
Engineering an Enhanced EGFR Engager: Humanization of Cetuximab for Improved Developability

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Humanization
2.2. Sequence Analysis
2.3. Expression and Purification
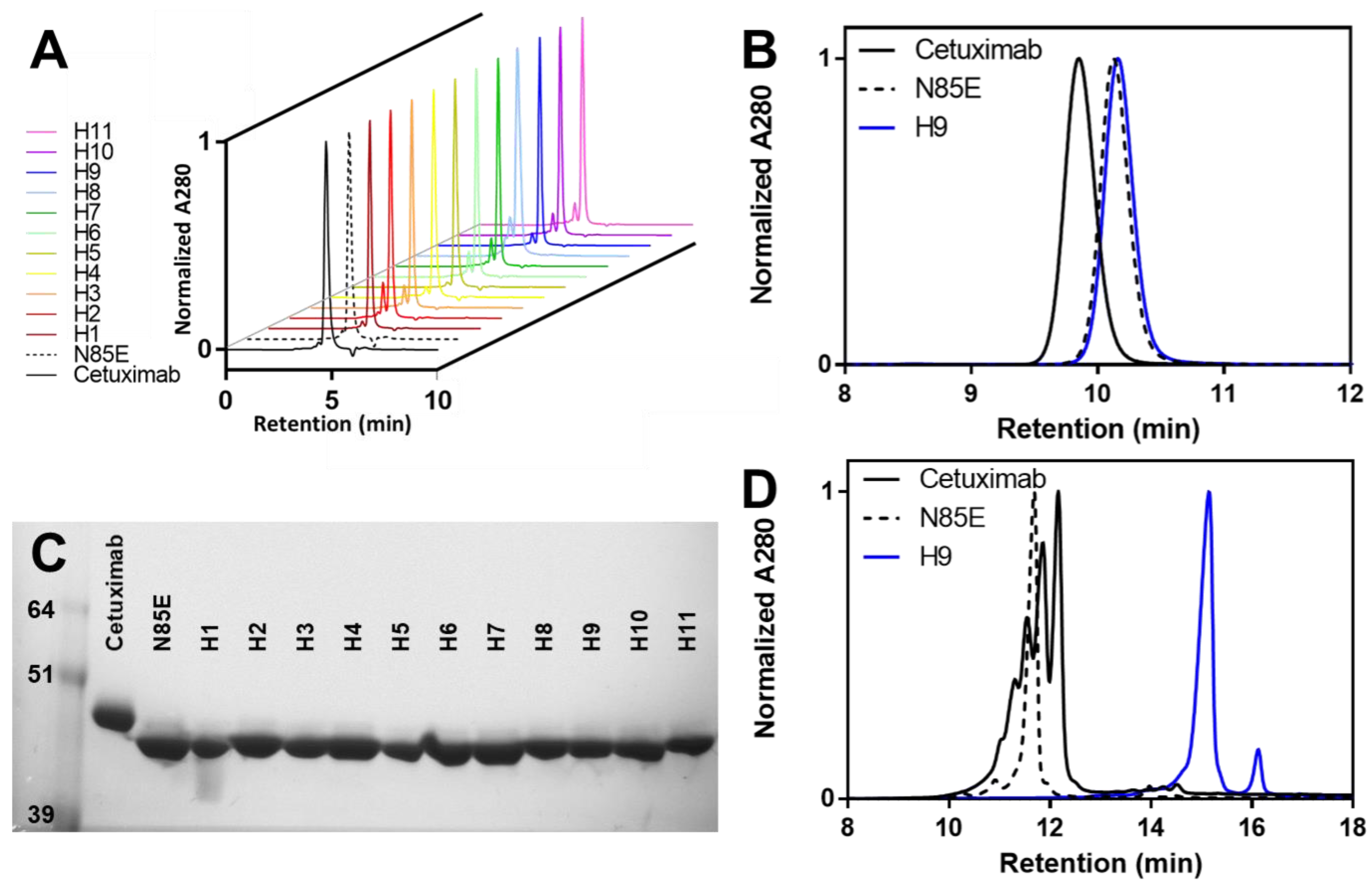
2.4. Analytical Size-Exclusion Chromatography
2.5. SDS-PAGE
2.6. Cation Exchange Chromatography
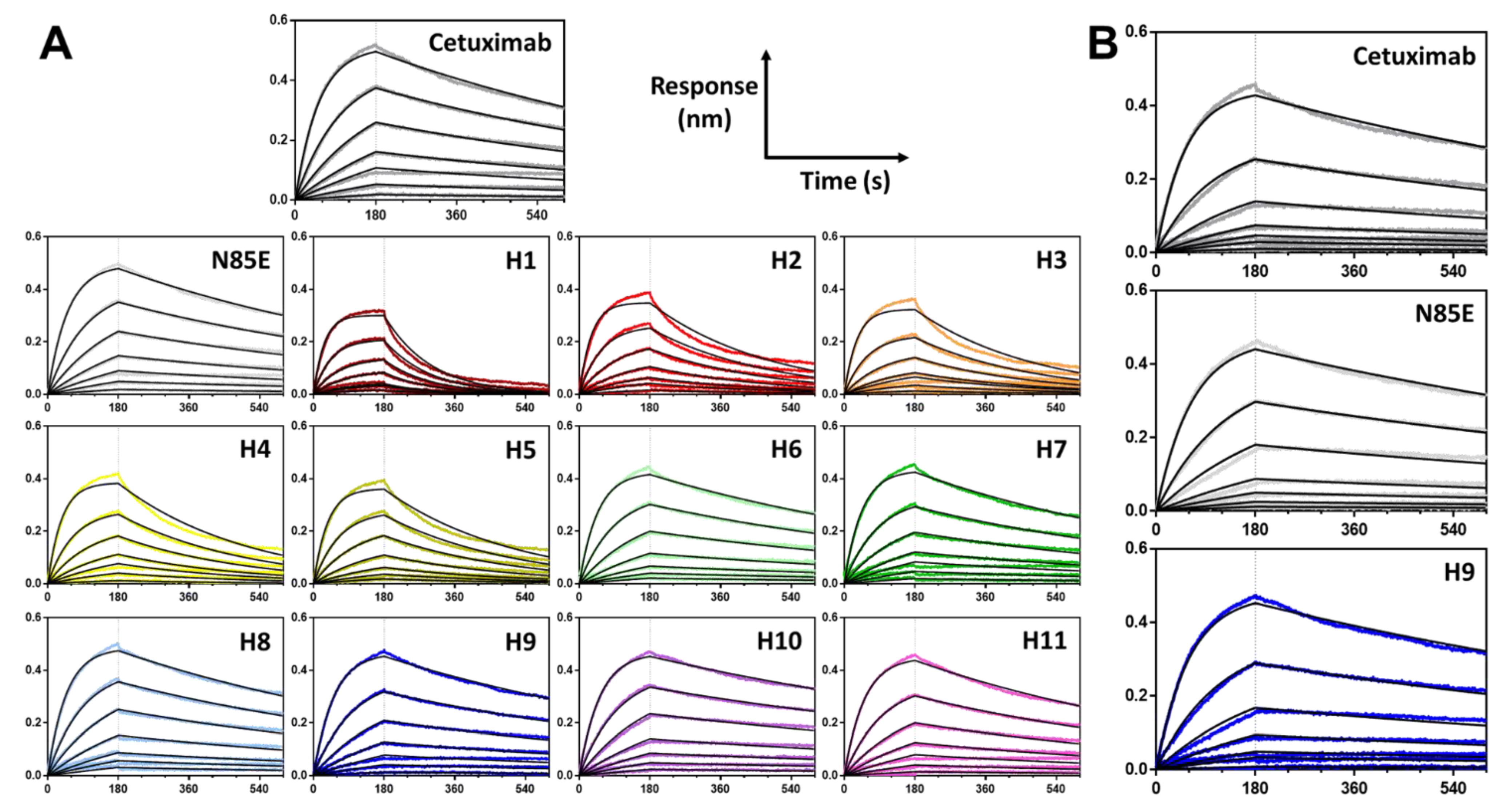
2.7. Biolayer Interferometry
2.8. Dynamic Light Scattering
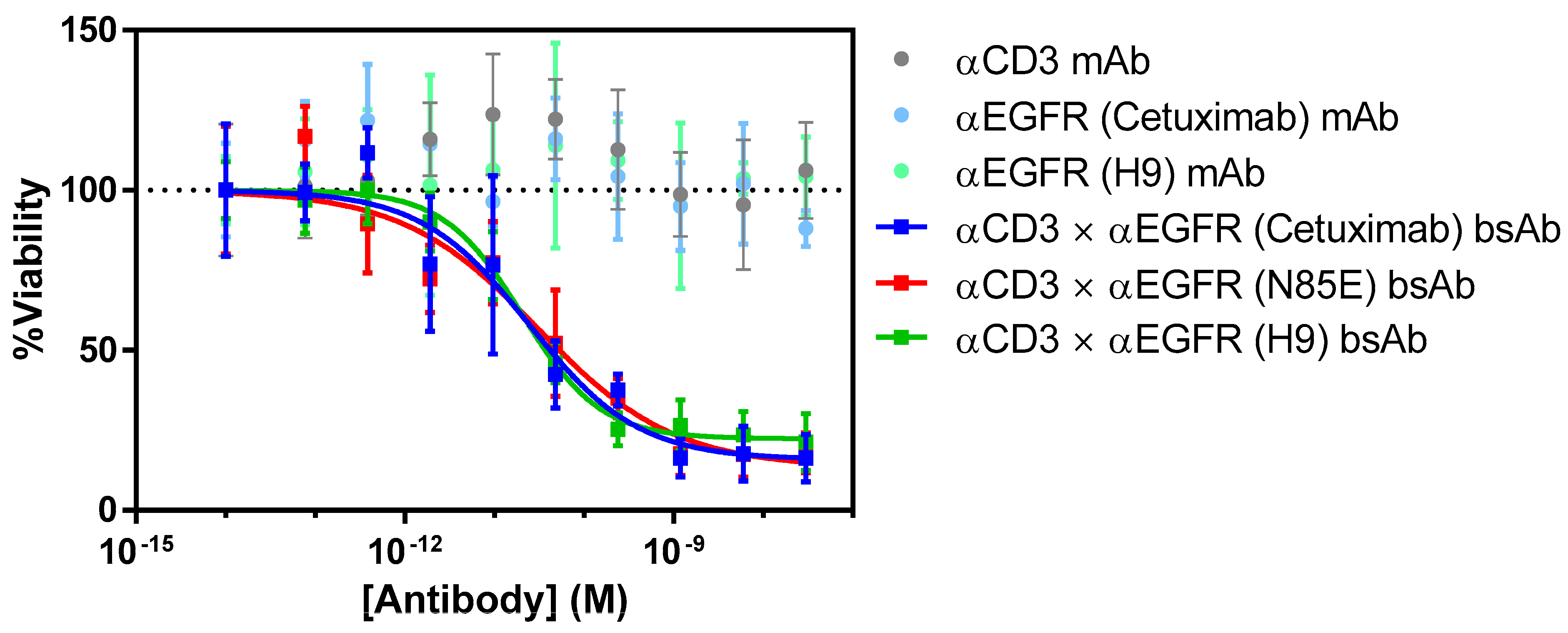
2.9. T Cell-Dependent Cellular Cytotoxicity
3. Results
3.1. Humanization Strategy
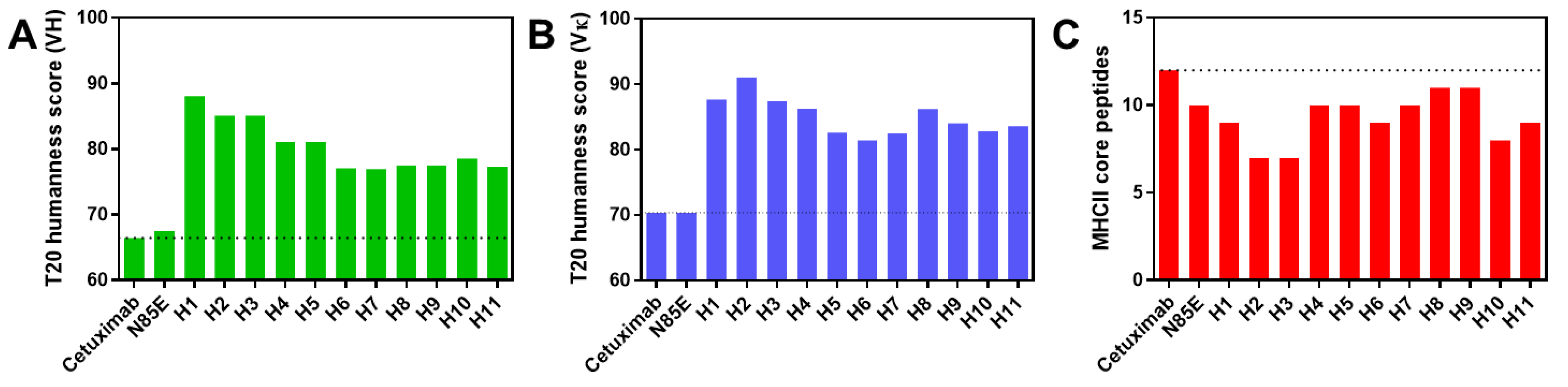
3.2. Sequence Analysis
3.3. Biophysical Properties of Humanized scFv Domains
3.4. Biophysical Properties of Humanized mAb
3.5. T Cell-Mediated Killing by αEGFR × αCD3 Bispecific Antibodies
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Graham, J.; Muhsin, M.; Kirkpatrick, P. Cetuximab. Nat. Rev. Drug Discov. 2004, 3, 549–550. [Google Scholar] [CrossRef]
- Bou-Assaly, W.; Mukherji, S. Cetuximab (Erbitux). Am. J. Neuroradiol. 2010, 31, 626–627. [Google Scholar] [CrossRef] [Green Version]
- Hanck-Silva, G.; Fatori Trevizan, L.N.; Petrilli, R.; de Lima, F.T.; Eloy, J.O.; Chorilli, M. A Critical Review of Properties and Analytical/Bioanalytical Methods for Characterization of Cetuximab. Crit. Rev. Anal. Chem. 2020, 50, 125–135. [Google Scholar] [CrossRef] [PubMed]
- García-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing Features of Cetuximab and Panitumumab in Colorectal Cancer and Other Solid Tumors. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. MAbs 2021, 13, 1860476. [Google Scholar] [CrossRef]
- Hwang, W.Y.K.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef]
