Genomic Analysis of Antimicrobial Resistance and Resistance Plasmids in Salmonella Serovars from Poultry in Nigeria

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic and Genotypic Resistance
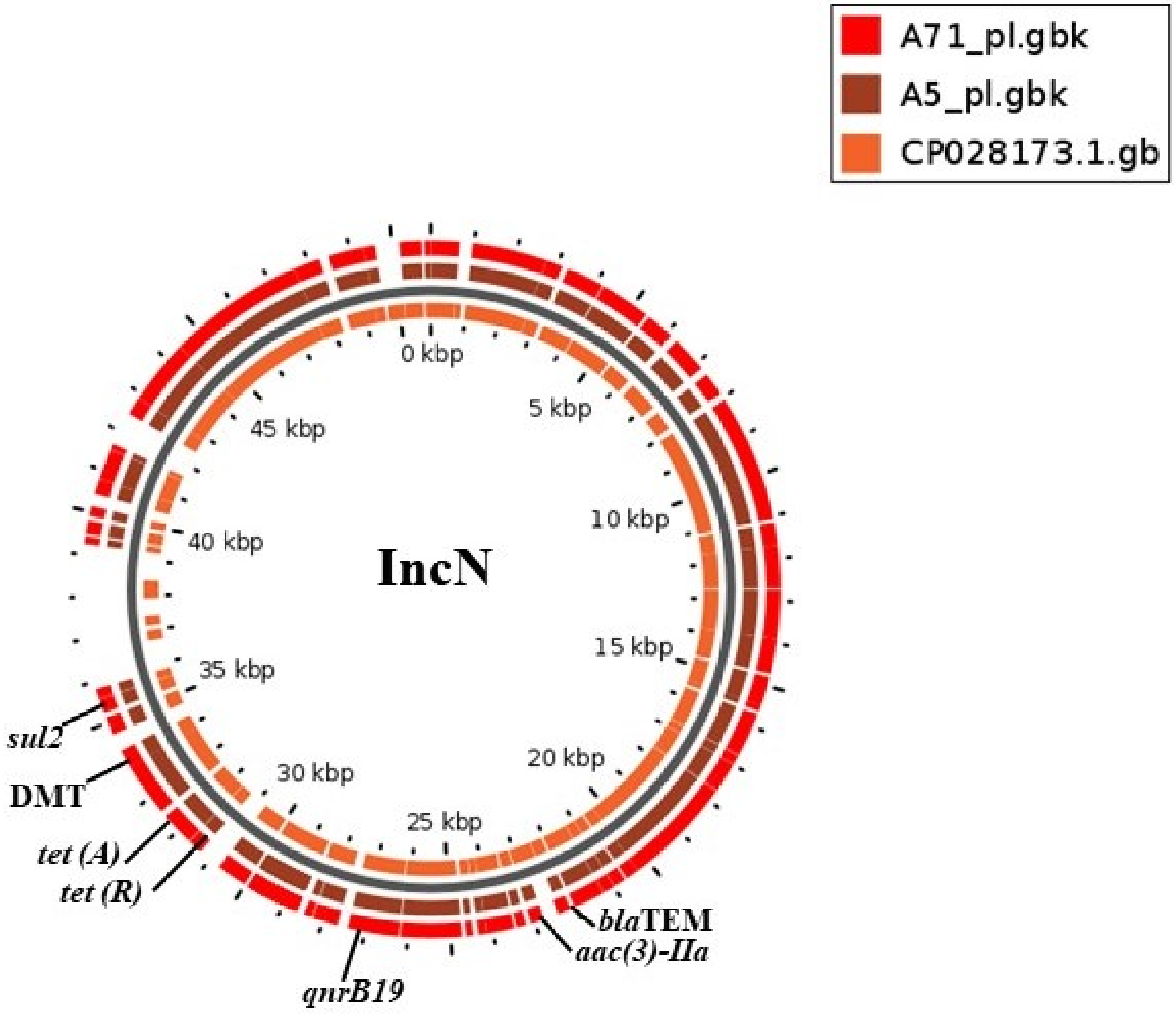
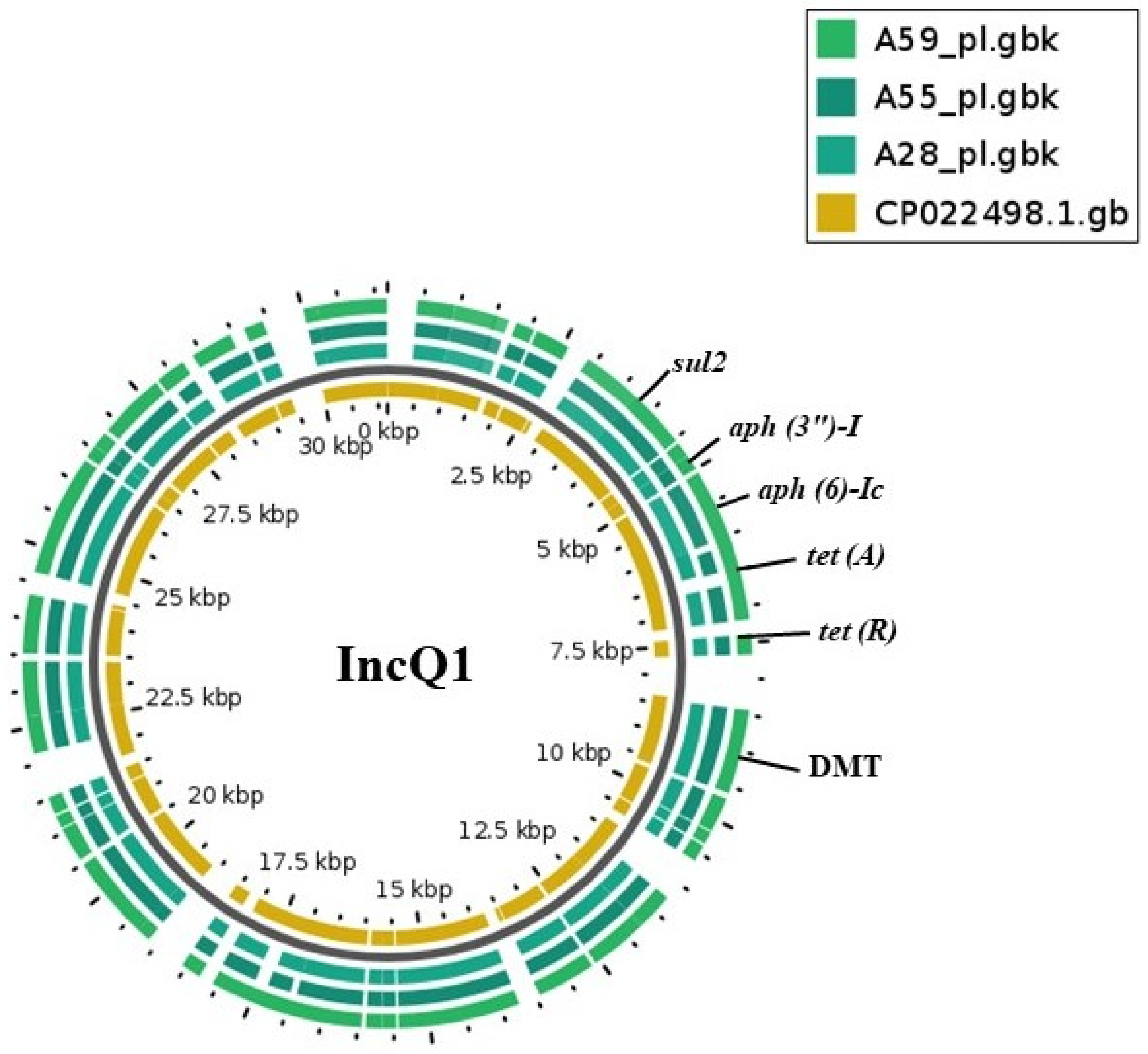
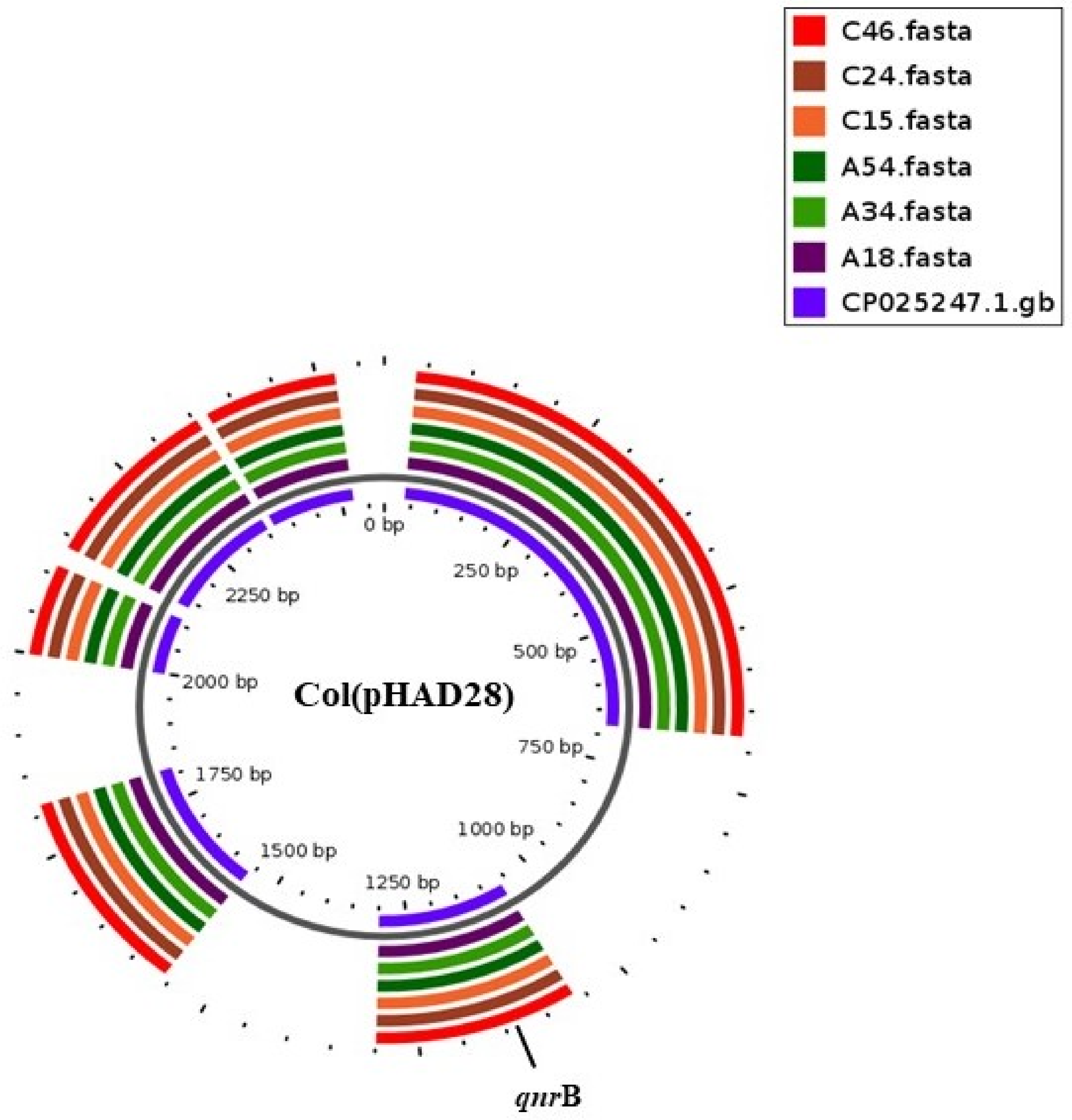
2.2. Plasmid Replicons and Association with Resistant Genes
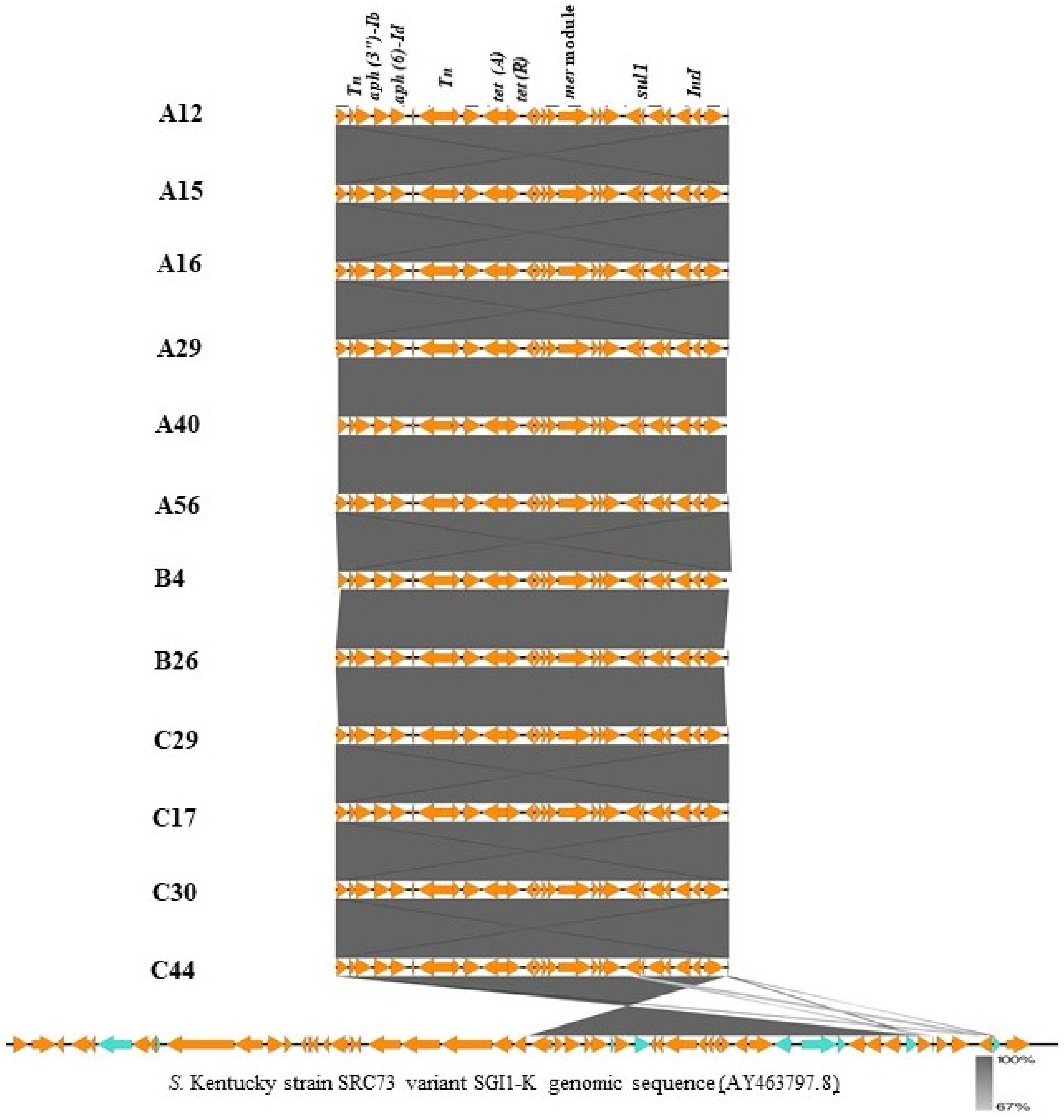
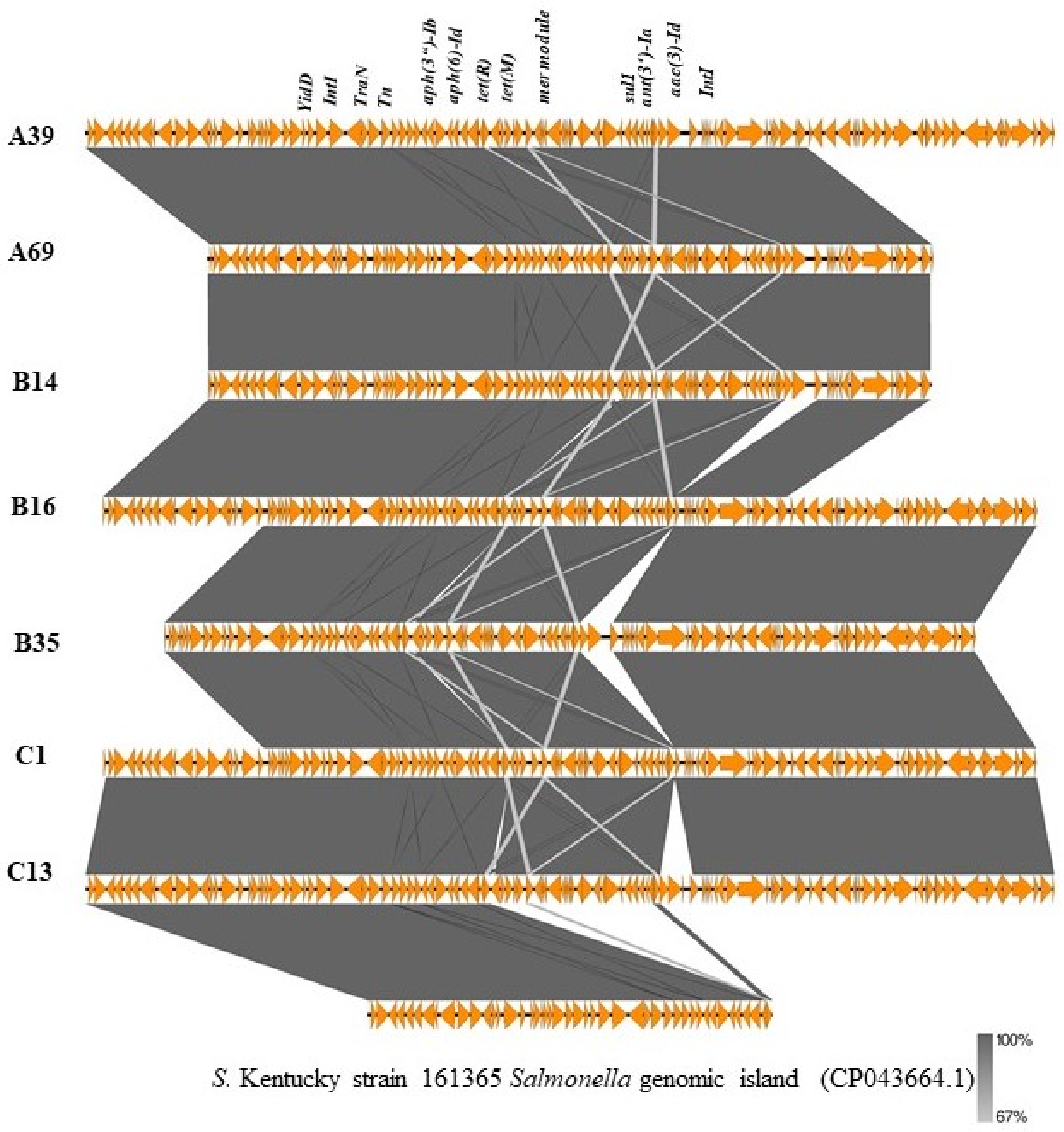
2.3. Genomic Islands Bearing Multiple Resistance Genes in S. Kentucky Strains
2.4. Virulence Genes
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Draft Genomes
4.2. Antimicrobial Screening and In Silico Prediction of Resistance Genes
4.3. Plasmid Extraction, In Silico Prediction and Reconstruction
4.4. Mapping of Resistance Genes on Plasmids and Genomic Islands
4.5. Identification of Salmonella Virulence
4.6. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobsen, A.; Hendriksen, R.S.; Aaresturp, F.M.; Ussery, D.W.; Friis, C. The Salmonella enterica Pan-genome. Microb. Ecol. 2011, 62, 487–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scallan, E.; Hoesktra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States-major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC). The European Union One Health 2018 Zoonoses Report. EFSA J. 2019, 17, e05926. [Google Scholar]
- Tack, B.; Vanaenrode, J.; Verbakel, J.Y.; Toelen, J.; Jacobs, J. Invasive non-typhoidal Salmonella infections in sub-Saharan Africa: A systematic review on antimicrobial resistance and treatment. BMC Med. 2020, 18, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Al-Mustapha, A.I.; Adetunji, V.O.; Heikinheimo, A. Risk Perceptions of Antibiotic Usage and Resistance: A Cross-Sectional Survey of Poultry Farmers in Kwara State, Nigeria. Antibiotics 2020, 9, 378. [Google Scholar] [CrossRef]
- Selaledi, L.; Hassan, Z.M.; Manyelo, T.G.; Mabelebele, M. The Current Status of the Alternative Use to Antibiotics in Poultry Production: An African Perspective. Antibiotics 2020, 9, 594. [Google Scholar] [CrossRef]
- Emond-Rheault, J.-G.; Hamel, J.; Jeukens, J.; Freschi, L.; Kukavica-Ibrulj, I.; Boyle, B.; Tamber, S.; Malo, D.; Franz, E.; Burnett, E.; et al. The Salmonella enterica Plasmidome as a Reservoir of Antibiotic Resistance. Microorganisms 2020, 8, 1016. [Google Scholar] [CrossRef]
- Carattoli, A. Plasmids and the spread of resistance. Int. J. Med. Microbiol. 2013, 303, 298–304. [Google Scholar] [CrossRef]
- San Millan, A. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol. 2018, 26, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Mathers, A.J.; Peirano, G.; Pitout, J.D.D. The Role of Epidemic Resistance Plasmids and International High-Risk Clones in the Spread of Multidrug-Resistant Enterobacteriaceae. Clin. Microbiol. Rev. 2015, 28, 565–591. [Google Scholar] [CrossRef] [Green Version]
- Andoh, L.A.; Dalsgaard, A.; Obiri-Danso, K.; Newman, M.J.; Barco, L.; Olsen, J.E. Prevalence and antimicrobial resistance of Salmonella serovars isolated from poultry in Ghana. Epidemiol. Infect. 2016, 144, 3288–3299. [Google Scholar] [CrossRef] [Green Version]
