
Delivery of Drugs into Cancer Cells Using Antibody–Drug Conjugates Based on Receptor-Mediated Endocytosis and the Enhanced Permeability and Retention Effect

Abstract
:1. Introduction
2. Discussion
2.1. Effective and Non-Invasive Cancer Cures
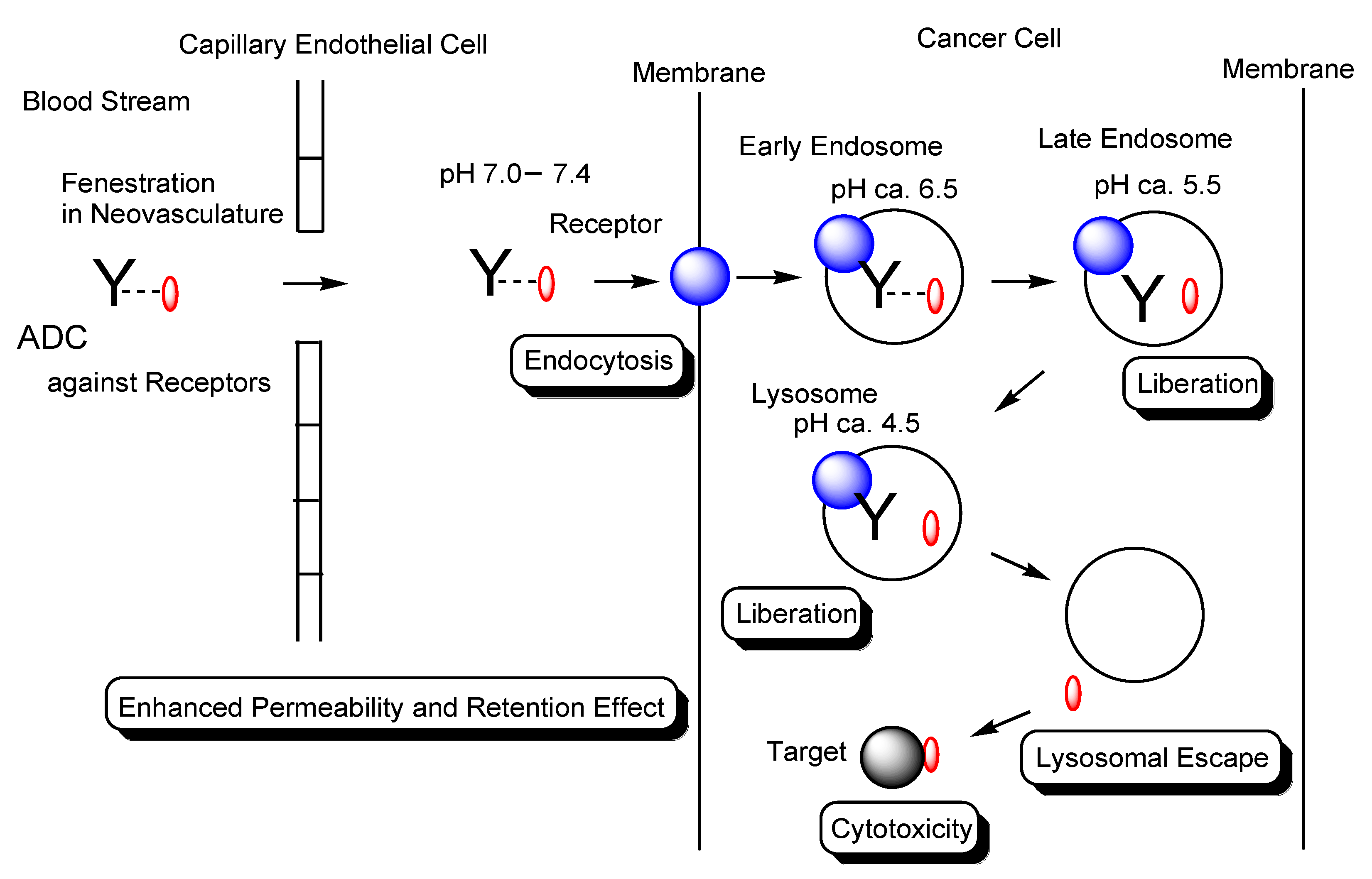
2.2. Endocytosis and Endosomal–Lysosomal System
2.3. Ab Drugs for Cancer Therapy
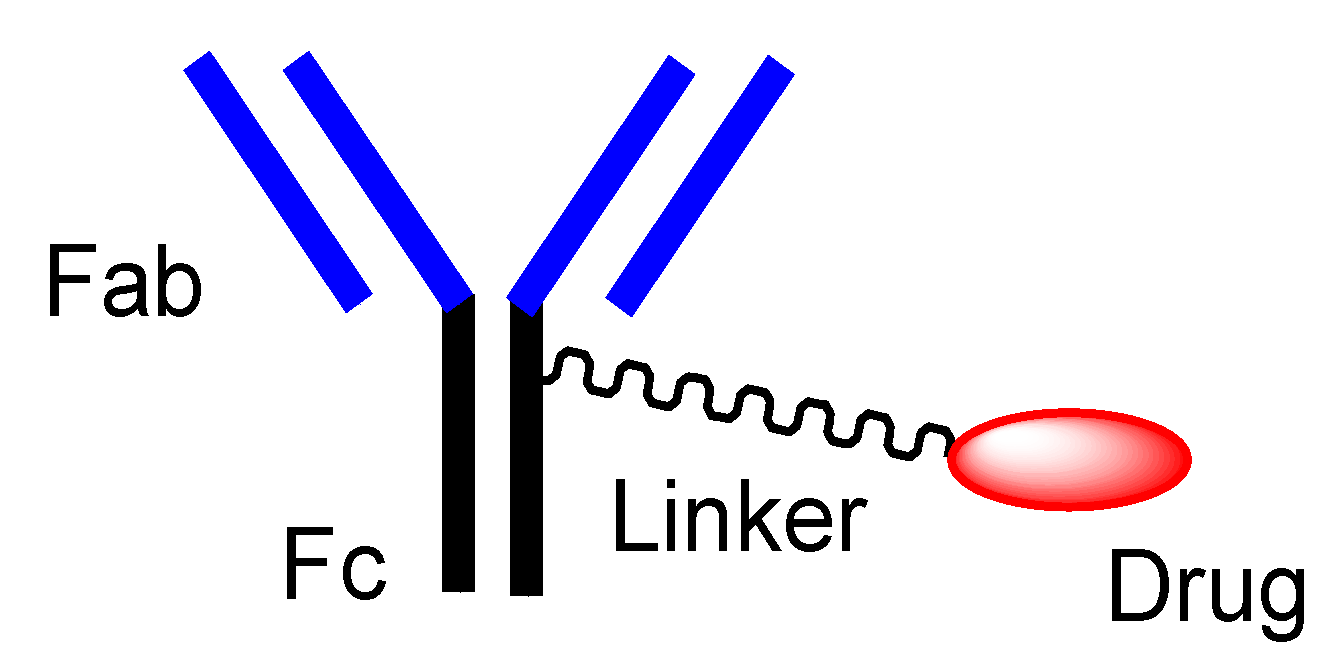
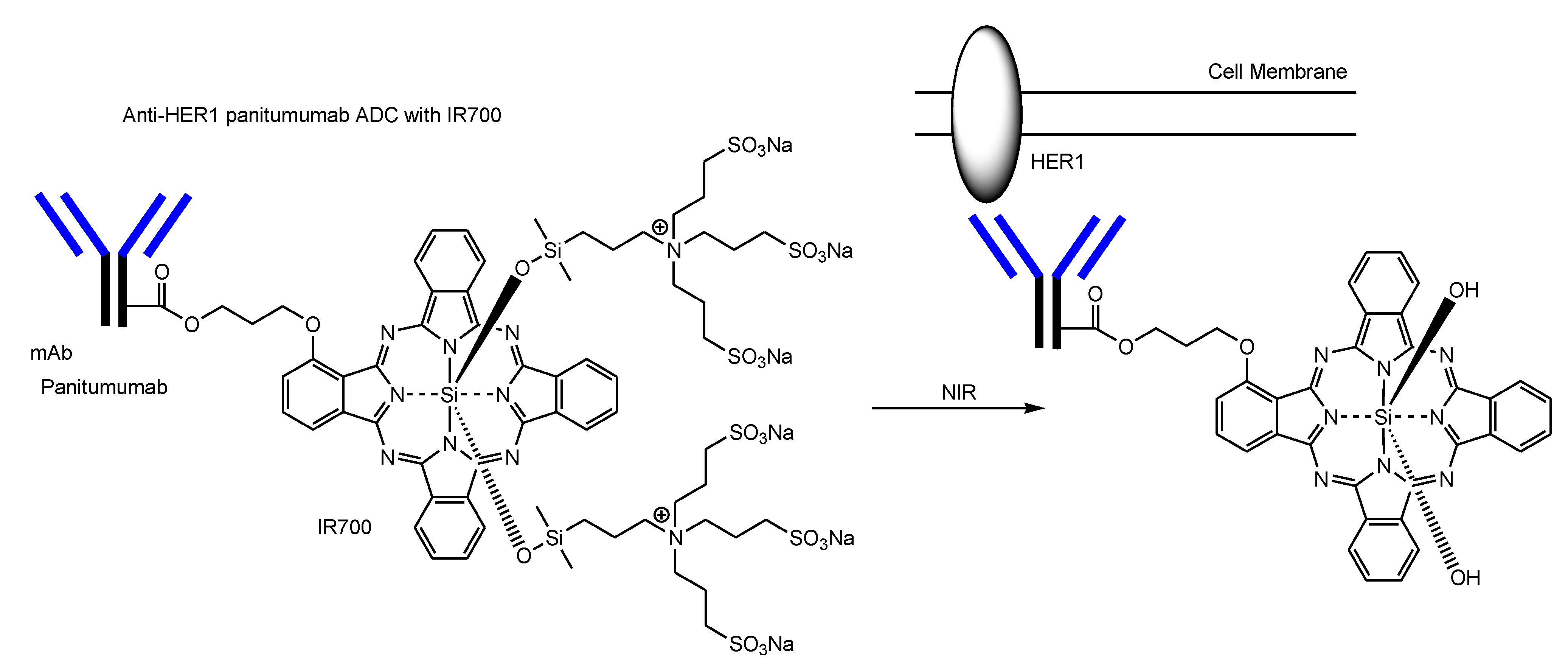
2.4. ADCs for Cancer Therapy
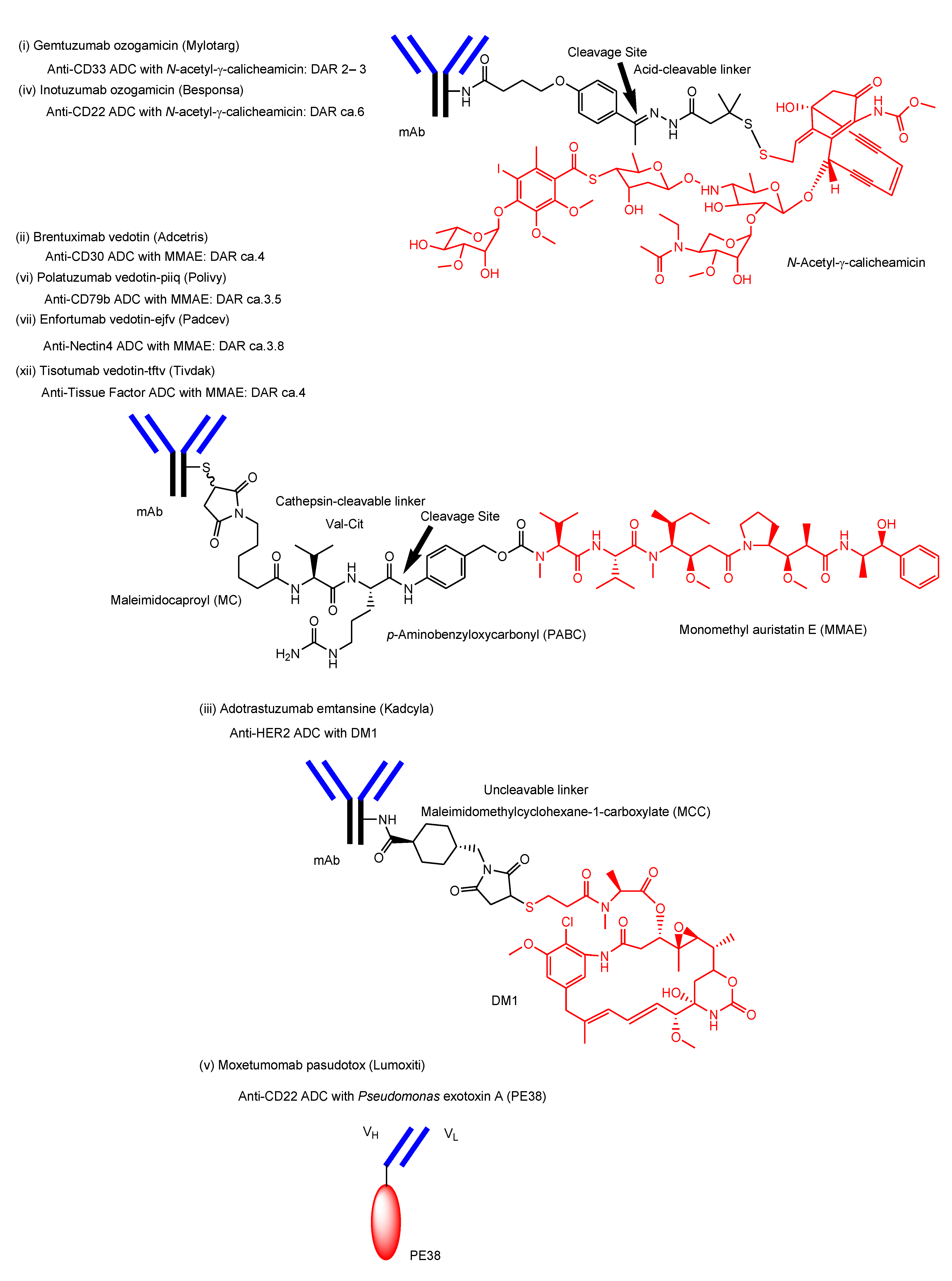
- i.
- Gemtuzumab ozogamicin (Mylotarg®) against CD33, approved for blood cancer in 2000 and 2017, was endocytosed through the endosome/lysosome pathway in CD33-expressing HL-60 cells [19]. The endosomal and/or lysosomal escape mechanism of N-acetyl-γ-calicheamicin (MW approximately 1410) was unclarified. Calicheamicin γ1, structurally related to N-acetyl-γ-calicheamicin, showed a 45-fold less efficient cleavage of cellular DNA at 0 °C, compared to 37 °C, due to poor cell permeability at a low temperature [61]. Thus, it was suggested that N-acetyl-γ-calicheamicin was unlikely to cross the plasma membrane via passive diffusion, while it might cross the membrane energy-dependently via carrier-mediated transport or via a type of endocytosis. Since the sugar residues of N-acetyl-γ-calicheamicin are involved in DNA interaction, it would retain sugar residues and alkynes after endosomal and/or lysosomal escape. Probably, N-acetyl-γ-calicheamicin is structurally stable under weak acid because of the usage of an acid-cleavable linker. Carrier-mediated transport might be its escape mechanism, although its molecular weight is relatively large, compared to lysine-MCC-DM1 (MW1089.69).
- ii.
- iii.
