
Profiling and Functional Analysis of mRNAs during Skeletal Muscle Differentiation in Goats

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. MuSC Culture and Differentiation
2.2. RNA Extraction and Sequencing
2.3. Differentially Expressed Gene Analysis
2.4. Expression Trend Analysis
2.5. GO and KEGG Pathway Analysis
2.6. Validation of the Sequencing Results by qRT-PCR
2.7. Statistical Analysis
3. Results
3.1. MuSC Differentiation Program
3.2. Temporally Regulated Transcription during Skeletal Myogenesis
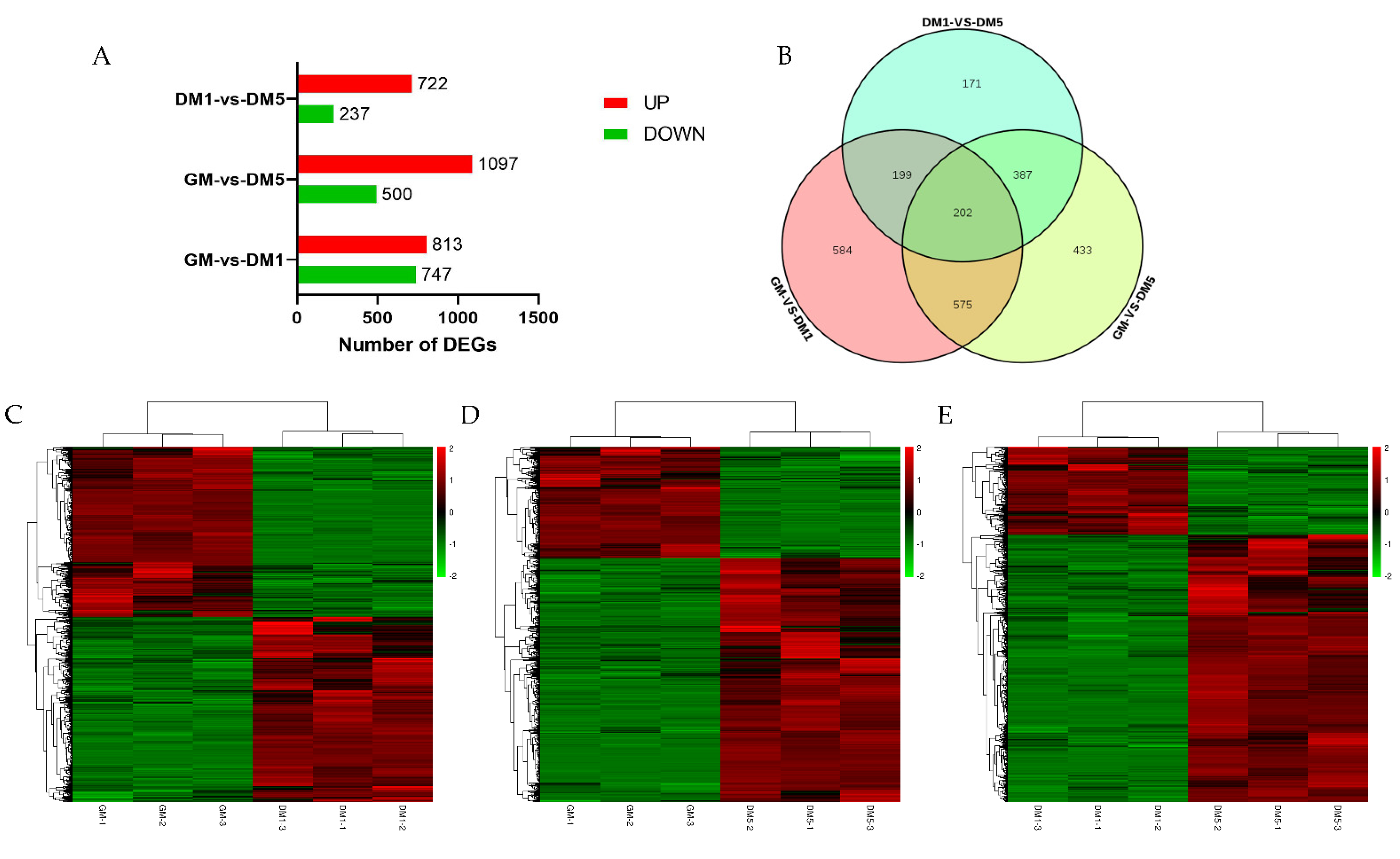
3.3. Differentially Expressed Genes (DEGs) Analysis
3.4. Expression Trend Analysis
3.5. GO Enrichment Analysis
3.6. KEGG Enrichment Analysis
3.7. Validation of the DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Costa, T.C.; Gionbelli, M.P.; Duarte, M.d.S. Fetal programming in ruminant animals: Understanding the skeletal muscle development to improve meat quality. Anim. Front. 2021, 11, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.; Sebastian, S.; Dilworth, F.J. The origin and fate of muscle satellite cells. Stem Cell Rev. Rep. 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, M.; Rigby, P.W. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev. Cell 2014, 28, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Hernandez, J.M.; Garcia-Gonzalez, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Kopantseva, E.E.; Belyavsky, A.V. Key regulators of skeletal myogenesis. Molecular. Biol. 2016, 50, 169–192. [Google Scholar] [CrossRef]
- Tang, Z.; Yang, Y.; Wang, Z.; Zhao, S.; Mu, Y.; Li, K. Integrated analysis of miRNA and mRNA paired expression profiling of prenatal skeletal muscle development in three genotype pigs. Sci. Rep. 2015, 5, 15544. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Bai, M.; Xiang, L.; Zhang, G.; Ma, W.; Jiang, H. Comparative transcriptome profiling of longissimus muscle tissues from Qianhua Mutton Merino and Small Tail Han sheep. Sci. Rep. 2016, 6, 33586. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, J.; Liu, H.; Xi, Y.; Xue, M.; Liu, W.; Zhuang, Z.; Lei, M. Dynamic transcriptome profiles of skeletal muscle tissue across 11 developmental stages for both Tongcheng and Yorkshire pigs. BMC Genom. 2015, 16, 377. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.H.; Zhang, C.L.; Plath, M.; Fang, X.T.; Lan, X.Y.; Zhou, Y.; Chen, H. Global transcriptional profiling of longissimus thoracis muscle tissue in fetal and juvenile domestic goat using RNA sequencing. Anim. Genet. 2015, 46, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, L.; Zhong, T.; Wang, L.; Guo, J.; Dong, Y.; Feng, J.; Song, T.; Li, L.; Zhang, H. The differential proliferation and differentiation ability of skeletal muscle satellite cell in Boer and Nanjiang brown goats. Small Rumin. Res. 2018, 169, 99–107. [Google Scholar] [CrossRef]