- Langata, L.M.; Maingi, J.M.; Musonye, H.A.; Kiiru, J.; Nyamache, A.K. Antimicrobial resistance genes in Salmonella and Escherichia coli isolates from chicken droppings in Nairobi, Kenya. BMC Res. Notes 2019, 12, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddese, D.; Tolosa, T.; Deresa, B.; Lakow, M.; Olani, A.; Shumi, E. Antibiograms and risk factors of Salmonella isolates from laying hens and eggs in Jimma Town, South Western Ethiopia. BMC Res. Notes 2019, 12, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makaya, P.V.; Matope, G.; Pfukenyi, D.M. Distribution of Salmonella serovars and antimicrobial susceptibility of Salmonella Enteritidis from poultry in Zimbabwe. Avian Pathol. 2012, 41, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Oloso, N.O.; Adeyemo, I.A.; Van Heerden, H.; Fasanmi, O.G.; Fasina, F.O. Antimicrobial drug administration and antimicrobial resistance of salmonella isolates originating from the broiler production value chain in Nigeria. Antibiotics 2019, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- WHO. Who Estimates of the Global Burden of Foodborne Diseases; WHO Press: Geneva, Switzerland, 2015; pp. 11–265. [Google Scholar]
- Ilyas, B.; Tsai, C.N.; Coombes, B.K. Evolution of Salmonella-Host Cell Interactions through a Dynamic Bacterial Genome. Front. Cell. Infect. Microbiol. 2017, 7, 428. [Google Scholar] [CrossRef]
- FAOSTAT. Statistics of the Food and Agriculture Organization of the United Nations; Food and Agriculture Organization of the United Nations: Rome, Italy, 2018; pp. 7–254. [Google Scholar]
- Mensah, N.; Tang, Y.; Cawthraw, S.; AbuOun, M.; Fenner, J.; Thomson, N.R.; Mather, A.E.; Petrovska-Holmes, L. Determining antimicrobial susceptibility in Salmonella enterica serovar Typhimurium through whole genome sequencing: A comparison against multiple phenotypic susceptibility testing methods. BMC Microbiol. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Oluwasile, B.B.; Agbaje, M.; Ojo, O.E.; Dipeolu, M.A. Antibiotic usage pattern in selected poultry farms in Ogun state. Sokoto J. Veter. Sci. 2014, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Clausen, P.T.L.C.; Zankari, E.; Aarestrup, F.M.; Lund, O. Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J. Antimicrob. Chemother. 2016, 71, 2484–2488. [Google Scholar] [CrossRef] [Green Version]
- Jaja, I.F.; Bhembe, N.L.; Green, E.; Oguttu, J.; Muchenje, V. Molecular characterisation of antibiotic-resistant Salmonella enterica isolates recovered from meat in South Africa. Acta Trop. 2019, 190, 129–136. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, E.A.; Gupta, S.K.; Williams, L.E.; Jové, T.; Hiott, L.M.; Woodley, T.A.; Barrett, J.B.; Jackson, C.R.; Wasilenko, J.L.; Simmons, M.; et al. Antimicrobial resistance genes, cassettes, and plasmids present in salmonella enterica associated with United States food animals. Front. Microbiol. 2019, 17, 832. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.; Seribelli, A.A.; Medeiros, M.I.C.; Rodrigues, D.D.P.; Varani, A.D.M.; Luo, Y.; Allard, M.W.; Falcão, J. Phylogenetic and antimicrobial resistance gene analysis of Salmonella Typhimurium strains isolated in Brazil by whole genome sequencing. PLoS ONE 2018, 13, e0201882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suleyman, G.; Perri, M.; Vager, D.; Samuel, L.; Zervos, M.J.; Alangaden, G.; Tibbetts, R.J. Characterization of Salmonella Isangi possessing a CTX-M15 ESBL associated with an outbreak in a US Hospital. Diagn. Microbiol. Infect. Dis. 2016, 85, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Veldman, K.T.; Cavaco, L.M.; Mevius, D.; Battisti, A.; Franco, A.; Botteldoorn, N.; Bruneau, M.; Perrin-Guyomard, A.; Cerny, T.; Escobar, C.D.F.; et al. International collaborative study on the occurrence of plasmid-mediated quinolone resistance in Salmonella enterica and Escherichia coli isolated from animals, humans, food and the environment in 13 European countries. J. Antimicrob. Chemother. 2011, 66, 1278–1286. [Google Scholar] [CrossRef]