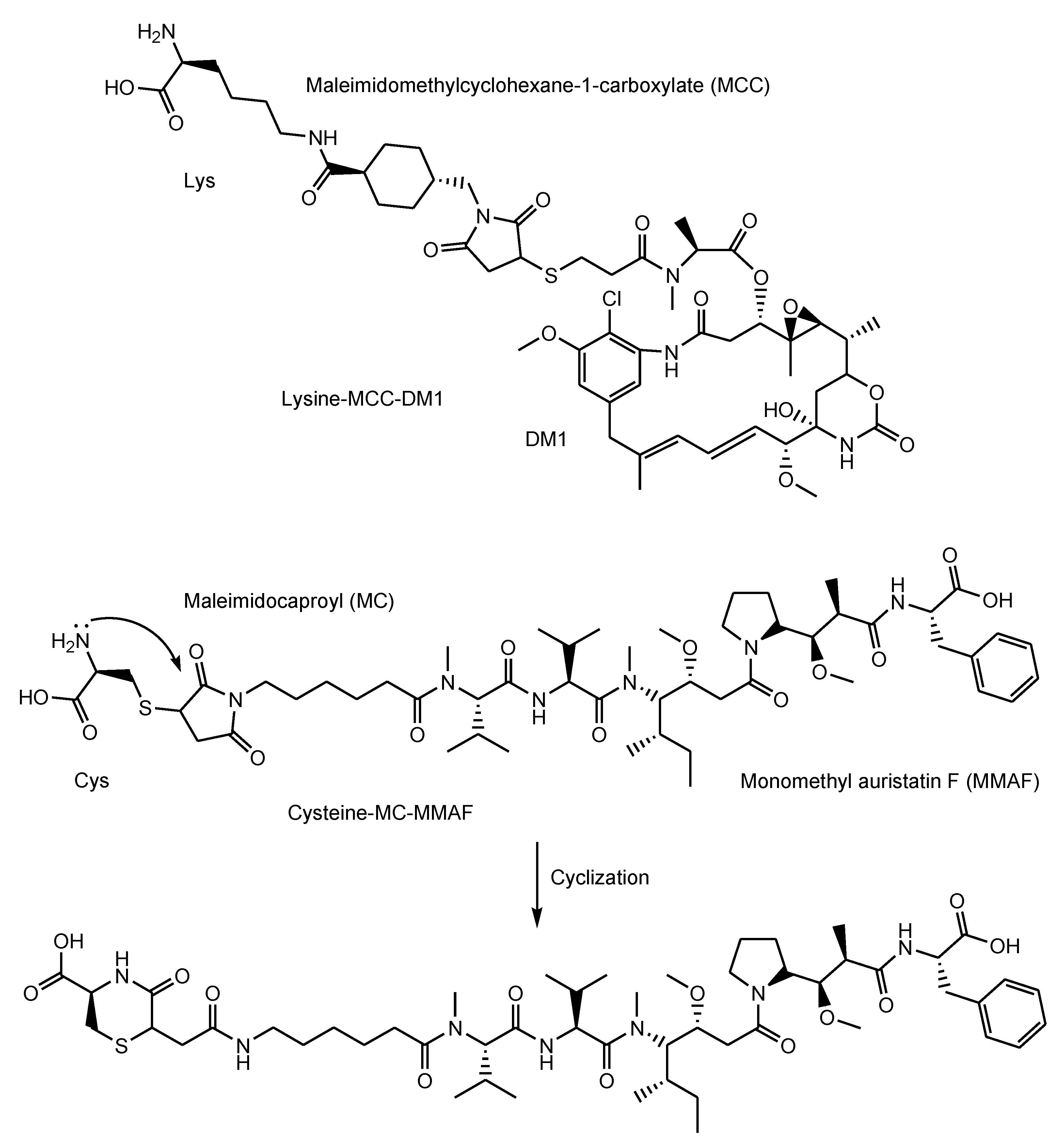
- Adotrastuzumab emtansine (Kadcyla®) against HER2, approved for solid cancers such as breast cancer in February 2013, entered the cell via RME and released lysine-MCC-DM1 (Figure 4) as a catabolite to the cytoplasm through carrier-mediated transport using the lysosomal transporter SLC46A3 [54] in the lysosomal escape process [24].
- iv.
- Inotuzumab ozogamicin (Besponsa®) against CD22 with an acid-cleavable linker, approved for blood cancer in 2017, was internalized into cells and released a potent cytotoxic agent, N-acetyl-γ-calicheamicin, to the cytoplasm and the nucleus after lysosomal escape [25].
- v.
- Moxetumomab pasudotox-tdfk (Lumoxiti®) against CD22, approved for blood cancer in 2018, was internalized through clathrin-coated pits into the endocytic compartment. This is structurally an anti-CD22 immunoglobulin variable domain genetically joined to Pseudomonas exotoxin (PE38) as a payload, although it is not a canonical ADC. PE38 was cleaved by the disulfide bond reduction in the endosome and was released to the cytoplasm by way of the endoplasmic reticulum [26]. It was unknown what the endosomal escape of the cleaved PE38 was like. In the future, different formats of Ab fragments and their derivatives, such as nanobodies (approximately 15 kDa) known as single-domain Abs or variable fragments of heavy-chain (VHH) domains, will be developed in ADCs [62].
- vi.
- Polatuzumab vedotin-piiq (Polivy®) against CD79b with an enzymatically cleavable linker, approved for blood cancer in 2019, entered cells and released a potent cytotoxic agent, MMAE, into the cytoplasm after lysosomal escape via passive diffusion [27].
- vii.
- Enfortumab vedotin-ejfv (Padcev®) against Nectin4, approved for solid cancers such as urothelial cancer in 2019, was intracellularly internalized by endocytosis and was degraded in a lysosome to subsequently release the cytotoxic payload MMAE [28].
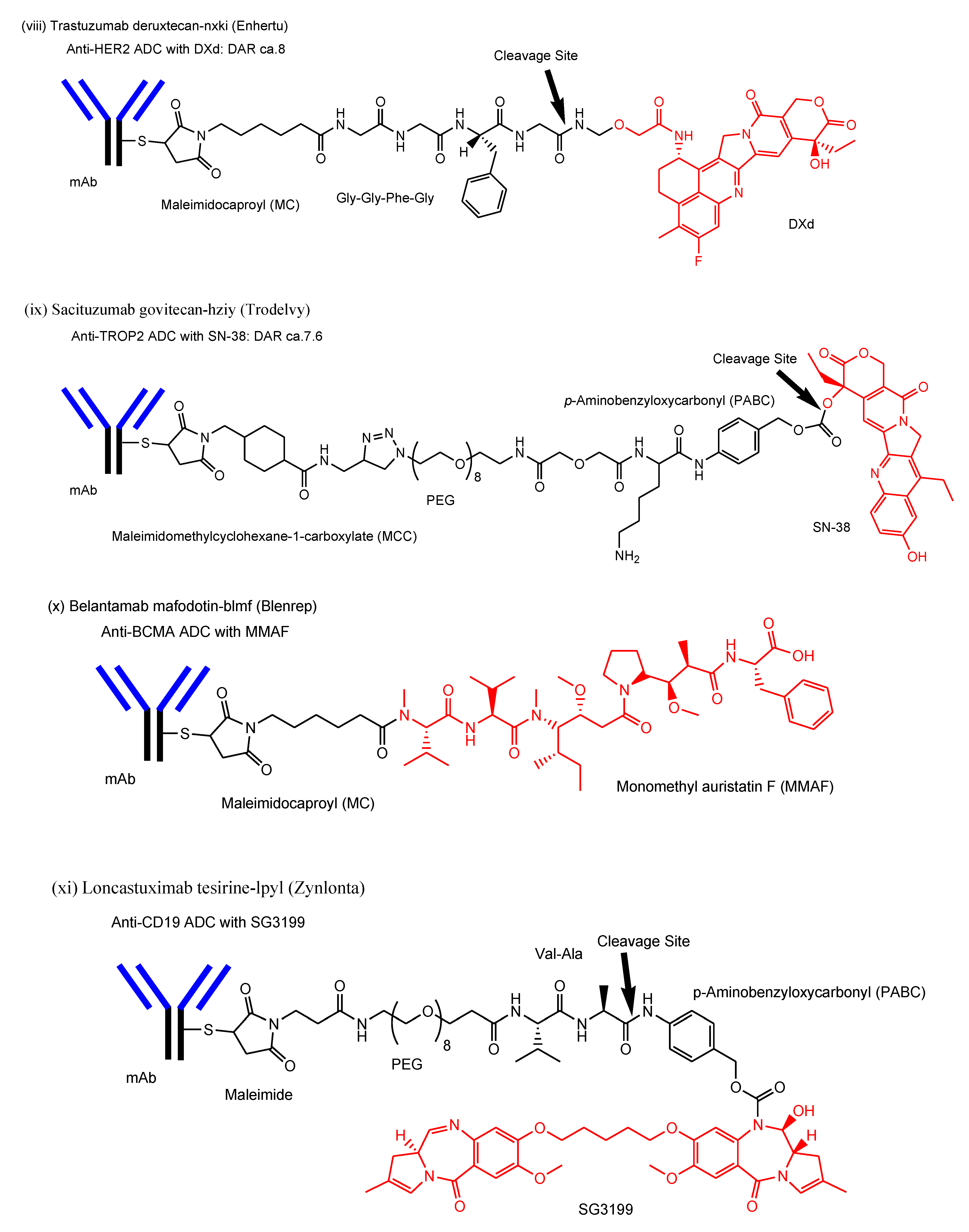
- viii.