- Park, S.S.; Kim, J.; Brandts, J.F.; Hong, H.J. Stability of murine, chimeric and humanized antibodies against pre-S2 surface antigen of hepatitis B virus. Biologicals 2003, 31, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, D.; Jabs, W.; Resemann, A.; Evers, W.; Evans, C.; Main, L.; Baessmann, C.; Wagner-Rousset, E.; Suckau, D.; Beck, A. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. MAbs 2013, 5, 699–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.-T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-Induced Anaphylaxis and IgE Specific for Galactose-α-1,3-Galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriakose, A.; Chirmule, N.; Nair, P. Immunogenicity of Biotherapeutics: Causes and Association with Posttranslational Modifications. J. Immunol. Res. 2016, 2016, 1298473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Woen, S.; Wang, T.; Liau, B.; Zhao, S.; Chen, C.; Yang, Y.; Song, Z.; Wormald, M.R.; Yu, C.; et al. Challenges of glycosylation analysis and control: An integrated approach to producing optimal and consistent therapeutic drugs. Drug Discov. Today 2016, 21, 740–765. [Google Scholar] [CrossRef] [Green Version]
- Borras, L.; Gunde, T.; Tietz, J.; Bauer, U.; Hulmann-Cottier, V.; Grimshaw, J.P.A.; Urech, D.M. Generic approach for the generation of stable humanized single-chain Fv fragments from rabbit monoclonal antibodies. J. Biol. Chem. 2010, 285, 9054–9066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.H.; Huang, K.; Tu, H.; Adler, A.S. Monoclonal antibody humanness score and its applications. BMC Biotechnol. 2013, 13, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racle, J.; Michaux, J.; Rockinger, G.A.; Arnaud, M.; Bobisse, S.; Chong, C.; Guillaume, P.; Coukos, G.; Harari, A.; Jandus, C.; et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019, 37, 1283–1286. [Google Scholar] [CrossRef]