- Li, L.; Chen, Y.; Nie, L.; Ding, X.; Zhang, X.; Zhao, W.; Xu, X.; Kyei, B.; Dai, D.; Zhan, S.; et al. MyoD-induced circular RNA CDR1as promotes myogenic differentiation of skeletal muscle satellite cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mohammadabadi, M.; Bordbar, F.; Jensen, J.; Du, M.; Guo, W. Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals. Animals 2021, 11, 835. [Google Scholar] [CrossRef]
- Moncaut, N.; Rigby, P.W.; Carvajal, J.J. Dial M(RF) for myogenesis. FEBS J. 2013, 280, 3980–3990. [Google Scholar] [CrossRef] [Green Version]
- Berkes, C.A.; Tapscott, S.J. MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 2005, 16, 585–595. [Google Scholar] [CrossRef]
- Naya, F.J.; Olson, E. MEF2: A transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr. Opin. Cell Biol. 1999, 11, 683–688. [Google Scholar] [CrossRef]
- Estrella, N.L.; Desjardins, C.A.; Nocco, S.E.; Clark, A.L.; Maksimenko, Y.; Naya, F.J. MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J. Biol. Chem. 2015, 290, 1256–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, S.; Soejima, M.; Iwanuma, O.; Saka, H.; Matsunaga, S.; Sakiyama, K.; Ide, Y. Expression of myostatin and follistatin in Mdx mice, an animal model for muscular dystrophy. Zoolog. Sci. 2009, 26, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Ren, H.; Gao, S. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins: Roles in skeletal muscle growth and differentiation. Gen. Comp. Endocrinol. 2010, 167, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Mavalli, M.D.; DiGirolamo, D.J.; Fan, Y.; Riddle, R.C.; Campbell, K.S.; van Groen, T.; Frank, S.J.; Sperling, M.A.; Esser, K.A.; Bamman, M.M.; et al. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J. Clin. Investig. 2010, 120, 4007–4020. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, M.; Sun, Y.; Cai, H.; Li, R.; Wei, X.; Lan, X.; Huang, Y.; Lei, C.; Chen, H. The developmental transcriptome landscape of bovine skeletal muscle defined by Ribo-Zero ribonucleic acid sequencing. J. Anim. Sci. 2015, 93, 5648–5658. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.K.; Patel, A.K.; Shah, R.K.; Patel, A.B.; Shah, T.M.; Bhatt, V.D.; Joshi, C.G. Transcriptomic dissection of myogenic differentiation signature in caprine by RNA-Seq. Mech. Dev. 2014, 132, 79–92. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, J.; Xiao, Z.; Zou, C.; An, Q.; Li, H.; Zhou, X.; Wu, Z.; Shi, D.; Deng, Y.; et al. The Expression Profiles of mRNAs and lncRNAs in Buffalo Muscle Stem Cells Driving Myogenic Differentiation. Front. Genet. 2021, 12, 643497. [Google Scholar] [CrossRef]
- Sheikh, F.; Lyon, R.C.; Chen, J. Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene 2015, 569, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Brocker, C.N.; Vasiliou, V.; Nebert, D.W. Evolutionary divergence and functions of the ADAM and ADAMTS gene families. Hum. Genom. 2009, 4, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Bar-Joseph, Z.; Gitter, A.; Simon, I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet. 2012, 13, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Nau, G.J.; Bar-Joseph, Z. Clustering short time series gene expression data. Bioinformatics 2005, 21 (Suppl. 