- Trastuzumab deruxtecan-nxki (Enhertu®) against HER2, approved for solid cancers such as breast cancer in 2019, underwent endocytosis by binding to HER2-positive tumor cells and released the payload deruxtecan (DXd) by lysosomal cathepsins [29]. DXd demonstrated passive diffusion across the membrane [63].
- ix.
- Sacituzumab govitecan-hziy (Trodelvy®) against TROP2, approved for solid cancers such as breast cancer in 2020, was internalized via RME. The payload SN-38 was released by double ester hydrolysis of the CL2A linker at low pH within lysosomes [30,31,32]. It was revealed that SN-38 crossed the plasma apical membrane via carrier-mediated transport using transporters different from organic anion-transporting polypeptides (OATP) and the monocarboxylate transporter (MCT) in Caco-2 cells [64]. Thus, it was suggested that SN-38 was transported from lysosomes into the cytoplasm via carrier-mediated transport.
- x.
- Belantamab mafodotin-blmf (Blenrep®) against BCMA, approved for blood cancer in 2020, was probably endocytosed clathrin-dependently by binding cell-surface BCMAs. It was supported by the fact that it possessed the cytotoxic payload auristatin F (MMAF) [33]. MMAF was released via proteolytic cleavage, as cysteine-maleimidocaproyl (MC)-MMAF (Figure 4) that became further the six-membered cyclic form derived from cysteine and maleimido by the intramolecular nucleophilic substitution of the amino group to the ketone (Figure 4) [65]. Positively charged cysteine-MC-MMAF under physiological pH is not thought to be membrane-permeable via passive diffusion [66]. Similarly, it would be positively charged under a pH of approximately 4.5 in lysosomes. However, the cyclic form of cysteine-MC-MMAF lost the amino group and could be transported across the lysosomal membrane via passive diffusion, although cysteine-MC-MMAF might be a substrate of arbitrary lysosomal transporters such as SLC46A3 for lysine-MCC-DM1.
- xi.
- Loncastuximab tesirine-lpyl (Zynlonta®) against CD19, approved for blood cancer in 2021, was internalized via RME and released the cytotoxic molecule SG3199 by lysosomal proteolysis and the subsequent self-motivated degradation of the linker [34]. MDR1 decreased the permeability of pyrrolobenzodiazepin (PBD) dimers such as SJG-136 and DRG-16, which were structurally related derivatives of SG3199, across the cell membrane in Caco-2 cells [67]. Thus, SG3199 was suggested to be transported across the membrane via passive diffusion.
- xii.
- Tisotumab vedotin-tftv (Tivdak®) against Tissue Factor, approved for solid cancers such as cervical cancer on 20 September 2021, was thought to be endocytosed and released the cytotoxic payload MMAE [35].
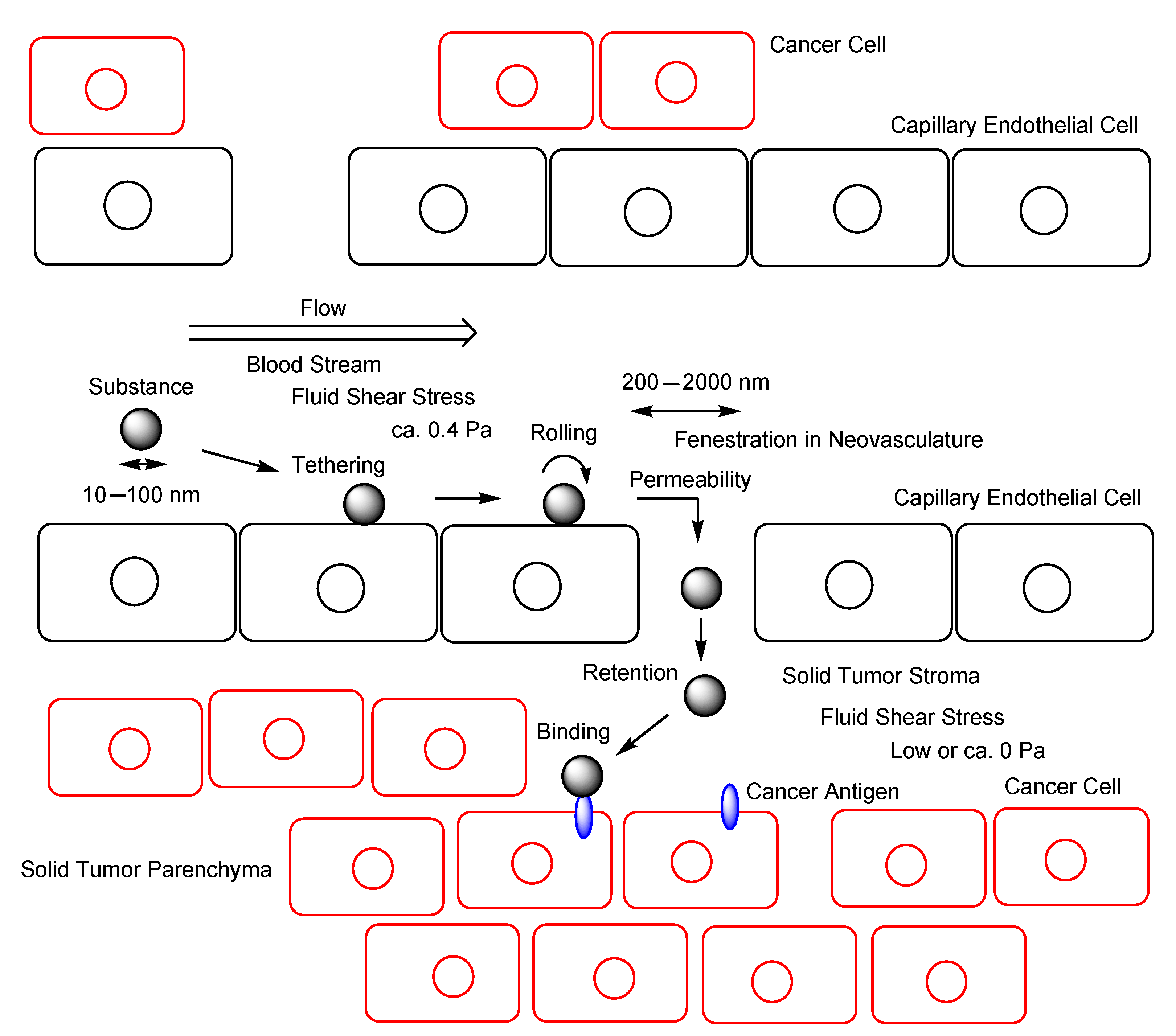
2.5. EPR Effect
2.6. Implementation of EPR Effect
2.7. Promising ADCs for Cancer Therapy
2.7.1. Approaches That Increase the Number of ADC Molecules in Solid Tumor Parenchyma
2.7.2. Approaches That Increase the ADC Molecular Size to Up the Probability of Collision
2.7.3. Approaches That Render Solid Tumor Parenchyma Leakier
2.8. Nanobody–Drug Conjugates
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Torrice, M. Does nanomedicine have a delivery problem? ACS Cent. Sci. 2016, 2, 434–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashima, T. Intriguing possibilities and beneficial aspects of transporter-conscious drug design. Bioorg. Med. Chem. 2015, 23, 4119–4131. [Google Scholar] [CrossRef] [PubMed]
- Tashima, T. Intelligent substance delivery into cells using cell-penetrating peptides. Bioorg. Med. Chem. Lett. 2017, 27, 121–130. [Google Scholar] [CrossRef]
- Tashima, T. Effective cancer therapy based on selective drug delivery into cells across their membrane using receptor-mediated endocytosis. Bioorg. Med. Chem. Lett. 2018, 28, 3015–3024. [Google Scholar] [CrossRef]
- Tashima, T. Smart Strategies for Therapeutic Agent Delivery into Brain across the Blood-Brain Barrier Using Receptor-Mediated Transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Tashima, T. Shortcut Approaches to Substance Delivery into the Brain Based on Intranasal Administration Using Nanodelivery Strategies for Insulin. Molecules 2020, 25, 5188. [Google Scholar] [CrossRef]
- Tashima, T. Delivery of Orally Administered Digestible Antibodies Using Nanoparticles. Int. J. Mol. Sci. 2021, 22, 3349. [Google Scholar] [CrossRef]
- Tashima, T. Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis. Pharmaceutics 2022, 14, 411. [Google Scholar] [CrossRef]
- Tashima, T. Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates. Biomedicines 2022, 10, 1597. [Google Scholar] [CrossRef]
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 28 November 2022).