- Wang, C.; Wu, Y.; Wang, L.; Hong, B.; Jin, Y.; Hu, D.; Chen, G.; Kong, Y.; Huang, A.; Hua, G.; et al. Engineered soluble monomeric IgG1 Fc with significantly decreased non-specific binding. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Ewert, S.; Huber, T.; Honegger, A.; Plückthun, A. Biophysical properties of human antibody variable domains. J. Mol. Biol. 2003, 325, 531–553. [Google Scholar] [CrossRef]
- Nieba, L.; Honegger, A.; Krebber, C.; Plückthun, A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: Improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. 1997, 10, 435–444. [Google Scholar] [CrossRef]
- Desplancq, D.; King, D.J.; Lawson, A.D.G.; Mountain, A. Multimerization behaviour of single chain fv variants for the tumour-binding antibody b72.3. Protein Eng. Des. Sel. 1994, 7, 1027–1033. [Google Scholar] [CrossRef]
- Luo, D.; Mah, N.; Krantz, M.; Wilde, K.; Wishart, D.; Zhang, Y.; Jacobs, F.; Martin, L. Vl-linker-vh orientation-dependent expression of single chain Fv containing an engineered disulfide-stabilized bond in the framework regions. J. Biochem. 1995, 118, 825–831. [Google Scholar] [CrossRef]
- Plückthun, A.; Pack, P. New protein engineering approaches to multivalent and bispecific antibody fragments. Immunotechnology 1997, 3, 83–105. [Google Scholar] [CrossRef]