- Hopkins, K.L.; Day, M.; Threlfall, E.J. Plasmid-mediated Quinolone Resistance in Salmonella enterica, United Kingdom. Emerg. Infect. Dis. 2008, 14, 340–342. [Google Scholar] [CrossRef]
- Monte, D.F.M.; Lincopan, N.; Berman, H.; Cerdeira, L.; Keelara, S.; Thakur, S.; Fedorka-Cray, P.J.; Landgraf, M. Genomic Features of High-Priority Salmonella enterica Serovars Circulating in the Food Production Chain, Brazil, 2000–2016. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Neuert, S.; Nair, S.; Day, M.R.; Doumith, M.; Ashton, P.M.; Mellor, K.C.; Jenkins, C.; Hopkins, K.L.; Woodford, N.; De Pinna, E.; et al. Prediction of Phenotypic Antimicrobial Resistance Profiles from Whole Genome Sequences of Non-typhoidal Salmonella enterica. Front. Microbiol. 2018, 9, 592. [Google Scholar] [CrossRef] [Green Version]
- Cummings, K.J.; Rodriguez-Rivera, L.D.; Norman, K.N.; Ohta, N.; Scott, H.M. Identification of a Plasmid-Mediated Quinolone Resistance Gene in Salmonella Isolates from Texas Dairy Farm Environmental Samples. Zoonoses Public Health 2016, 64, 305–307. [Google Scholar] [CrossRef]
- Van Duijkeren, E.; Schink, A.K.; Roberts, M.C.; Wang, Y.; Schwarz, S. Mechanisms of Bacterial Resistance to Antimicrobial Agents. Microbiol. Spectr. 2018, 6, 51–82. [Google Scholar]
- Hooda, Y.; Sajib, M.S.I.; Rahman, H.; Luby, S.P.; Bondy-Denomy, J.; Santosham, M.; Andrews, J.R.; Saha, S.K.; Saha, S. Molecular mechanism of azithromycin resistance among typhoidal Salmonella strains in Bangladesh identified through passive pediatric surveillance. PLoS Negl. Trop. Dis. 2019, 13, e0007868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, M.A.; Yin, X.; Persaud-Lachhman, M.G.; Diarra, M.S. First Detection of a Fosfomycin Resistance Gene, fosA7, in Salmonella enterica Serovar Heidelberg Isolated from Broiler Chickens. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Mathema, B.; Chavda, K.D.; DeLeo, F.R.; Bonomo, R.A.; Kreiswirth, B.N. Carbapenemase-producing Klebsiella pneumoniae: Molecular and genetic decoding. Trends Microbiol. 2014, 22, 686–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, H.-K.; Pan, Q.; Lo, W.-U.; Liu, X.; Law, C.O.K.; Chan, T.-F.; Ho, P.L.; Lau, T.C.K. Fine-tuning carbapenem resistance by reducing porin permeability of bacteria activated in the selection process of conjugation. Sci. Rep. 2018, 8, 15248. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Garg, R.; Kumaraswamy, K.; Datta, P.; Mohi, G.K.; Chander, J. Phenotypic and genotypic characterization of carbapenem resistance mechanisms in Klebsiella pneumoniae from blood culture specimens: A study from North India. J. Lab. Physicians 2018, 10, 125–129. [Google Scholar] [CrossRef]
- Mohan, B.; Hallur, V.; Singh, G.; Sandhu, H.K.; Appannanavar, S.B.; Taneja, N. Occurrence of blaNDM-1 & absence of blaKPC genes encoding carbapenem resistance in uropathogens from a tertiary care centre from north India. Indian J. Med. Res. 2015, 142, 336–343. [Google Scholar]