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, C.D.; D’Aquili, E.G. Biogenetic Structuralism; Columbia University Press: New York, NY, USA, 1974. [Google Scholar]
- Leavy, S.A. Biogenetic Structuralism. Yale J. Biol. Med. 1976, 49, 420–421. [Google Scholar]
- Cheng, J.; Liang, M.; Carvalho, M.F.; Tigue, N.; Faggioni, R.; Roskos, L.K.; Vainshtein, I. Molecular Mechanism of HER2 Rapid Internalization and Redirected Trafficking Induced by Anti-HER2 Biparatopic Antibody. Antibodies 2020, 9, 49. [Google Scholar] [CrossRef]
- Jones, S.; King, P.J.; Antonescu, C.N.; Sugiyama, M.G.; Bhamra, A.; Surinova, S.; Angelopoulos, N.; Kragh, M.; Pedersen, M.W.; Hartley, J.A.; et al. Targeting of EGFR by a combination of antibodies mediates unconventional EGFR trafficking and degradation. Sci. Rep. 2020, 10, 663. [Google Scholar] [CrossRef] [Green Version]
- Gunturi, A.; McDermott, D.F. Nivolumab for the treatment of cancer. Expert Opin. Investig. Drugs 2015, 24, 253–260. [Google Scholar] [CrossRef]
- Kozani, P.S.; Naseri, A.; Kozani, P.S.; Khatami, S.; Sheikhi, A. Monoclonal Antibodies (mAbs) Approved for Cancer Treatment in the 2020s. Trends Med. Sci. 2021, 1, e116686. [Google Scholar] [CrossRef]
- Mylotarg European Public Assessment Report EMA/155284/2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/mylotarg-epar-public-assessment-report_en.pdf (accessed on 3 August 2022).
- Diefenbach, C.S.; Leonard, J.P. Targeting CD30 in Hodgkin Lymphoma: Antibody-Drug Conjugates Make a Difference. Am. Soc. Clin. Oncol. Educ. Book 2012, 32, 162–166. [Google Scholar] [CrossRef]
- Donk, N.W.V.D.; Dhimolea, E. Brentuximab vedotin. MAbs 2012, 4, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Powell Gray, B.; Kelly, L.; Ahrens, D.P.; Barry, A.P.; Kratschmer, C.; Levy, M.; Sullenger, B.A. Tunable cytotoxic aptamer–drug conjugates for the treatment of prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 4761–4766. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 is required to transport catabolites of noncleavable antibody maytansine conjugates from the lysosome to the cytoplasm. Cancer Res. 2015, 75, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Thota, S.; Advani, A. Inotuzumab ozogamicin in relapsed B-cell acute lymphoblastic leukemia. Eur. J. Haematol. 2017, 98, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.Y.; Dinner, N.S. Moxetumomab pasudotox for hairy cell leukemia: Preclinical development to FDA approval. Blood Adv. 2019, 3, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawalha, Y.; Maddocks, K. Profile of Polatuzumab Vedotin in the Treatment of Patients with Relapsed/Refractory Non-Hodgkin Lymphoma: A Brief Report on the Emerging Clinical Data. Onco Targets Ther. 2020, 13, 5123–5133. [Google Scholar] [CrossRef]
- Halford, Z.; Anderson, M.K.; Clark, M.D. Enfortumab Vedotin-ejfv: A First-in-Class Anti–Nectin-4 Antibody-Drug Conjugate for the Management of Urothelial Carcinoma. Ann. Pharmacother. 2021, 55, 772–782. [Google Scholar] [CrossRef]
- Azar, I.; Alkassis, S.; Fukui, J.; Alsawah, F.; Fedak, K.; Al Hallak, M.N.; Sukari, A.; Nagasaka, M. Spotlight on Trastuzumab Deruxtecan (DS-8201,T-DXd) for HER2 Mutation Positive Non-Small Cell Lung Cancer. Lung Cancer 2021, 12, 103–114. [Google Scholar] [CrossRef]
- Nonneville, A.D.; Goncalves, A.; Mamessier, E.; Bertucci, F. Sacituzumab govitecan in triple-negative breast cancer. Ann. Transl. Med. 2022, 10, 647. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Tolaney, S.M. Clinical Development of New Antibody–Drug Conjugates in Breast Cancer: To Infnity and Beyond. BioDrugs 2021, 35, 159–174. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- Calabretta, E.; Hamadani, M.; Zinzani, P.L.; Caimi, P.; Carlo-Stella, C. The antibody-drug conjugate loncastuximab tesirine for the treatment of diffuse large B-cell lymphoma. Blood 2022, 140, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; Toki, T.; Yamaguchi, J.; Kitamura, M.; Kamei, R.; et al. Datopotamab Deruxtecan, a Novel TROP2-directed Antibody-drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Mol. Cancer Ther. 2021, 20, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Baik, C.; Su, W.C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2022, 12, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Johnson, M.L.; Falchook, G.S.; Doi, T.; Friedman, C.F.; Piha-Paul, S.A.; Gutierrez, M.; Shimizu, T.; Cheng, B.; Qian, M.; et al. DS-7300 (B7-H3 DXd-ADC) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): A subgroup analysis of a phase 1/2 multicenter study. J. Clin. Oncol. 2022, 40, 87. [Google Scholar] [CrossRef]
- Murakami, M.; Tokui, T.; Nakamaru, K.; Cogswell, J.; Ford, S.K.; Gallant, G. Expanded precision medicine capabilities for strengthening oncology drug development at Daiichi Sankyo. J. Precis. Med. 2021, 7, 14–19. Available online: https://www.thejournalofprecisionmedicine.com/wp-content/uploads/expanded-precision.pdf (accessed on 19 October 2022).