1), i159–i168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, M.M.; Runci, A.; Russo, M.A.; Tafani, M. Muscle Lim Protein (MLP)/CSRP3 at the crossroad between mechanotransduction and autophagy. Cell Death Dis. 2015, 6, e1940. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Han, S.; Tang, S.; He, H.; Shen, X.; Zhao, J.; Chen, Y.; Wei, Y.; Wang, Y.; Zhu, Q.; et al. The Autophagy Regulatory Molecule CSRP3 Interacts with LC3 and Protects Against Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 749. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Flick, M.J.; Kudla, A.J.; Konieczny, S.F. Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Mol. Cell Biol. 1997, 17, 4750–4760. [Google Scholar] [CrossRef] [Green Version]
- Filomena, M.C.; Yamamoto, D.L.; Caremani, M.; Kadarla, V.K.; Mastrototaro, G.; Serio, S.; Vydyanath, A.; Mutarelli, M.; Garofalo, A.; Pertici, I.; et al. Myopalladin promotes muscle growth through modulation of the serum response factor pathway. J. Cachexia Sarcopenia Muscle 2020, 11, 169–194. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.C.; Chung, E.R. Identification of differentially expressed genes between high and low marbling score grades of the longissimus lumborum muscle in Hanwoo (Korean cattle). Meat. Sci. 2016, 121, 114–118. [Google Scholar] [CrossRef]
- Wenping, H.; Yuan, Z.; Jie, S.; Lijun, Z.; Zhezhi, W. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 2011, 98, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Xie, S.J.; Li, J.H.; Chen, H.F.; Tan, Y.Y.; Liu, S.R.; Zhang, Y.; Xu, H.; Yang, J.H.; Liu, S.; Zheng, L.L.; et al. Inhibition of the JNK/MAPK signaling pathway by myogenesis-associated miRNAs is required for skeletal muscle development. Cell Death Differ. 2018, 25, 1581–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Amerongen, R.; Berns, A. Knockout mouse models to study Wnt signal transduction. Trends Genet. 2006, 22, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, A.; Schmidt, C.; Luke, G.; Allen, S.; Valasek, P.; Muntoni, F.; Lawrence-Watt, D.; Patel, K. Canonical Wnt signalling induces satellite-cell proliferation during adult skeletal muscle regeneration. J. Cell Sci. 2008, 121, 2939–2950. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.V.; Hausman, G.J.; Guan, L.; Du, M.; Rasmussen, T.P.; Poulos, S.P.; Mir, P.; Bergen, W.G.; Fernyhough, M.E.; McFarland, D.C.; et al. Skeletal muscle stem cells from animals I. Basic cell biology. Int. J. Biol. Sci. 2010, 6, 465–474. [Google Scholar] [CrossRef]
- Ren, Y.; Wu, H.; Ma, Y.; Yuan, J.; Liang, H.; Liu, D. Potential of adipose-derived mesenchymal stem cells and skeletal muscle-derived satellite cells for somatic cell nuclear transfer mediated transgenesis in Arbas Cashmere goats. PLoS ONE 2014, 9, e93583. [Google Scholar] [CrossRef]