- Carattoli, A.; Villa, L.; Feudi, C.; Curcio, L.; Orsini, S.; Luppi, A.; Pezzotti, G.; Magistrali, C.F. Novel plasmid-mediated colistin resistance mcr-4 gene in Salmonella and Escherichia coli, Italy 2013, Spain and Belgium, 2015 to 2016. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- Orlek, A.; Stoesser, N.; Anjum, M.F.; Doumith, M.; Ellington, M.J.; Peto, T.; Crook, D.; Woodford, N.; Walker, A.S.; Phan, H.; et al. Plasmid Classification in an Era of Whole-Genome Sequencing: Application in Studies of Antibiotic Resistance Epidemiology. Front. Microbiol. 2017, 8, 182. [Google Scholar] [CrossRef]
- Arredondo-Alonso, S.; Willems, R.J.; Van Schaik, W.; Schürch, A.C. On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microb. Genomics. 2017, 3, e000128. [Google Scholar] [CrossRef]
- Laczny, C.C.; Galata, V.; Plum, A.; Posch, A.E.; Keller, A. Assessing the heterogeneity of in silico plasmid predictions based on whole-genome-sequenced clinical isolates. Brief. Bioinform. 2019, 20, 857–865. [Google Scholar] [CrossRef]
- Kudirkiene, E.; Andoh, L.A.; Ahmed, S.; Herrero-Fresno, A.; Dalsgaard, A.; Obiri-Danso, K.; Olsen, J.E. The Use of a Combined Bioinformatics Approach to Locate Antibiotic Resistance Genes on Plasmids from Whole Genome Sequences of Salmonella enterica Serovars From Humans in Ghana. Front. Microbiol. 2018, 9, 1010. [Google Scholar] [CrossRef] [PubMed]
- Ragupathi, N.K.D.; Sethuvel, D.P.M.; Shankar, B.A.; Munusamy, E.; Anandan, S.; Veeraraghavan, B. Draft genome sequence of blaTEM-1-mediated cephalosporin-resistant Salmonella enterica serovar Typhi from bloodstream infection. J. Glob. Antimicrob. Resist. 2016, 7, 11–12. [Google Scholar] [CrossRef]
- Oliva, M.; Monno, R.; D’Addabbo, P.; Pesole, G.; Dionisi, A.; Scrascia, M.; Chiara, M.; Horner, D.; Manzari, C.; Luzzi, I.; et al. A novel group of IncQ1 plasmids conferring multidrug resistance. Plasmid 2017, 89, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Fiegen, U.; Klein, G.; De Jong, A.; Kehrenberg, C. Detection of a Novel qnrB19-Carrying Plasmid Variant Mediating Decreased Fluoroquinolone Susceptibility in Salmonella enterica Serovar Hadar. Microb. Drug Resist. 2017, 23, 280–284. [Google Scholar] [CrossRef]
- Rahman, T.; Yarnall, B.; Doyle, D.A. Efflux drug transporters at the forefront of antimicrobial resistance. Eur. Biophys. J. 2017, 46, 647–653. [Google Scholar] [CrossRef]
- Levings, R.S.; Partridge, S.R.; Djordjevic, S.P.; Hall, R.M. SGI1-K, a Variant of the SGI1 Genomic Island Carrying a Mercury Resistance Region, in Salmonella enterica Serovar Kentucky. Antimicrob. Agents Chemother. 2006, 51, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.; Davidovich, M.; Rokney, A.; Valinsky, L.; Rahav, G.; Gal-Mor, O. Emergence of new variants of antibiotic resistance genomic islands among multidrug-resistant Salmonella enterica in poultry. Environ. Microbiol. 2019, 22, 413–432. [Google Scholar] [CrossRef]
- Levings, R.S.; Lightfoot, D.; Partridge, S.R.; Hall, R.M.; Djordjevic, S.P. The Genomic Island SGI1, Containing the Multiple Antibiotic Resistance Region of Salmonella enterica Serovar Typhimurium DT104 or Variants of It, Is Widely Distributed in Other S. enterica Serovars. J. Bacteriol. 2005, 187, 4401–4409. [Google Scholar] [CrossRef] [Green Version]
- Hensel, M. Evolution of pathogenicity islands of Salmonella enterica. Int. J. Med. Microbiol. 2004, 294, 95–102. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Wang, T.; Deng, X.; Chu, X. Natural compound sanguinarine chloride targets the type III secretion system of Salmonella enterica Serovar Typhimurium. Biochem. Biophys. Rep. 2018, 14, 149–154. [Google Scholar] [CrossRef]