- Kimoto, A.; Watanabe, A.; Yamamoto, E.; Higashi, T.; Kato, M. Rapid Analysis of DOXIL Stability and Drug Release from DOXIL by HPLC Using a Glycidyl Methacrylate-Coated Monolithic Column. Chem. Pharm. Bull. 2017, 65, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Takahara, T.; Mukai, H. History of the development of DOXIL. Drug Deliv. Syst. 2013, 28, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.-C.; Burnouf, P.-A.; Chung, K.-H.; Chen, B.-M.; Cheng, T.-L.; Roffler, S.R. Conditional internalization of PEGylated nanomedicines by PEG engagers for triple negative breast cancer therapy. Nat. Commun. 2017, 8, 15507. [Google Scholar] [CrossRef] [Green Version]
- Xenaki, K.T.; Dorrestijn, B.; Muns, J.A.; Adamzek, K.; Doulkeridou, S.; Houthoff, H.; Oliveira, S.; van Bergen En Henegouwen, P.M. Homogeneous tumor targeting with a single dose of HER2-targeted albumin-binding domain-fused nanobody-drug conjugates results in long-lasting tumor remission in mice. Theranostics 2021, 11, 5525–5538. [Google Scholar] [CrossRef]
- Wouters, Y.; Jaspers, T.; Strooper, B.D.; Dewilde, M. Identification and in vivo charac-terization of a brain-penetrating nano-body. Fluids Barriers CNS 2020, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and Function of Immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankarious, S.; Lee, M.; Fischer, S.; Pyun, K.H.; Ochs, H.D.; Oxelius, V.A.; Wedgwood, J.W. The half-lives of IgG subclasses and specific antibodies in patients with primary immunodeficiency who are receiving intravenously administered immunoglobulin. J. Lab. Clin. Med. 1988, 112, 634–640. [Google Scholar]
- Vattepu, R.; Sneed, S.L.; Anthony, R.M. Sialylation as an Important Regulator of Antibody Function. Front. Immunol. 2022, 13, 818736. [Google Scholar] [CrossRef]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs 2018, 10, 693–711. [Google Scholar] [CrossRef] [Green Version]
- Bas, M.; Terrier, A.; Jacque, E.; Dehenne, A.; Pochet-Béghin, V.; Beghin, C.; Dezetter, A.S.; Dupont, G.; Engrand, A.; Beaufils, B.; et al. Fc Sialylation Prolongs Serum Half-Life of Therapeutic Antibodies. J. Immunol. 2019, 202, 1582–1594. [Google Scholar] [CrossRef] [Green Version]
- Wada, W.; Matsui, M.; Kawasaki, N. Influence of N-glycosylation on effector functions and thermal stability of glycoengi-neered IgG1 monoclonal antibody with homogeneous glycoforms. MAbs 2019, 11, 350–372. [Google Scholar] [CrossRef]
- Gelpi, A.; Gilbertson, A.; Tucker, J.D. Magic bullet: Paul Ehrlich, Salvarsan and the birth of venereology. Sex. Transm. Infect. 2015, 91, 68–69. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T. Current situations and the future prospect of monoclonal antibody products. Bull. Natl. Inst. Health Sci. 2014, 132, 36–46. Available online: http://www.nihs.go.jp/library/eikenhoukoku/2014/036-046.pdf (accessed on 1 August 2022).
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.H.; Chen, X. Monoclonal antibodies for cancer therapy approved by FDA. MOJ Immunol. 2016, 4, 11–12. [Google Scholar] [CrossRef] [Green Version]
- Goydel, R.S.; Rader, C. Antibody-based cancer therapy. Oncogene 2021, 40, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)—A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef] [PubMed]
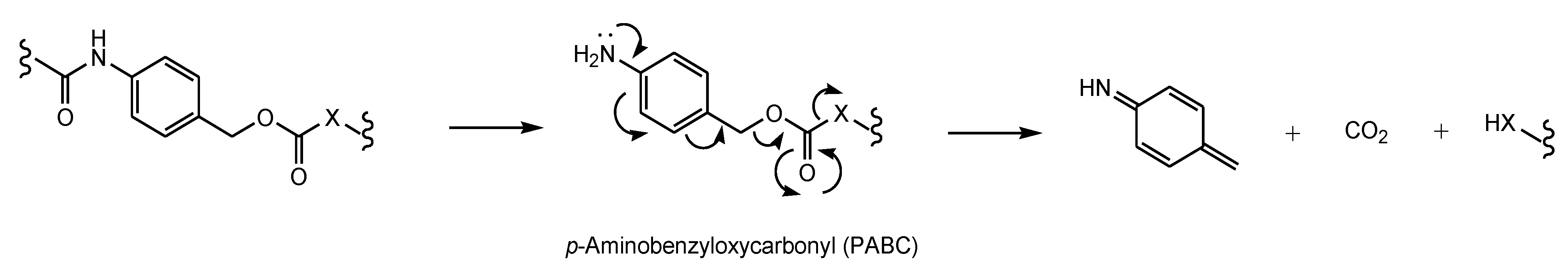
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Edupuganti, V.V.S.R.; Tyndall, J.D.; Gamble, A.B. Self-immolative Linkers in Prodrugs and Antibody Drug Conjugates in Cancer Treatment. Recent Pat. Anti Cancer Drug Discov. 2021, 16, 479–497. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Elmroth, K.; Nygren, J.; Mårtensson, S.; Ismail, I.H.; Hammarsten, O. Cleavage of cellular DNA by calicheamicin gamma1. DNA Repair 2003, 2, 363–374. [Google Scholar] [CrossRef]
- Kang, W.; Ding, C.; Zheng, D.; Ma, X.; Yi, L.; Tong, X.; Wu, C.; Xue, C.; Yu, Y.; Zhou, Q. Nanobody Conjugates for Targeted Cancer Therapy and Imaging. Technol. Cancer Res. Treat. 2021, 20, 15330338211010117. [Google Scholar] [CrossRef]
- Conilh, L.; Fournet, G.; Fourmaux, E.; Murcia, A.; Matera, E.-L.; Joseph, B.; Dumontet, C.; Viricel, W. Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform. Pharmaceuticals 2021, 14, 247. [Google Scholar] [CrossRef]
- Itagaki, S. Intestinal Absorption and Secretion Mechanism of Carboxylate Drugs. Yakugaku Zasshi 2009, 129, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.P.; Licea-Perez, H.; Boram, S.; Pannullo, K.E.; Kehler, J.; Evans, C.A. Overcoming challenges associated with the bioanalysis of cysteine-conjugated metabolites in the presence of antibody–drug conjugates. Bioanalysis 2021, 13, 18. [Google Scholar] [CrossRef]
- Thompson, J.A.; Motzer, R.J.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; Kim, S.K.; et al. Phase I Trials of Anti-ENPP3 Antibody-Drug Conjugates in Advanced Refractory Renal Cell Carcinomas. Clin. Cancer Res. 2018, 24, 4399–4406. [Google Scholar] [CrossRef] [Green Version]
- Kaliszczak, M.; Antonow, D.; Patel, K.I.; Howard, P.; Jodrell, D.I.; Guichard, S.M.; Thurston, D.E. Optimization of the Antitumor Activity of Sequence-specific Pyrrolobenzodiazepine Derivatives Based on their Affinity for ABC Transporters. AAPS J. 2010, 12, 617–627. [Google Scholar] [CrossRef]
- The FDA. Novel Drug Approvals for 2021. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2021 (accessed on 1 August 2022).
- The FDA. Novel Drug Approvals for 2022. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2022 (accessed on 26 October 2022).
