
Drug Repurposing Targeting Pseudomonas aeruginosa MvfR Using Docking, Virtual Screening, Molecular Dynamics, and Free-Energy Calculations




Abstract
:1. Introduction
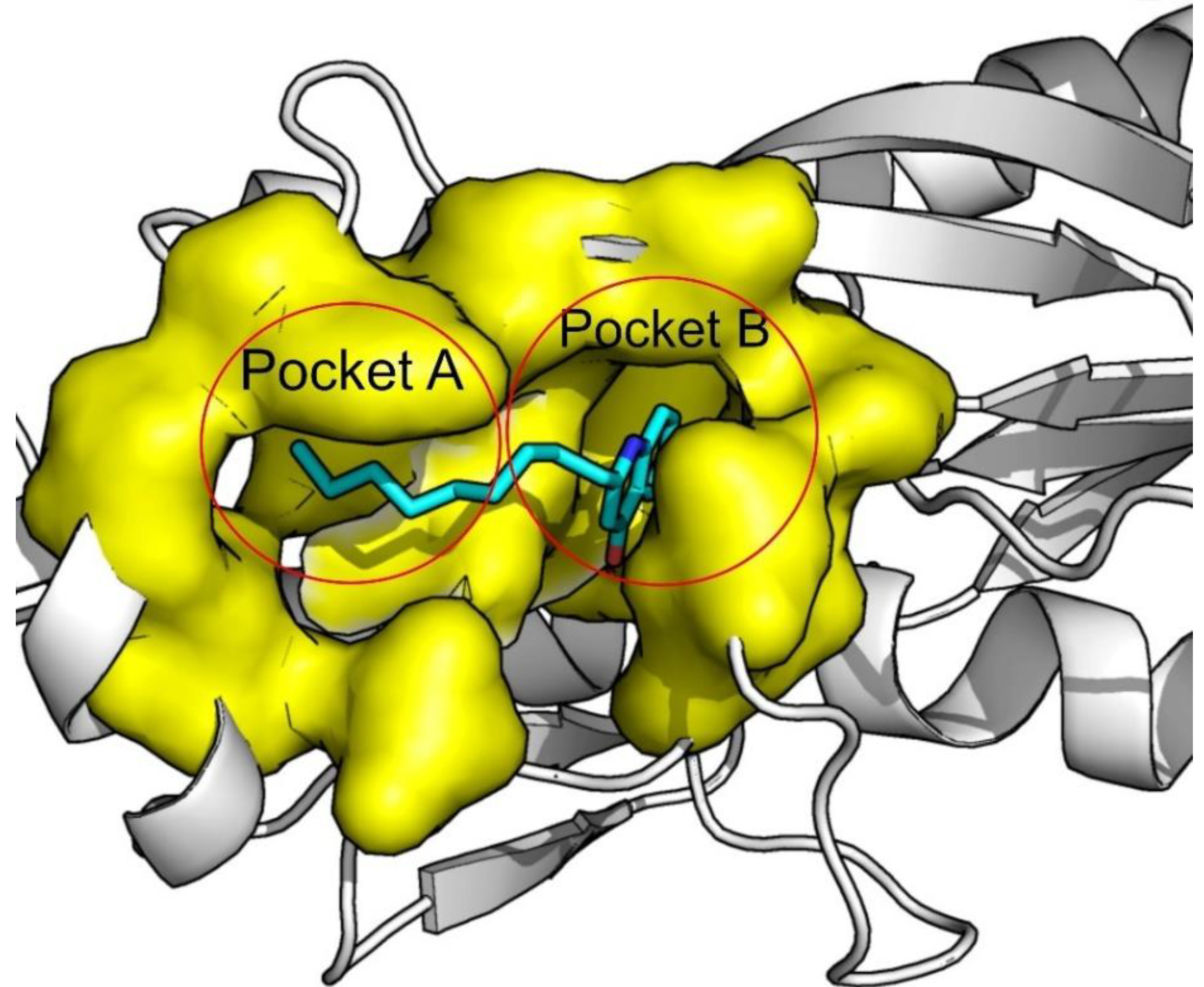
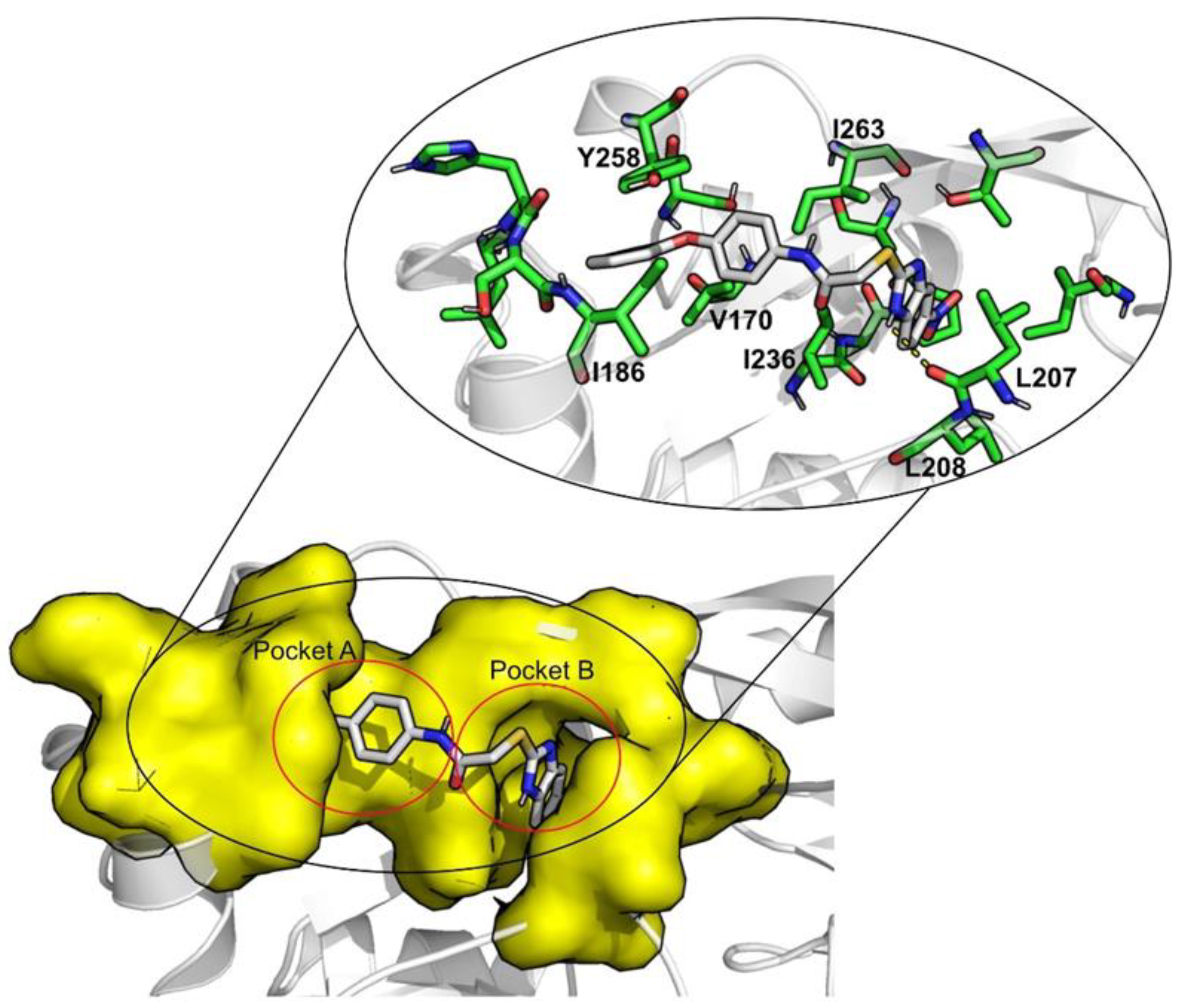