- Weatherill, E.E.; Cain, K.L.; Heywood, S.P.; Compson, J.E.; Heads, J.T.; Adams, R.; Humphreys, D.P. Towards a universal disulphide stabilised single chain Fv format: Importance of interchain disulphide bond location and vLvH orientation. Protein Eng. Des. Sel. 2012, 25, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Package Leaflet: Erbitux 5 mg/mL Solution for Infusion. Available online: https://www.medicines.org.uk/emc/product/317/smpc (accessed on 11 December 2021).
- Ulitzka, M.; Carrara, S.; Grzeschik, J.; Kornmann, H.; Hock, B.; Kolmar, H. Engineering therapeutic antibodies for patient safety: Tackling the immunogenicity problem. Protein Eng. Des. Sel. 2020, 33, gzaa025. [Google Scholar] [CrossRef]
- Fussl, F.; Trappe, A.; Carillo, S.; Jakes, C.; Bones, J. Comparative Elucidation of Cetuximab Heterogeneity on the Intact Protein Level by Cation Exchange Chromatography and Capillary Electrophoresis Coupled to Mass Spectrometry. Anal. Chem. 2020, 92, 5431–5438. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, A.; Safdari, Y.; Kianmehr, A.; Pourafshar, M. Production of a germline-humanized cetuximab scFv and evaluation of its activity in recognizing EGFR- overexpressing cancer cells. Hum. Vaccines Immunother. 2018, 14, 856–863. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Ndong, C.; Griswold, K.E.; Bailey-Kellogg, C. Computationally driven antibody engineering enables simultaneous humanization and thermostabilization. Protein Eng. Des. Sel. 2016, 29, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Giddens, J.P.; Lomino, J.V.; DiLillo, D.J.; Ravetch, J.V.; Wang, L.X. Site-selective chemoenzymatic glycoengineering of Fab and Fc glycans of a therapeutic antibody. Proc. Natl. Acad. Sci. USA 2018, 115, 12023–12027. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Humanized Version | Method | Basis | Starting Sequence/Model | Modification |