- Zhao, P.; Zhang, Y.; Li, W.; Jeanty, C.; Xiang, G.; Dong, Y. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm. Sin. B 2020, 10, 1589–1600. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving Conventional Enhanced Permeability and Retention (EPR) Effects; What Is the Appropriate Target? Theranostics 2014, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Roberts, W.G.; Palade, G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57, 765–772. [Google Scholar]
- Yadav, K.S.; Mishra, D.K.; Deshpande, A.; Pethe, A.M. Chapter 7—Levels of Drug Targeting. In Basic Fundamentals of Drug Delivery; Academic Press: Cambridge, MA, USA, 2019; pp. 269–305. [Google Scholar] [CrossRef]
- Shurbaji, S.; Anlar, G.G.; Hussein, E.A.; Elzatahry, A.; Yalcin, H.C. Effect of Flow-Induced Shear Stress in Nanomaterial Uptake by Cells: Focus on Targeted Anti-Cancer Therapy. Cancers 2020, 12, 1916. [Google Scholar] [CrossRef]
- Samuel, S.P.; Jain, N.; O’Dowd, F.; Paul, T.; Kashanin, D.; Gerard, V.A.; Gun’ko, Y.K.; Prina-Mello, A.; Volkov, Y. Multifactorial determinants that govern nanoparticle uptake by human endothelial cells under flow. Int. J. Nanomed. 2012, 7, 2943–2956. [Google Scholar] [CrossRef] [Green Version]
- Papademetriou, I.; Vedula, E.; Charest, J.; Porter, T. Effect of flow on targeting and penetration of angiopep-decorated nanoparticles in a microfluidic model blood-brain barrier. PLoS ONE 2018, 13, e0205158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, C.F.; Verbridge, S.S.; Vlachos, P.P.; Rylander, M.N. Flow shear stress regulates endothelial barrier function and expression of angiogenic factors in a 3D microfluidic tumor vascular model. Cell Adhes. Migr. 2014, 8, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasunaga, M.; Manabe, S.; Matsumura, Y. Tumor stromal barrier and cancer stromal targeting therapy. Microvasc. Rev. Commun. 2013, 6, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Speelmans, G.; Staffhorst, R.W.; Steenbergen, H.G.; Kruijff, B.D. Transport of the anti-cancer drug doxorubicin across cytoplasmic membranes and membranes composed of phospholipids derived from Escherichia coli occurs via a similar mechanism. Biochim. Biophys. Acta 1996, 1284, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Creemers, S.G.; Koetsveld, P.M.V.; Herder, W.W.D.; Dogan, F.; Franssen, G.J.H.; Feelders, R.A.; Hofland, L.J. MDR1 inhibition increases sensitivity to doxorubicin and etoposide in adrenocortical cancer. Endocr. Relat. Cancer 2019, 26, 367–378. [Google Scholar] [CrossRef]
- Jobin, M.-L.; Alves, I.D. On the importance of electrostatic interactions between cell penetrating peptides and membranes: A pathway toward tumor cell selectivity? Biochimie 2014, 107, 154–159. [Google Scholar] [CrossRef]
- Bechara, C.; Pallerla, M.; Zaltsman, Y.; Burlina, F.; Alves, I.D.; Lequin, O.; Sagan, S. Tryptophan within basic peptide sequences triggersglycosaminoglycan-dependent endocytosis. FASEB J. 2013, 27, 738–749. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Wu, F.-G. Antibody-Incorporated Nanomedicines for Cancer Therapy. Adv. Mater. 2022, 34, e2109210. [Google Scholar] [CrossRef]
- Roberts, M.J.; Bentley, M.D.; Harris, J.M. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 2012, 64, 116–127. [Google Scholar] [CrossRef]
- Adhipandito, C.F.; Cheung, S.-H.; Lin, Y.-H.; Wu, S.-H. Atypical Renal Clearance of Nanoparticles Larger than the Kidney Filtration Threshold. Int. J. Mol. Sci. 2021, 22, 11182. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Berghoff, K.; Kress, H. Phagosomal transport depends strongly on phagosome size. Sci. Rep. 2017, 7, 17068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef] [PubMed]
- Doshi, N.; Mitragotri, S. Macrophages recognize size and shape of their targets. PLoS ONE 2010, 5, e10051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef]
- Chapman, A.P. PEGylated antibodies and antibody fragments for improved therapy: A review. Adv. Drug Deliv. Rev. 2002, 54, 531–545. [Google Scholar] [CrossRef]
- Natarajan, A.; DeNardo, S.J. PEGylation of Antibody Fragments to Improve Pharmacodynamics and Pharmacokinetics. In Antibody Engineering; Springer Protocols Handbooks, Kontermann, R., Dübel, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 191–205. [Google Scholar] [CrossRef]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [Green Version]
- Rozga, J.; Piątek, T.; Małkowski, P. Human albumin: Old, new, and emerging applications. Ann. Transplant. 2013, 18, 205–217. [Google Scholar] [CrossRef]
- Katona, G.; Balogh, G.T.; Dargó, G.; Gáspár, R.; Márki, Á.; Ducza, E.; Sztojkov-Ivanov, A.; Tömösi, F.; Kecskeméti, G.; Janáky, T.; et al. Development of Meloxicam-Human Serum Albumin Nanoparticles for Nose-to-Brain Delivery via Application of a Quality by Design Approach. Pharmaceutics 2020, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, S.F.; Sharma, N.; Woodward, S.C. Disulfide linking of albumin to the hinge region of immunoglobulin G in normal human serum. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzymol. 1983, 749, 47–51. [Google Scholar] [CrossRef]
- Kawakami, A.; Kubota, K.; Yamada, N.; Tagami, U.; Takehana, T.; Sonaka, I.; Suzuki, I.; Hirayama, K. Identification and characterization of oxidized human serum albumin. FEBS J. 2006, 273, 3346–3357. [Google Scholar] [CrossRef] [PubMed]
- Turell, L.; Carballal, L.; Botti, H.; Radi, R.; Alvarez, B. Oxidation of the albumin thiol to sulfenic acid and its implications in the intravascular compartment. Braz. J. Med. Biol. Res. 2009, 42, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundaktepe, B.P.; Sozer, V.; Durmus, S.; Kocael, P.C.; Kundaktepe, F.O.; Papila, C.; Gelisgen, R.; Uzun, H. The evaluation of oxidative stress parameters in breast and colon cancer. Medicine 2021, 100, e25104. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell–selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. A Mechanism of Cancer Cell Cytotoxicity of Near-Infrared Photoimmunotherapy. J. Jpn. Soc. Laser Surg. Med. 2020, 41, 104–109. [Google Scholar] [CrossRef]
- Westphal, D.; Kluck, R.M.; Dewson, G. Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014, 21, 196–205. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Zhuang, X.; Yang, X.; Xu, Y.; Zhou, Z.; Pan, L.; Chen, S. A multivalent biparatopic EGFR-targeting nanobody drug conjugate displays potent anticancer activity in solid tumor models. Signal Transduct. Target. Ther. 2021, 6, 320. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Administrated Drug | Formulation | Cancer Antigen | Disease | Vector | Payload | Linker | Status | References |
|---|---|---|---|---|---|---|---|---|---|
| (1) | Anti-HER2 bispecific mAb | Anti-HER2 bispecific mAb targeting two non-overlapping epitopes on HER2 | HER2 | - | Anti-HER2 bispecific mAb | - | - | Basic research | [15] |
| (2) | Sym004 | The mixture of two anti-EGFR mAbs | EGFR | - | anti-EGFR mAb | - | - | Basic research | [16] |
| (3) | Nivolumab (Opdivo®) | Anti-PD-1 mAb | PD-1 | Metastatic lung squamous cell carcinoma | - | Nivolumab | - | Launched in 2014 | [17] |
| (4) | Isatuximab (Sarclisa®) | Anti-CD38 mAb | CD38 | Multiple myeloma | - | Isatuximab | - | Launched in 2020 | [18] |