- Lou, L.; Zhang, P.; Piao, R.; Wang, Y. Salmonella Pathogenicity Island 1 (SPI-1) and Its Complex Regulatory Network. Front. Cell. Infect. Microbiol. 2019, 9, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueira, R.; Watson, K.G.; Holden, D.W.; Helaine, S. Identification of Salmonella Pathogenicity Island-2 Type III Secretion System Effectors Involved in Intramacrophage Replication of S. enterica Serovar Typhimurium: Implications for Rational Vaccine Design. mBio 2013, 4, e00065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuge, X.; Sun, Y.; Xue, F.; Tang, F.; Ren, J.; Li, D.; Wang, J.; Jiang, M.; Dai, J. A Novel PhoP/PhoQ Regulation Pathway Modulates the Survival of Extraintestinal Pathogenic Escherichia coli in Macrophages. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.A.; Eade, C.R.; Wiedmann, M. Embracing diversity: Differences in virulence mechanisms, disease severity, and host adaptations contribute to the success of nontyphoidal salmonellas a foodborne pathogen. Front. Microbiol. 2019, 10, 1368. [Google Scholar] [CrossRef] [Green Version]
- Haley, B.J.; Luo, Y.; Wang, C.; Brown, E.; Allard, M.; Karns, J.S.; Van Kessel, J.A.S. Genome Sequences of Salmonella enterica subsp. enterica Serovar Kentucky Sequence Type 152 Isolated from Dairy Cows in the United States. Genome Announc. 2017, 5, e01119-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Hardt, W.D.; Galán, J.E. Salmonella typhimurium encodes a putative iron transport system within the centisome 63 pathogenicity island. Infect Immun. 1999, 67, 1974–1981. [Google Scholar] [CrossRef]
- Yue, M.; Rankin, S.C.; Blanchet, R.T.; Nulton, J.D.; Edwards, R.A.; Schifferli, D.M. Diversification of the Salmonella Fimbriae: A Model of Macro- and Microevolution. PLoS ONE 2012, 7, e38596. [Google Scholar] [CrossRef]
- Blondel, C.J.; Jiménez, J.C.; Leiva, L.E.; Álvarez, S.A.; Pinto, B.I.; Contreras, F.; Pezoa, D.; Santiviago, C.A.; Contreras, I. The Type VI Secretion System Encoded in Salmonella Pathogenicity Island 19 Is Required for Salmonella enterica Serotype Gallinarum Survival within Infected Macrophages. Infect. Immun. 2015, 83, 4175–4176. [Google Scholar] [CrossRef] [Green Version]
- Sana, T.G.; Flaugnatti, N.; Lugo, K.A.; Lam, L.H.; Jacobson, A.; Baylot, V.; Durand, E.; Journet, L.; Cascales, E.; Monack, D.M. SalmonellaTyphimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proc. Natl. Acad. Sci. USA 2016, 113, E5044–E5051. [Google Scholar] [CrossRef] [Green Version]
- Schroll, C.; Huang, K.; Ahmed, S.; Kristensen, B.M.; Pors, S.E.; Jelsbak, L.; Lemire, S.; Thomsen, L.E.; Christensen, J.P.; Jensen, P.R.; et al. The SPI-19 encoded type-six secretion-systems (T6SS) of Salmonella enterica serovars Gallinarum and Dublin play different roles during infection. Veter. Microbiol. 2019, 230, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Morales, C.; Lee, M.D.; Hofacre, C.; Maurer, J.J. Detection of a novel virulence gene and a Salmonella virulence homologue among Escherichia coli isolated from broiler chickens. Foodborne Pathog. Dis. 2004, 1, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, D.M.; Heffernan, E.J.; Wu, L.; Harwood, J.; Fierer, J.; Guiney, D.G. Identification of a domain in Rck, a product of the Salmonella typhimurium virulence plasmid, required for both serum resistance and cell invasion. Infect. Immun. 