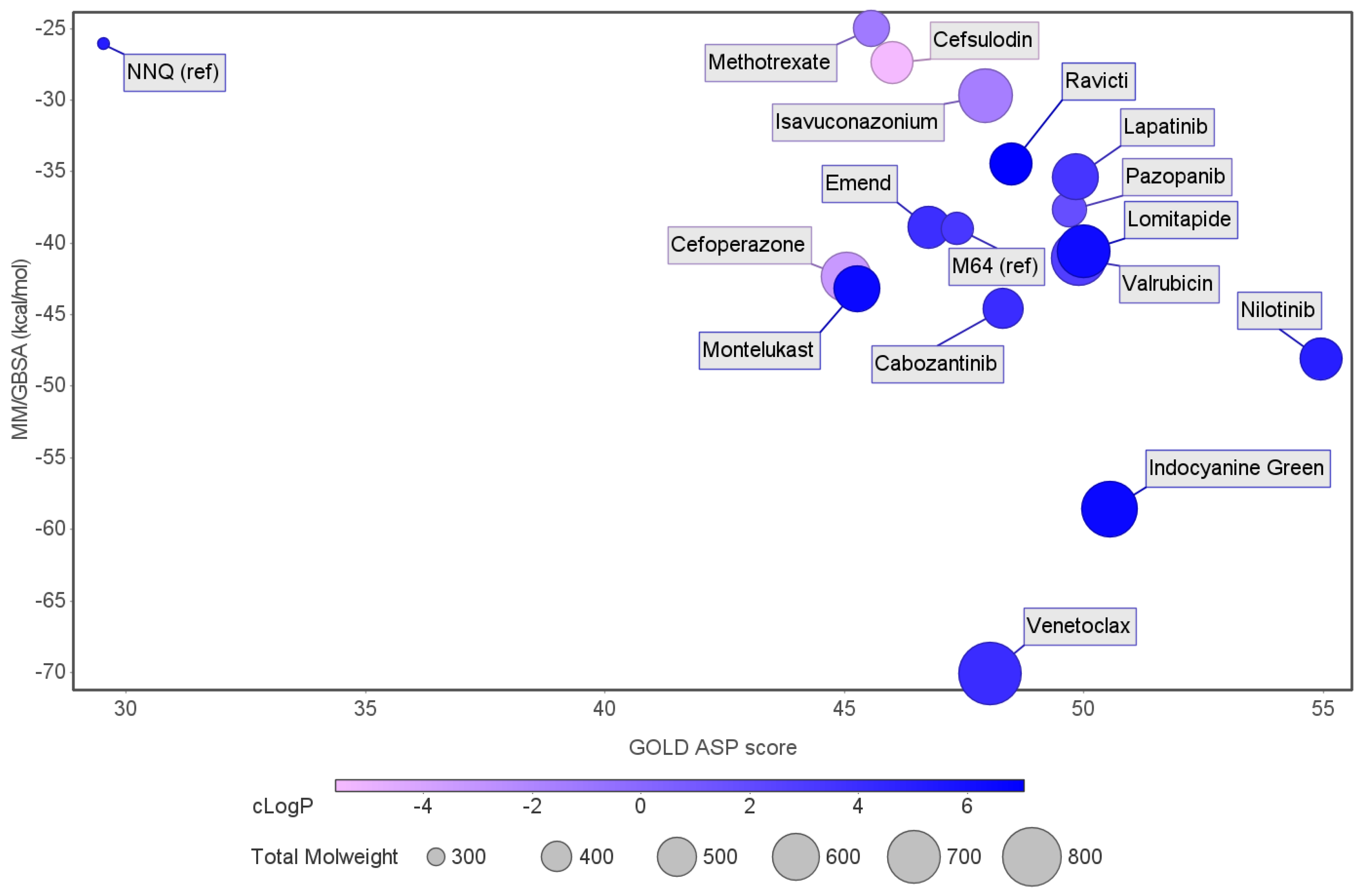
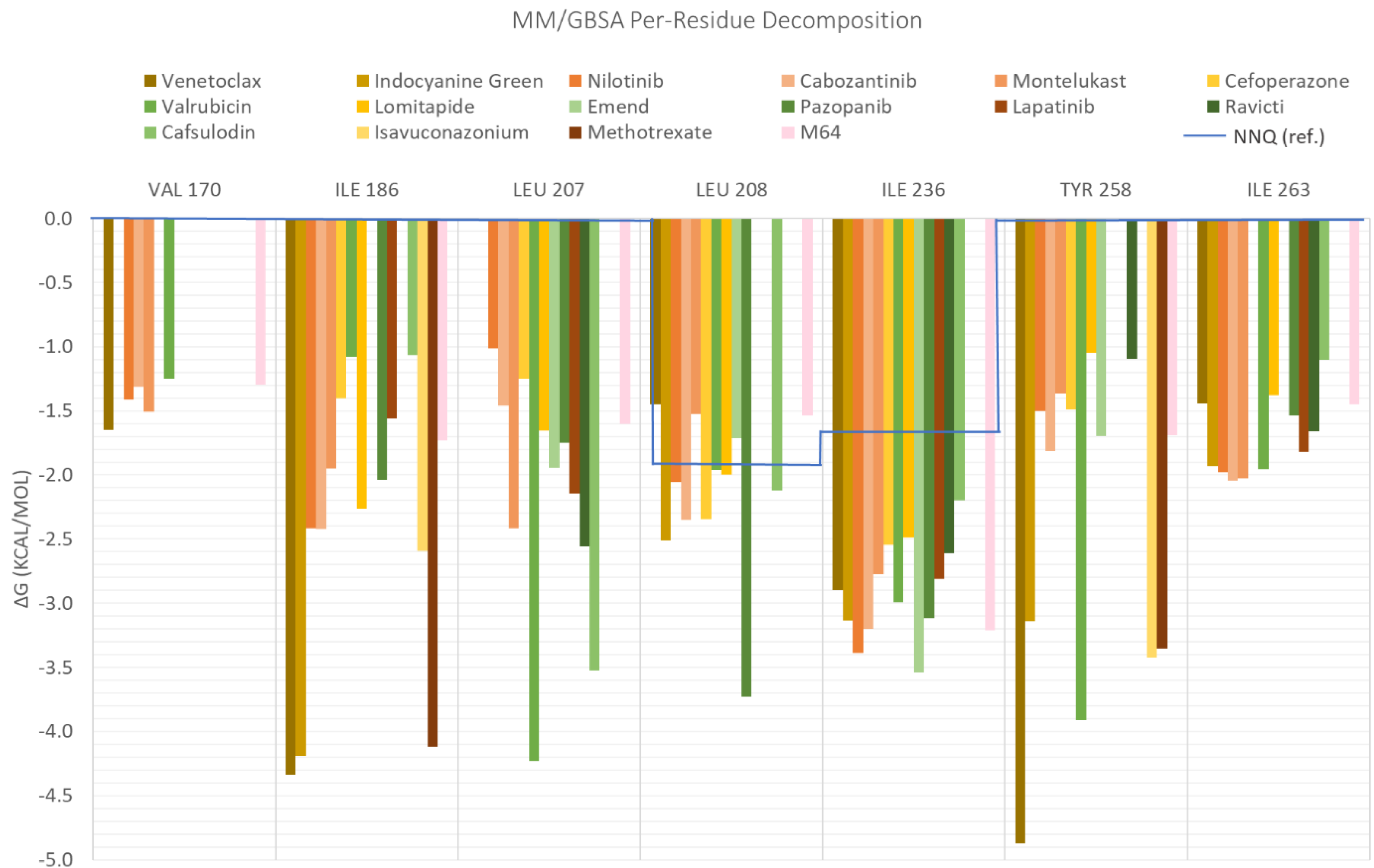