|---|---|---|---|---|
| H1 | CDR graft onto stable framework | Sequence | Cetuximab, FW1.4gen | None |
| H2 | DS germline substitutions | Sequence | Cetuximab | None |
| H3 | DS germline substitutions | Sequence | Cetuximab | Lambda J |
| H4 | DS frequent residue substitutions | Sequence | Cetuximab | None |
| H5 | DS frequent residue substitutions | Sequence | Cetuximab | Lambda J |
| H6 | DS best single mutations | Model | PDB 1YY9 | None |
| H7 | DS best single mutations | Model | Cetuximab model | None |
| H8 | DS best single mutations | Model | Cetuximab model (TVSS) | None |
| H9 | DS best single mutations | Model | Cetuximab model (TVSS) | Lambda J |
| H10 | DS best single mutations | Model | Cetuximab model (LTVL) | None |
| H11 | DS best single mutations | Model | Cetuximab model (TVSS-LTVL) | None |
| Version | Titer (µg/mL) | aSEC %POI | DLS Temp (°C) | EGFR KD (nM) | ka [1/(M·s)] × 105 | kd (1/s) × 10−3 |
|---|---|---|---|---|---|---|
| Cetuximab | 163 ± 39 | 93.6 ± 2.6 | 47.2 ± 1.0 | 3.18 ± 0.18 | 4.44 ± 1.13 | 1.42 ± 0.44 |
| N85E | 116 ± 6 | 94.9 ± 3.1 | 44.5 ± 1.3 | 3.16 ± 0.35 | 4.44 ± 0.91 | 1.42 ± 0.44 |
| H1 | 309 ± 26 | 91.5 ± 7.1 | 46.2 ± 0.9 | 13.1 ± 1.2 | 7.18 ± 1.68 | 9.27 ± 1.34 |
| H2 | 308 ± 3 | 83.7 ± 1.9 | 52.6 ± 1.3 | 6.77 ± 2.10 | 6.65 ± 0.73 | 4.58 ± 1.89 |
| H3 | 376 ± 12 | 88.6 ± 2.4 | 53.0 ± 1.3 | 6.74 ± 1.61 | 6.61 ± 1.11 | 4.54 ± 1.81 |
| H4 | 220 ± 3 | 92.2 ± 0.0 | 51.3 ± 1.1 | 6.79 ± 1.35 | 5.71 ± 0.78 | 3.93 ± 1.30 |
| H5 | 211 ± 22 | 97.4 ± 0.9 | 48.7 ± 1.3 | 6.92 ± 1.58 | 5.63 ± 0.67 | 3.95 ± 1.35 |
| H6 | 411 ± 59 | 86.8 ± 2.1 | 52.4 ± 2.1 | 3.08 ± 0.68 | 4.81 ± 0.91 | 1.51 ± 0.61 |
| H7 | 418 ± 8 | 88.1 ± 2.7 | 52.3 ± 2.0 | 3.45 ± 0.81 | 5.04 ± 0.92 | 1.77 ± 0.73 |