1996, 64, 2019–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Shin, D.; Ryu, S. Implication of Quorum Sensing in Salmonella enterica Serovar Typhimurium Virulence: The luxS Gene Is Necessary for Expression of Genes in Pathogenicity Island 1. Infect. Immun. 2007, 75, 4885–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.A.; Betteken, M.I.; Guo, X.; Altier, C.; Duhamel, G.E.; Wiedmann, M. The Typhoid Toxin Produced by the Nontyphoidal Salmonella enterica Serotype Javiana Is Required for Induction of a DNA Damage Response In Vitro and Systemic Spread In Vivo. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mambu, J.; Virlogeux-Payant, I.; Holbert, S.; Grépinet, O.; Velge, P.; Wiedemann, A. An Updated View on the Rck Invasin of Salmonella: Still Much to Discover. Front. Cell. Infect. Microbiol. 2017, 7, 500. [Google Scholar] [CrossRef]
- Jibril, A.H.; Okeke, I.N.; Dalsgaard, A.; Kudirkiene, E.; Akinlabi, O.C.; Bello, M.B.; Jibril, A.H. Prevalence and risk factors of Salmonella in commercial poultry farms in Nigeria. PLoS ONE 2020, 15, e0238190. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. M100: Performance Standards for Antimicrobial Susceptibility Testing, 27th ed.; Clinical & Laboratory Standards Institute: Annapolis Junction, MD, USA, 2017; pp. 16–282. [Google Scholar]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Hartwick, N.; Shen, M.; Raiko, M.; Lapidus, A.; Pevzner, P.A. plasmidSPAdes: Assembling plasmids from whole genome sequencing data. Bioinformatics 2016, 32, 3380–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. F1000Prime recommendation of: QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Kado, C.I.; Liu, S.T. Rapid procedure for detection and isolation of large and small plasmids. J. Bacteriol. 1981, 145, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrina, F.L.; Kopecko, D.J.; Jones, K.R.; Ayers, D.J.; McCowen, S.M. A multiple plasmid-containing Escherichia coli strain: Convenient source of size reference plasmid molecules. Plasmid 1978, 1, 417–420. [Google Scholar] [CrossRef]
- Threlfall, E.J.; Rowe, B.; Ferguson, J.L.; Ward, L.R. Characterization of plasmids conferring resistance to gentamicin and apramycin in strains of Salmonella typhimurium phage type 204c isolated in Britain. J. Hyg. 1986, 97, 419–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- McHugh, M.L. Interrater reliability: The kappa statistic. Biochem. Medica. 2012, 22, 276–282. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serovar | N | Number of Isolates Resistant to Antimicrobials * | MDR ** | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AMP | GEN | KAN | CTX | CIP | SUL | TET | CHL | TMP | NAL | MEM | N | (%) | ||
| Abadina | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| Aberdeen | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 100 |
| Alachua | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Birmingham | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bradford | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Chester | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 1 | 0 | 0 | 1 | 0 | 1 | 50 |
| Chomedey | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Colindale | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Corvalis | 2 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 50 |
| Essen | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 100 |
| Give | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Isangi | 8 | 0 | 5 | 2 | 0 | 5 | 4 | 5 | 5 | 5 | 8 | 0 | 5 | 62.5 |
| Ituri | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 2 | 0 | 0 | 0 |
