [Google Scholar] [CrossRef]
- Moreira, G.C.M.; Salvian, M.; Boschiero, C.; Cesar, A.S.M.; Reecy, J.M.; Godoy, T.F.; Ledur, M.C.; Garrick, D.; Mourão, G.B.; Coutinho, L.L. Genome-wide association scan for QTL and their positional candidate genes associated with internal organ traits in chickens. BMC Genom. 2019, 20, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Ramón, M.; Calvo, J.H.; Jiménez, M.Á.; Freire, F.; Vázquez, J.M.; Arranz, J.J. Genome-wide association studies for sperm traits in Assaf sheep breed. Animal 2020, 15, 100065. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Guzman, J.L.; Lázaro, S.F.; do Nascimento, A.V.; de Abreu Santos, D.J.; Cardoso, D.F.; Becker Scalez, D.C.; Galvão de Albuquerque, L.; Hurtado Lugo, N.A.; Tonhati, H. Genome-wide association study applied to type traits related to milk yield in water buffaloes (Bubalus bubalis). J. Dairy Sci. 2020, 103, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Tiezzil, F.; Parker-Gaddis, K.L.; Cole, J.B.; Clay, J.S.; Maltecca, C. A genome-wide association study for clinical mastitis in first parity US Holstein cows using single-step approach and genomic matrix re-weighting procedure. PLoS ONE 2015, 10, e0114919. [Google Scholar] [CrossRef] [Green Version]
- Nayeri, S.; Kroezen, V.; Sargolzaei, M.; Fleming, A.; Schenkel, F.S.; Baes, C.; Canovas, A.; Squires, J.; Miglior, F. Genome-wide Association Analysis for β-hydroxybutyrate Concentration in Milk Using Mid-Infrared Spectroscopy in North American Holstein Cattle. BMC Genet. 2019, 20, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flórez, J.C.R.; Herrera, A.L.; Zuluaga, J.J.E. Genome-wide association study using the Bayes C method for important traits in dairy yield in Colombian Holstein cattle. Acta Sci. Anim. Sci. 2018, 40, 1–12. [Google Scholar] [CrossRef]
- Saravanan, K.A.; Panigrahi, M.; Kumar, H.; Parida, S.; Bhushan, B.; Gaur, G.K.; Dutt, T.; Mishra, B.P.; Singh, R.K. Genomic scans for selection signatures revealed candidate genes for adaptation and production traits in a variety of cattle breeds. Genomics 2021, 113, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yao, Y.; Ma, P.; Wang, Q.; Pan, Y. Haplotype-based genome-wide association study identifies loci and candidate genes for milk yield in Holsteins. PLoS ONE 2018, 13, e0192695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm-Perry, A.K.; Kuehn, L.A.; Smith, T.P.L.; Ferrell, C.L.; Jenkins, T.G.; Freetly, H.C.; Snelling, W.M. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle. Anim. Genet. 2012, 43, 216–219. [Google Scholar] [CrossRef] [PubMed]
- De Las Heras-Saldana, S.; Clark, S.A.; Duijvesteijn, N.; Gondro, C.; Van Der Werf, J.H.J.; Chen, Y. Combining information from genome-wide association and multi-tissue gene expression studies to elucidate factors underlying genetic variation for residual feed intake in Australian Angus cattle. BMC Genom. 2019, 20, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuiyan, M.S.A.; Lim, D.; Park, M.; Lee, S.; Kim, Y.; Gondro, C.; Park, B.; Lee, S. Functional partitioning of genomic variance and genome-wide association study for carcass traits in Korean hanwoo cattle using imputed sequence level SNP data. Front. Genet. 2018, 9, 217. [Google Scholar] [CrossRef]
- Srivastava, S.; Srikanth, K.; Won, S.; Son, J.H.; Park, J.E.; Park, W.; Chai, H.H.; Lim, D. Haplotype-based genome-wide association study and identification of candidate genes associated with carcass traits in Hanwoo cattle. Genes 2020, 11, 551. [Google Scholar] [CrossRef]