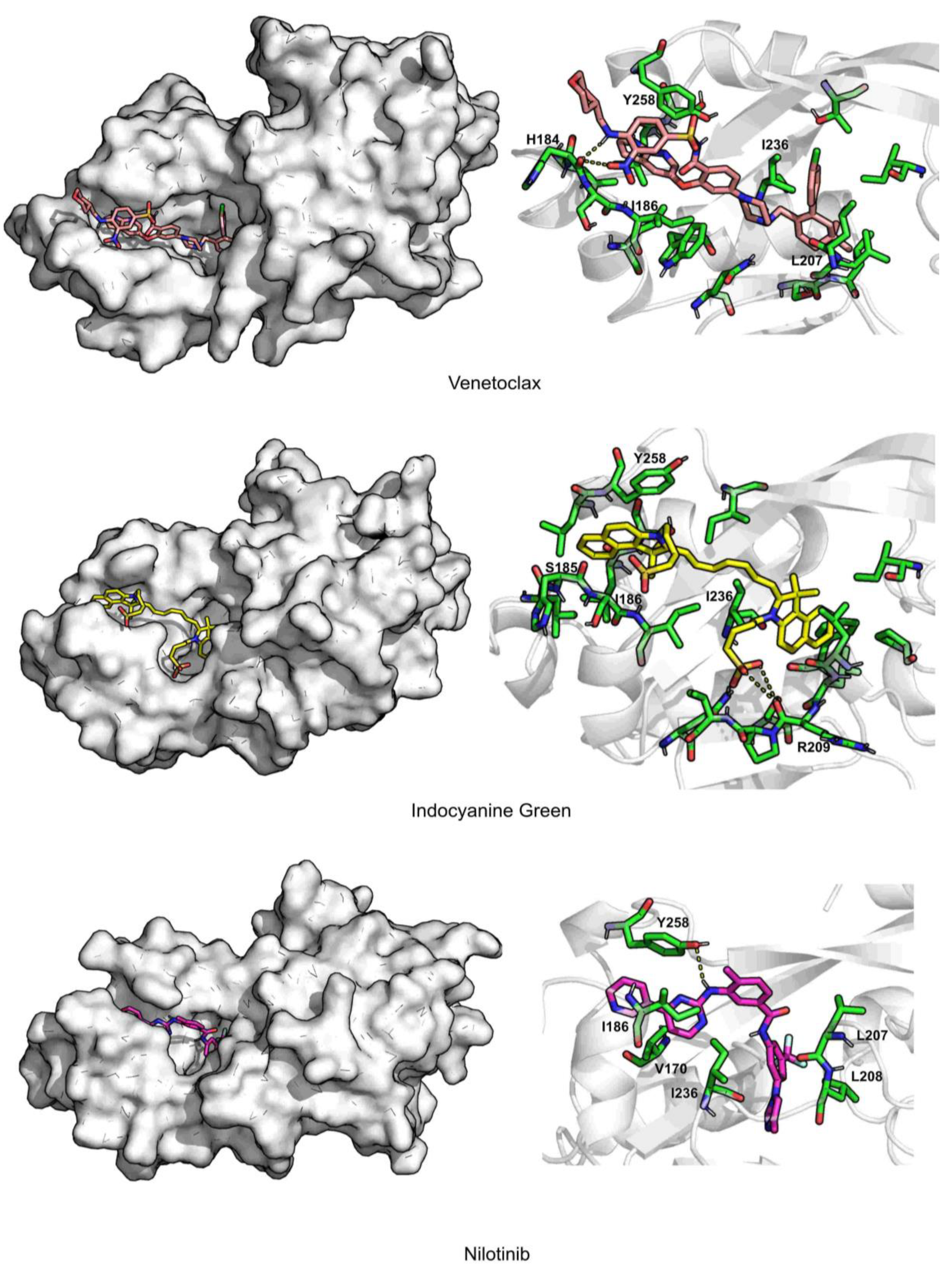