| Kentucky | 24 | 3 | 21 | 21 | 4 | 23 | 21 | 22 | 3 | 1 | 23 | 0 | 21 | 87.5 |
| Larochelle | 4 | 0 | 1 | 1 | 0 | 2 | 1 | 1 | 1 | 1 | 4 | 1 | 2 | 50 |
| Menston | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 100 |
| Muenster | 4 | 0 | 0 | 1 | 1 | 1 | 4 | 3 | 1 | 0 | 4 | 0 | 3 | 75 |
| Poona | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Schwarzengrund | 4 | 4 | 4 | 2 | 1 | 1 | 4 | 4 | 2 | 0 | 4 | 0 | 4 | 100 |
| Takoradi | 6 | 0 | 1 | 4 | 0 | 1 | 4 | 1 | 0 | 0 | 1 | 0 | 1 | 16.7 |
| Telelkebir | 3 | 0 | 1 | 2 | 0 | 1 | 2 | 1 | 1 | 1 | 2 | 0 | 1 | 33.3 |
| Virchow | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Waycross | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| :z13,z28:I,z13,z28 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 74 | 7 | 36 | 37 | 7 | 37 | 53 | 44 | 16 | 9 | 59 | 1 | 42 | 56.8 |
| AMP | GEN | KAN | CIP | NAL | SUL | TMP | TET | CHL | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Serovar | No | blaTEM | aac(3)-Id aac(3)-IIa aac(3)-Iva | aac(6′)-Iaa, aph(3′)-Ia,b | T57S:S80I and S83F:D87Y | T57S:S80, S83F:D87Y | sul 1,2,3 | dfrA (14, 15, 17) | tet(A) tet(M) | cat, cml, floR |
| Abadina | 2 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 |
| Aberdeen | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Alachua | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Birmingham | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bradford | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Chester * | 2 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 0 |
| Chomedey | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 |
| Colindale | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Corvalis | 2 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 |
| Essen | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Give | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Isangi | 8 | 0 | 5 | 8 | 0 | 0 | 5 | 5 | 5 | 5 |
| Ituri | 2 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 0 |
| Kentucky * | 24 | 1 | 20 | 23 | 21 | 21 | 21 | 1 | 21 | 1 |
| Larochelle * | 4 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 |
| Menston § | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Muenster | 4 | 0 | 0 | 4 | 3 | 3 | 3 | 0 | 3 | 0 |
| Poona | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Schwarzengrund | 4 | 3 | 3 | 3 | 0 | 3 | 3 | 0 | 4 | 0 |
| Takoradi | 6 | 0 | 0 | 6 | 1 | 5 | 0 | 0 | 0 | 0 |
| Telelkebir | 3 | 0 | 0 | 3 | 0 | 2 | 0 | 0 | 0 | 0 |
| Virchow | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Waycross | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| :z13,z28:I,z13,z28 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Total | 74 | 4 | 28 | 70 | 26 | 52 | 33 | 6 | 34 | 6 |
| Serovar and Strains | Plasmid Replicons | In Silico | * Base Pairs (kb) | # Electro-Phoresis | Plasmid Size (kb) |
|---|---|---|---|---|---|
| S. Isangi | |||||
| A18 | Col(pHAD28) | + | 2.7 | + | 2.6 |
| A34 | Col(pHAD28), Col440I | +/+ | 2.7/2.5 | +/+ | 2.6/2 |
| A54 | Col(pHAD28) | + | 2.7 | + | 2.6 |
| C15 | Col(pHAD28) | + | 2.7 | + | 2.6 |
| C24 | Col(pHAD28) | + | 2.7 | + | 2.6 |
| C46 | Col(pHAD28), IncHI2/IncHI2A | +/+ | 2.7/14.5 | +/- | 2.6/ND |
| S. Kentucky | |||||
| A16 | IncFIIpCRY | + | 41 | + | 36 |
| A39 | ColpVC | + | 1.9 | + | 2 |
| A40 | IncFIIpCRY | + | 41 | + | 36 |
| A56 | IncFIIpCRY | + | 41 | + | 36 |
| A61 | ColpVC | + | 1.9 | + | 2 |
| A69 | ColpVC | + | 1.9 | + | 2 |