| H8 | 412 ± 5 | 82.6 ± 5.5 | 49.7 ± 1.4 | 3.05 ± 0.62 | 4.50 ± 0.54 | 1.39 ± 0.44 |
| H9 | 506 ± 14 | 92.7 ± 2.3 | 50.5 ± 2.4 | 3.14 ± 0.53 | 4.47 ± 0.97 | 1.43 ± 0.54 |
| H10 | 394 ± 23 | 85.6 ± 6.2 | 50.2 ± 1.5 | 2.27 ± 0.38 | 4.74 ± 1.31 | 1.10 ± 0.48 |
| H11 | 372 ± 73 | 88.0 ± 3.4 | 52.2 ± 2.0 | 3.42 ± 0.36 | 4.76 ± 1.33 | 1.65 ± 0.62 |
| Version | Titer (µg/mL) | aSEC %POI | DLS Temp (°C) | EGFR KD (nM) | ka [1/(M·s)] × 105 | kd (1/s) × 10−4 | * EC50 (pM) |
|---|---|---|---|---|---|---|---|
| Cetuximab | 203 ± 13 | 99.9 ± 0.2 | 68.3 ± 0.1 | 2.68 ± 0.07 | 3.46 ± 0.27 | 9.26 ± 0.48 | 24.6 [10.4–58.4] |
| N85E | 209 ± 9 | 99.4 ± 0.3 | 68.1 ± 0.9 | 2.46 ± 0.28 | 3.26 ± 0.41 | 7.96 ± 0.09 | 30.7 [11.3–83.8] |
| H9 | 249 ± 1 | 99.1 ± 0.4 | 72.5 ± 1.2 | 2.41 ± 0.15 | 3.37 ± 0.20 | 8.12 ± 0.05 | 20.3 [13.9–29.9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goulet, D.R.; Chatterjee, S.; Lee, W.-P.; Waight, A.B.; Zhu, Y.; Mak, A.N.-S. Engineering an Enhanced EGFR Engager: Humanization of Cetuximab for Improved Developability. Antibodies 2022, 11, 6. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11010006
Goulet DR, Chatterjee S, Lee W-P, Waight AB, Zhu Y, Mak AN-S. Engineering an Enhanced EGFR Engager: Humanization of Cetuximab for Improved Developability. Antibodies. 2022; 11(1):6. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11010006
Chicago/Turabian StyleGoulet, Dennis R., Soumili Chatterjee, Wai-Ping Lee, Andrew B. Waight, Yi Zhu, and Amanda Nga-Sze Mak. 2022. "Engineering an Enhanced EGFR Engager: Humanization of Cetuximab for Improved Developability" Antibodies 11, no. 1: 6. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11010006