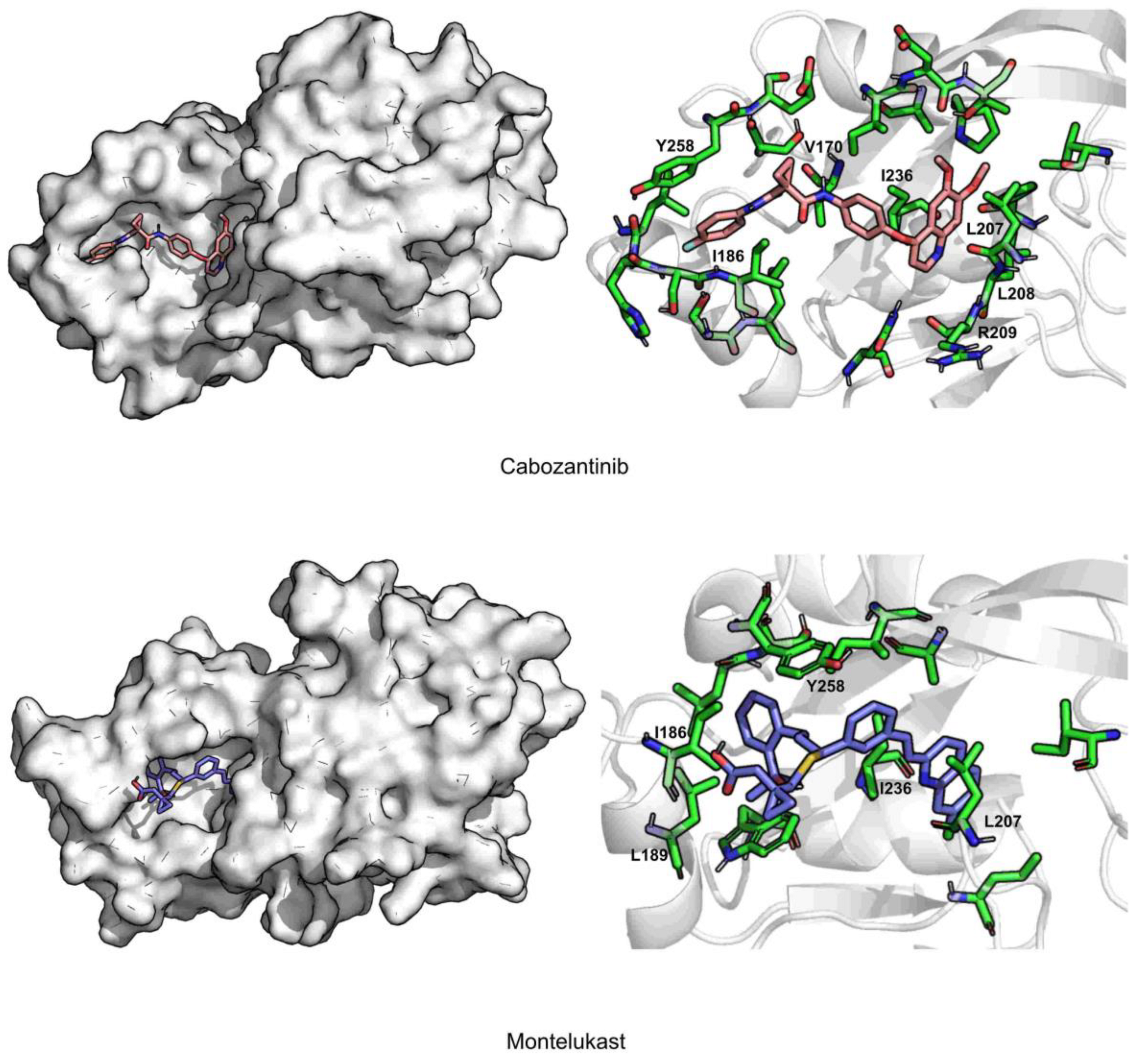
2. Results and Discussion
3. Materials and Methods