- Medeiros De Oliveira Silva, R.; Stafuzza, N.B.; Fragomeni, B.D.O.; Ferreira De Camargo, G.M.; Ceacero, T.M.; Cyrillo, J.N.D.S.G.; Baldi, F.; Boligon, A.A.; Mercadante, M.E.Z.; Lourenco, D.L.; et al. Genome-wide association study for carcass traits in an experimental nelore cattle population. PLoS ONE 2017, 12, e169860. [Google Scholar] [CrossRef]
- Porto Neto, L.R.; Bunch, R.J.; Harrison, B.E.; Barendse, W. Variation in the XKR4 gene was significantly associated with subcutaneous rump fat thickness in indicine and composite cattle. Anim. Genet. 2012, 43, 785–789. [Google Scholar] [CrossRef]
- Cisternas, F.A.; Vincent, J.B.; Scherer, S.W.; Ray, P.N. Cloning and characterization of human CADPS and CADPS2, new members of the Ca2+-dependent activator for secretion protein family. Genomics 2003, 81, 279–291. [Google Scholar] [CrossRef]
- Kim, S.; Cheong, H.S.; Shin, H.D.; Lee, S.S.; Roh, H.J.; Jeon, D.Y.; Cho, C.Y. Genetic diversity and divergence among Korean cattle breeds assessed using a BovineHD single-nucleotide polymorphism chip. Asian-Australas. J. Anim. Sci. 2018, 31, 1691–1699. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, P.; Wang, K.; Chen, D.; Zhou, J.; Ma, J.; Li, M.; Xiao, W.; Jiang, A.; Jiang, Y.; et al. SNPs associated with body weight and backfat thickness in two pig breeds identified by a genome-wide association study. Genomics 2019, 111, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- He, W.B.; Tan, C.; Zhang, Y.X.; Meng, L.L.; Gong, F.; Lu, G.X.; Lin, G.; Du, J.; Tan, Y.Q. Homozygous variants in SYCP2L cause premature ovarian insufficiency. J. Med. Genet. 2020, 58, 168–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Stein, P.; Leu, N.A.; Chmátal, L.; Xue, J.; Ma, J.; Huang, X.; Lampson, M.A.; Schultz, R.M.; Wang, P.J. Accelerated reproductive aging in females lacking a novel centromere protein SYCP2L. Hum. Mol. Genet. 2015, 24, 6505–6514. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.; Li, Y.; Liu, X.; Berihulay, H.; Abied, A.; Gebreselassie, G.; Ma, Q.; Ma, Y. Genome-wide runs of homozygosity, effective population size, and detection of positive selection signatures in six chinese goat breeds. Genes 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucenas, S.; Li, Z.; Cox, J.A.; Egan, T.M.; Voigt, M.M. Molecular characterization of the zebrafish P2X receptor subunit gene family. Neuroscience 2003, 121, 935–945. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, H.; Li, H.; Guo, J.; Ouyang, B.; Wang, M.; Xu, Q.; Wang, J.; Lv, M.; Guo, X.; et al. The White-Spotted Bamboo Shark Genome Reveals Chromosome Rearrangements and Fast-Evolving Immune Genes of Cartilaginous Fish; Elsevier: Amsterdam, The Netherlands, 2020; Volume 23, ISBN 0000017191. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Mean | Std. Error | Minimum | Maximum | Std. Deviation |
|---|---|---|---|---|---|
| HD | 7.04 | 0.051 | 2.00 | 9.00 | 1.61 |
| BQ | 6.03 | 0.028 | 2.00 | 9.00 | 0.87 |
| RLSV | 3.92 | 0.048 | 1.00 | 9.00 | 1.50 |
| RLRV | 5.60 | 0.045 | 2.00 | 9.00 | 1.57 |
| Traits | HD | BQ | RLSV | RLRV |
|---|---|---|---|---|
| HD | 0.15 | 0.67 | −0.03 | −0.36 |
| BQ | −0.08 ** | 0.05 | 0.84 | 0.10 |
| RLSV | −0.07 * | −0.08 * | 0.17 | −0.09 |
| RLRV | −0.09 ** | 0.1 ** | −0.39 ** | 0.15 |
| Traits | SNP Name | rs. SNP Name | Chr. | Position | MAF | p Value | Nearest Gene Name | Distance |
|---|---|---|---|---|---|---|---|---|
| Bone quality | Hapmap54208-rs29015846 | rs29015846 | 8 | 55190206 | 0.29 | 1.99 × 10−7 | LOC112447952 | 100 kb |