| (5) | Tafasitamab (Monjuvi®) | Anti-CD19 mAb | CD19 | Diffuse large B cell lymphoma | - | Tafasitamab | - | Launched in 2020 | [18] |
| (6) | Naxitamab (Danyelza®) | Anti-GD2 mAb | GD2 | High-risk neuroblastoma and refractory osteomedullary disease | - | Naxitamab | - | Launched in 2020 | [18] |
| (7) | Dostarlimab (Jemperli®) | Anti-PD-1 mAb | PD-1 | Endometrial cancer | - | Dostarlimab | - | Launched in 2021 | [18] |
| (8) | Gemtuzumab ozogamicin (Mylotarg®) | Anti-CD33 ADC with N-acetyl-γ-calicheamicin | CD33 | Blood cancer | Gemtuzumab | N-acetyl-γ-calicheamicin | Acid-cleavable linker | Launched in 2000 and 2017 | [19] |
| (9) | Brentuximab vedotin (Adcetris®) | Anti-CD30 ADC with MMAE | CD30 | Blood cancer | Brentuximab | MMAE | Enzymatically cleavable linker | Launched in 2011 | [20,21,22] |
| (10) | Adotrastuzumab emtansine (Kadcyla®) | Anti-HER2 ADC with DM1 | HER2 | Breast cancer | Adotrastuzumab | DM1 | Uncleavable linker | Launched in 2013 | [23,24] |
| (11) | Inotuzumab ozogamicin (Besponsa®) | Anti-CD22 ADC with N-acetyl-γ-calicheamicin | CD22 | Blood cancer | Inotuzumab | N-acetyl-γ-calicheamicin | Acid-cleavable linker | Launched in 2017 | [25] |
| (12) | Moxetumomab pasudotox-tdfk (Lumoxiti®) | Anti-CD22 ADC with PE38 | CD22 | Blood cancer | Moxetumomab | PE38 | Reductively cleavable linker | Launched in 2018 | [26] |
| (13) | Polatuzumab vedotin-piiq (Polivy®) | Anti-CD79b ADC with MMAE | CD79b | Blood cancer | Polatuzumab | MMAE | Enzymatically cleavable linker | Launched in 2019 | [27] |
| (14) | Enfortumab vedotin-ejfv (Padcev®) | Anti-Nectin4 ADC with MMAE | Nectin4 | Urothelial cancer | Enfortumab | MMAE | Enzymatically cleavable linker | Launched in 2019 | [28] |
| (15) | Trastuzumab deruxtecan-nxki (Enhertu®) | Anti-HER2 ADC with DXd | HER2 | Breast cancer | Trastuzumab | DXd | Enzymatically cleavable linker | Launched in 2019 | [29] |
| (16) | Sacituzumab govitecan-hziy (Trodelvy®) | Anti-TROP2 ADC with SN-38 | TROP2 | Breast cancer | Sacituzumab | SN-38 | Acid-cleavable linker | Launched in 2020 | [30,31,32] |
| (17) | Belantamab mafodotin-blmf (Blenrep®) | Anti-BCMA ADC with MMAF | BCMA | Blood cancer | Belantamab | MMAF | Uncleavable linker | Launched in 2020 | [33] |
| (18) | Loncastuximab tesirine-lpyl (Zynlonta®) | Anti-CD19 ADC with SG3199 | CD19 | Blood cancer | Loncastuximab | SG3199 | Enzymatically cleavable linker | Launched in 2021 | [34] |
| (19) | Tisotumab vedotin-tftv (Tivdak®) | Anti-Tissue Factor ADC withMMAE | Tissue Factor | Cervical cancer | Tisotumab | MMAE | Enzymatically cleavable linker | Launched in 2021 | [35] |
| (20) | Datopotamab deruxtecan (Dato-DXd) | Anti-ROP2 ADC | ROP2 | Solid cancer | Anti-ROP2 mAb | DXd | Linker | Clinical trial | [36] |
| (21) | Patritumab deruxtecan (HER3-DXd) | Anti-HER3 ADC | HER3 | Solid cancer | Anti-HER3 mAb | DXd | Linker | Clinical trial | [37] |
| (22) | DS-7300 | Anti-B7-H3 ADC | B7-H3 | Solid cancer | Anti-HER3 mAb | DXd | Linker | Clinical trial | [38] |
| (23) | DS-6000 | Anti-CDH6 ADC | CDH6 | Solid cancer | Anti-CDH6 | DXd | Linker | Clinical trial | [39] |
| (24) | DS-3939 | Anti-TA-MUC1 ADC | TA-MUC1 | Solid cancer | Anti-TA-MUC1 | DXd | Linker | Clinical trial | [39] |
| (25) | BYON3521 | Anti-c-MET receptor ADC | c-MET receptor | Solid cancer | Anti-c-MET receptor mAb | Duocarmycin | Cathepsin-cleavable linker | Phase1 (NCT05323045) | - |
| (26) | STRO-002 | Anti-folate receptor α ADC | Folate receptor α | Solid cancer | Anti-folate receptor α mAb | 3-Aminophenyl hemiasterlin | Cathepsin-cleavable linker | Phase1 (NCT03748186) | - |
| (27) | STI-6129 | Anti-CD38 ADC | CD38 | Solid cancer | Anti- CD38 mAb | Duostatin 5.2 | Non-polyethylene glycol linker | Phase1 (NCT05584709) | - |
| (28) | ARX788 | Anti-HER2 ADC | HER2 | Solid cancer | Anti-HER2 mAb | MMAF | Non-natural amino acid linker | Phase2 (NCT04983121) | - |
| (29) | MORAb-202 | Anti-folate receptor α ADC | Folate receptor α | Solid cancer | Anti-folate receptor α mAb | Eribulin | Cathepsin-cleavable linker | Phase2 (NCT05577715) | - |
| (30) | SYD985 | Anti-HER2 ADC | HER2 | Solid cancer | Anti-HER2 mAb | Duocarmycin | Cathepsin-cleavable linker | Phase2 (NCT04205630) | - |
| (31) | RC48 (disitamab vedotin) | Anti-HER2 ADC | HER2 | Solid cancer | Anti-HER2 mAb | Auristatin E | Cathepsin-cleavable linker | Phase2 (NCT04329429) | - |
| (32) | MRG002 | Anti-HER2 ADC | HER2 | Solid cancer | Anti-HER2 mAb | MMAE | Cathepsin-cleavable linker | Phase2 (NCT05263869) | - |
| (33) | XMT-1536 (upifitamab rilsodotin) | Anti-NaPi2b ADC | NaPi2b | Solid cancer | Anti-NaPi2b mAb | Auristatin F | Hydrophilic polymer linker | Phase3 (NCT05329545) | - |
| (34) | IMGN-853 (mirvetuximab soravtansine) | Anti-folate receptor α ADC | Folate receptor α | Solid cancer | Anti-folate receptor α mAb | DM4 | Disulfide-containing cleavable linker | Phase3 (NCT04296890) | - |
| (35) | Doxil® | Doxorubicin-encapsulated liposome coated with PEG | - | Ovarian cancer and breast cancer | - | Doxorubicin | - | Launched in 1999 and 2003 | [40,41] |
| (36) | PEG engagerEGFR, Doxisome | Anti-EGFR and anti-PEG bispecific Ab, PEGylated liposomes containing doxorubicin | EGFR | Solid cancer | Anti-EGFR and anti-PEG bispecific Ab | Doxorubicin | - | Basic research | [42] |
| (37) | Anti-HER2 nanobody 11A4 fused to an albumin-binding domain-maleimide-auristatin F | Anti-HER2 nanobody 11A4 fused to an albumin-binding domain with auristatin F | HER2 | Solid cancer | Anti-HER2 nanobody 11A4 | Auristatin F | Maleimide | Basic research | [43] |
| (38) | Anti-transferrin receptor nanobodies with neurotensin | Anti-transferrin receptor nanobodies with neurotensin | - | - | Anti-transferrin receptor nanobodies | Neurotensin | - | Basic research | [44] |
| (39) | Anti-EGFR nanobodies-drug | Anti-EGFR nanobodies with MMAE | EGFR | Solid cancer | Anti-EGFR nanobodies | MMAE | - | Basic research | - |
| (40) | ADC–albumin complex | ADC with or without PEGs | Arbitrary | Solid cancer | Arbitrary | Arbitrary | - | Under analysis in Tashima lab | - |
| (41) | mAb-loaded nanoparticles containing payloads | mAb-loaded nanoparticles containing payloads | Arbitrary | Solid cancer | Arbitrary | Arbitrary | - | Under analysis in Tashima lab | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashima, T. Delivery of Drugs into Cancer Cells Using Antibody–Drug Conjugates Based on Receptor-Mediated Endocytosis and the Enhanced Permeability and Retention Effect. Antibodies 2022, 11, 78. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11040078
Tashima T. Delivery of Drugs into Cancer Cells Using Antibody–Drug Conjugates Based on Receptor-Mediated Endocytosis and the Enhanced Permeability and Retention Effect. Antibodies. 2022; 11(4):78. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11040078
Chicago/Turabian StyleTashima, Toshihiko. 2022. "Delivery of Drugs into Cancer Cells Using Antibody–Drug Conjugates Based on Receptor-Mediated Endocytosis and the Enhanced Permeability and Retention Effect" Antibodies 11, no. 4: 78. https://0-doi-org.brum.beds.ac.uk/10.3390/antib11040078