| B14 | ColpVC | + | 1.9 | + | 2 |
| B17 | IncM1 | + | 61 | + | 63 |
| C29 | Col(pHAD28) | + | 2.7 | - | ND |
| C36 | IncHI2A | + | 14.5 | - | ND |
| C44 | IncFIIpCRY | + | 41 | + | 36 |
| S. Larochelle | |||||
| C33 | Col(pHAD28) | + | 2.7 | + | 2.6 |
| C34 | Col(pHAD28), Col(Ye4449) | +/+ | 2.7/4.2 | +/+ | 2.6/3.5 |
| C37 | Col(pHAD28), Col(Ye4449) | +/+ | 2.7/4.2 | +/+ | 2.6/3.5 |
| C50 | Col(pHAD28), Col(Ye4449) | +/+ | 2.7/4.2 | +/+ | 2.6/3.5 |
| S. Menston | |||||
| C10 | IncI1 gamma | + | 88 | - | ND |
| S. Muenster | |||||
| A28 | IncQ1 | + | 12 | + | 14 |
| A55 | IncQ1 | + | 12 | - | ND |
| A59 | IncQ1 | + | 12 | + | 14 |
| S. Schwarzengrund | |||||
| A5 | IncN | + | 56 | + | 62 |
| A71 | IncN | + | 56 | + | 62 |
| S. Takoradi | |||||
| B21 | IncL | + | 55 | + | 62 |
| B28 | IncL | + | 55 | + | 62 |
| B36 | IncL | + | 55 | + | 62 |
| Serovars | Number | ST Type | SPI Profile |
|---|---|---|---|
| S. Abadina | 2 | 3899 | SPI-1 *, C63PI |
| S. Aberdeen | 1 | 3320 | SPI-1, C63PI C63PI SPI-3, SPI-5, SPI-13, SPI-14, C63PI |
| S. Alachua | 1 | 7743 | SPI-1, SPI-2, SPI-3, SPI-4, C63PI |
| S. Birmingham | 1 | 7749 | SPI-3, C63PI |
| S. Bradford | 1 | 7746 | SPI-1, SPI-2, SPI-3, C63PI |
| S. Chester | 2 | 441 | SPI-1, SPI-2, SPI-3, SPI-4 *, SPI-13, SPI-14, C63PI |
| S. Chomedey | 1 | 3961 | SPI-1, SPI-3, SPI-4, C63PI |
| S. Colindale | 1 | 584 | SPI-1, SPI-3, SPI-13, SPI-14, C63PI |
| S. Corvalis | 2 | 7744 | SPI-1 *, SPI-2 *, SPI-3, SPI-4 *, SPI-5, C63PI |
| S. Essen | 1 | 7747 | SPI-1, SPI-3, C63PI |
| S. Give | 1 | 524 | SPI-1, SPI-2, SPI-3, SPI-4, SPI-13, SPI-14, C63PI |
| S. Isangi | 8 | 216 | SPI-1 §, SPI-2 §, SPI-3 §, SPI-4, SPI-5, SPI-13, SPI-14, C63PI § |
| S. Ituri | 2 | 4498 | SPI-1, SPI-2 §, SPI-3, SPI-5 §, C63PI § |
| S. Kentucky | 24 | 198 | SPI-1, SPI-2, SPI-3, SPI-4, SPI-14, C63PI § |
| S. Larochelle | 4 | 22 | SPI-1, SPI-3, SPI-4, SPI-13 §, SPI-14 §, C63PI § |
| S. Menston | 1 | 7742 | SPI-1, SPI-2, C63PI |
| S. Muenster | 4 | 321 | SPI-1 §, SPI-2 *, SPI-13, SPI-14, C63PI |
| S. Poona | 1 | 308 | SPI-1, SPI-13, SPI-14, C63PI |
| S. Schwarzengrund | 4 | 96 | SPI-2, SPI-3 §, SPI-4, SPI-13, SPI-14, C63PI |
| S. Takoradi | 6 | 531 | SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, SPI-13 §, C63PI § |
| S. Telelkebir | 3 | 2222 | SPI-1 *, SPI-2 *, SPI-3 *, C63PI § |
| S. Virchow | 1 | 6166 | SPI-1, SPI-2, SPI-3, SPI-4, SPI-13, SPI-14 |
| S. Waycross | 1 | 7745 | SPI-1, SPI-2, SPI-3, SPI-4, SPI-8, C63PI |
| :z13,z28:I,z13,z28 | 1 | - | SPI-3, SPI-5, C63PI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jibril, A.H.; Okeke, I.N.; Dalsgaard, A.; Menéndez, V.G.; Olsen, J.E. Genomic Analysis of Antimicrobial Resistance and Resistance Plasmids in Salmonella Serovars from Poultry in Nigeria. Antibiotics 2021, 10, 99. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020099
Jibril AH, Okeke IN, Dalsgaard A, Menéndez VG, Olsen JE. Genomic Analysis of Antimicrobial Resistance and Resistance Plasmids in Salmonella Serovars from Poultry in Nigeria. Antibiotics. 2021; 10(2):99. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020099
Chicago/Turabian StyleJibril, Abdurrahman Hassan, Iruka N. Okeke, Anders Dalsgaard, Vanesa García Menéndez, and John Elmerdahl Olsen. 2021. "Genomic Analysis of Antimicrobial Resistance and Resistance Plasmids in Salmonella Serovars from Poultry in Nigeria" Antibiotics 10, no. 2: 99. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020099