- Feve, B. Adipogenesis: Cellular and molecular aspects. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 483–499. [Google Scholar] [CrossRef]
- Gnanalingham, M.G.; Mostyn, A.; Symonds, M.E.; Stephenson, T. Ontogeny and nutritional programming of adiposity in sheep: Potential role of glucocorticoid action and uncoupling protein-2. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1407–R1415. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence(5′-3′) | Tm/°C | Product Size/bp |
|---|---|---|---|
| MYL2 | F: GGAGTGCTCAAGGCTGATTATG R: GGCGAACATCTGCTCAATCTC | 63.3 | 87 |
| DES | F: GCCGGATCAACCTCCCTATC R: ATGGACCTCAGAACCCCTCT | 63.3 | 83 |
| MYOG | F: GGACCCTACAGATGCCCACAA R: TTGGTATGGTTTCATCTGGG | 59.0 | 101 |
| FAP | F: CGACCTTACAAACGGGGAGT R: TTTACTCCCAACAGGCGACC | 65.0 | 85 |
| PLK2 | F: TTCAGTGGGTCACGAAGTGG R: TTGTTCAGGGGCATCTGTGG | 55.7 | 191 |
| ADAM | F: TCCAGTTGCACAAAGGTGGT R: GGCAGTGAATCTGGTCTGGT | 55.0 | 135 |
| WWC1 | F: CGGATGCTGTGTCTGCTCTGTT R: GGTCTGCGTGCTGCTCCTTT | 63.3 | 80 |
| PRDX1 | F: AGCCTAGCTGACTACAAAGGAA R: GTGTTGATCCATGCCAGGT | 59.0 | 182 |
| GAPDH | F: GCAAGTTCCACGGCACAG R: GGTTCACGCCCATCACAA | 59.0 | 249 |
| Samples | Raw Reads | Clean Reads (%) | Unique-Mapped Reads | GC Content | Q30 Value |
|---|---|---|---|---|---|
| GM-1 | 79,898,070 | 79,694,066 (99.74%) | 77,518,765 (90.59%) | 49.96% | 93.74% |
| GM-2 | 82,449,876 | 82,218,204 (99.72%) | 70,235,626 (90.44%) | 50.41% | 94.15% |
| GM-3 | 78,770,454 | 78,549,636 (99.72%) | 70,114,545 (89.61%) | 49.92% | 93.90% |
| DM1-1 | 85,834,132 | 85,610,188 (99.74%) | 71,993,906 (90.73%) | 50.31% | 94.18% |
| DM1-2 | 77,919,438 | 77,705,900 (99.73%) | 71,317,809 (90.66%) | 50.49% | 94.27% |
| DM1-3 | 78,474,104 | 78,271,608 (99.74%) | 72,775,979 (91.04%) | 50.82% | 94.06% |
| DM5-1 | 79,580,920 | 79,392,054 (99.76%) | 72,103,450 (90.51%) | 49.86% | 94.20% |
| DM5-2 | 78,868,496 | 78,701,026 (99.79%) | 74,380,716 (90.51%) | 51.30% | 94.01% |
| DM5-3 | 80,154,802 | 79,979,248 (99.78%) | 71,321,563 (90.84%) | 49.57% | 93.85% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, S.; Zhai, H.; Tang, M.; Xue, Y.; Li, D.; Wang, L.; Zhong, T.; Dai, D.; Cao, J.; Guo, J.; et al. Profiling and Functional Analysis of mRNAs during Skeletal Muscle Differentiation in Goats. Animals 2022, 12, 1048. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12081048
Zhan S, Zhai H, Tang M, Xue Y, Li D, Wang L, Zhong T, Dai D, Cao J, Guo J, et al. Profiling and Functional Analysis of mRNAs during Skeletal Muscle Differentiation in Goats. Animals. 2022; 12(8):1048. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12081048
Chicago/Turabian StyleZhan, Siyuan, Hongfan Zhai, Min Tang, Yanan Xue, Dandan Li, Linjie Wang, Tao Zhong, Dinghui Dai, Jiaxue Cao, Jiazhong Guo, and et al. 2022. "Profiling and Functional Analysis of mRNAs during Skeletal Muscle Differentiation in Goats" Animals 12, no. 8: 1048. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12081048