| BovineHD0400024774 | rs133088614 | 4 | 88636425 | 0.46 | 2.25 × 10−7 | TMEM229A | 100 kb | |
| ARS-BFGL-NGS-81828 | rs41845981 | 17 | 44543822 | 0.35 | 3.14 × 10−7 | POLE | within | |
| BovineHD1400007035 | rs136017102 | 14 | 22608072 | 0.13 | 6.22 × 10−7 | XKR4 | 50 kb | |
| BovineHD2200011035 | rs110949452 | 22 | 38556301 | 0.34 | 7.67 × 10−7 | CADPS | within | |
| Heel depth | BovineHD2000006450 | rs137022628 | 20 | 21510411 | 0.27 | 3.03 × 10−9 | ACTBL2 | 200 kb |
| ARS-BFGL-BAC-36389 | rs109652453 | 23 | 45295465 | 0.37 | 4.22 × 10−8 | SYCP2L | within | |
| BovineHD2700002886 | rs42110372 | 27 | 10498902 | 0.10 | 3.11 × 10−7 | LOC112444670 | 100 kb | |
| ARS-BFGL-NGS-116157 | rs109601642 | 20 | 16333125 | 0.27 | 6.46 × 10−7 | LOC101907219 | 100 kb | |
| ARS-BFGL-NGS-73835 | rs41577664 | 15 | 80365537 | 0.48 | 7.43 × 10−7 | LOC112441589 | 200 bp | |
| BovineHD1400023839 | rs134726669 | 14 | 81745515 | 0.31 | 7.59 × 10−7 | MRPL13 | 200 bp | |
| Rear legs rear view | BovineHD0300015960 | rs134130409 | 3 | 52695086 | 0.41 | 6.72 × 10−8 | BARHL2 | 100 kb |
| BovineHD4100014792 | rs134139959 | 20 | 57471508 | 0.19 | 1.11 × 10−7 | FBXL7 | within | |
| Hapmap48798-BTA-51401 | rs41638134 | 1 | 131310669 | 0.35 | 6.11 × 10−7 | LOC107132214 | 20 kb | |
| Rear legs side view | ARS-BFGL-NGS-91167 | rs41565304 | 5 | 108222067 | 0.19 | 1.11 × 10−9 | ADIPOR2 | within |
| BovineHD0300022142 | rs43350216 | 3 | 76168946 | 0.38 | 4.05 × 10−9 | - | - | |
| BovineHD1100001372 | rs43656945 | 11 | 3761839 | 0.07 | 2.32 × 10−7 | INPP4A | within | |
| BovineHD1100021155 | rs136593856 | 11 | 74048825 | 0.44 | 5.07 × 10−7 | DNMT3A | within | |
| BTB-01677645 | rs42791722 | 10 | 52217450 | 0.09 | 7.40 × 10−7 | ALDH1A2 | within | |
| BTB-01518251 | rs42639670 | 6 | 49588438 | 0.09 | 9.65 × 10−7 | PCDH7 | 200 kb |
| Term | Description | Gene Count | % | p-Value | Genes |
|---|---|---|---|---|---|
| bta00531: | Glycosaminoglycan degradation | 3 | 2.9 | 0.00469386 | LOC527125, LOC509761, SPAM1 |
| bta01100: | Metabolic pathways | 12 | 11.4 | 0.016110131 | INPP4A, LOC527125, LIPC, LOC520336, MGAT4A, ALDH1A2, DNMT3A, LOC100848875, LOC509761, POLE, GALNT9, SPAM1 |
| bta04020: | Calcium signaling pathway | 4 | 3.8 | 0.052382966 | P2RX3, P2RX2, ADCY3, CACNA1C |
| bta05414: | Dilated cardiomyopathy | 3 | 2.9 | 0.056875544 | ADCY3, CACNA2D4, CACNA1C |
| bta04916: | Melanogenesis | 3 | 2.9 | 0.072824808 | POMC, WNT5B, DCY3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdalla, I.M.; Lu, X.; Nazar, M.; Arbab, A.A.I.; Xu, T.; Yousif, M.H.; Mao, Y.; Yang, Z. Genome-Wide Association Study Identifies Candidate Genes Associated with Feet and Leg Conformation Traits in Chinese Holstein Cattle. Animals 2021, 11, 2259. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11082259
Abdalla IM, Lu X, Nazar M, Arbab AAI, Xu T, Yousif MH, Mao Y, Yang Z. Genome-Wide Association Study Identifies Candidate Genes Associated with Feet and Leg Conformation Traits in Chinese Holstein Cattle. Animals. 2021; 11(8):2259. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11082259
Chicago/Turabian StyleAbdalla, Ismail Mohamed, Xubin Lu, Mudasir Nazar, Abdelaziz Adam Idriss Arbab, Tianle Xu, Mohammed Husien Yousif, Yongjiang Mao, and Zhangping Yang. 2021. "Genome-Wide Association Study Identifies Candidate Genes Associated with Feet and Leg Conformation Traits in Chinese Holstein Cattle" Animals 11, no. 8: 2259. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11082259