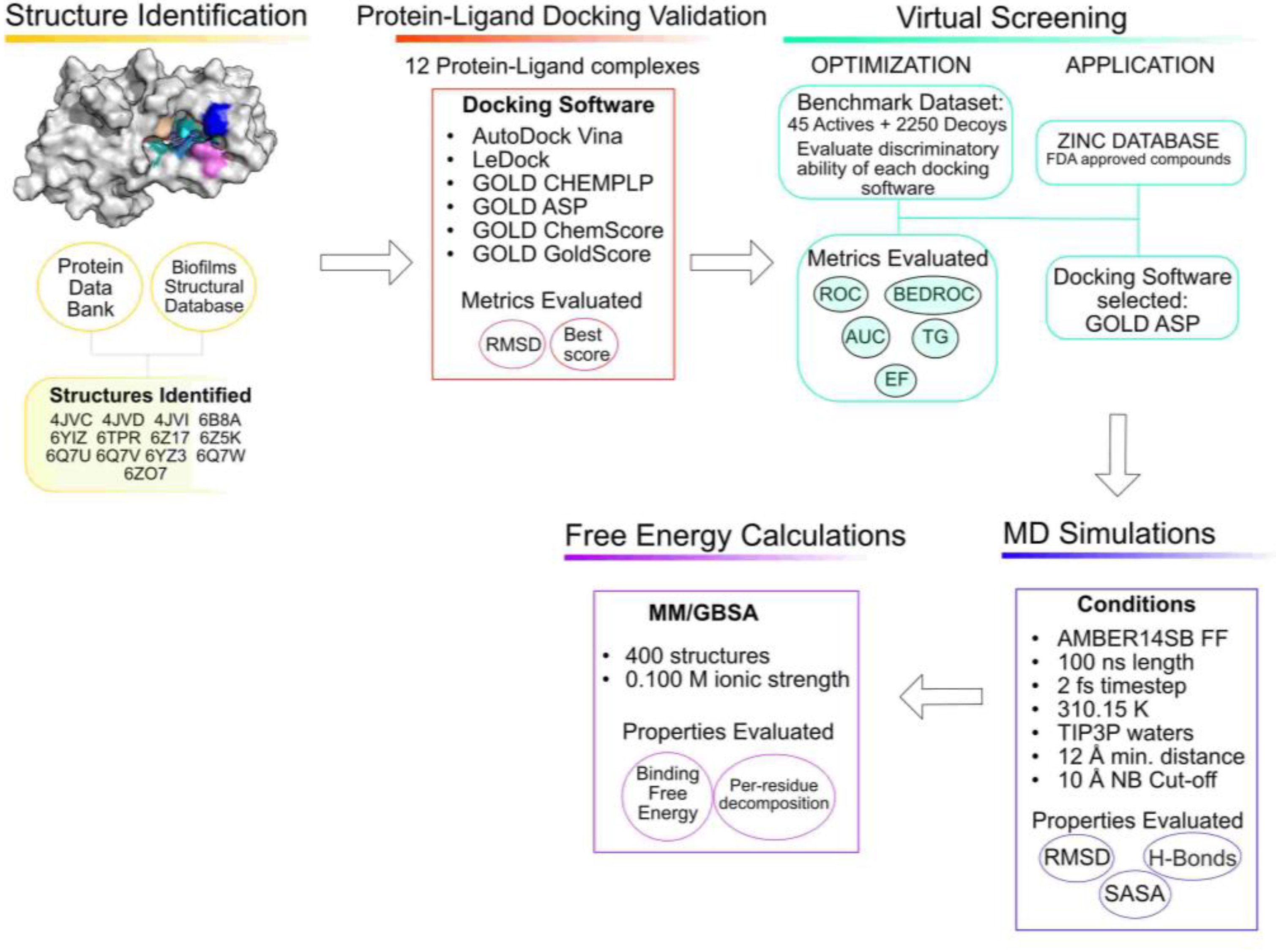
3.1. Structure Identification and Analysis
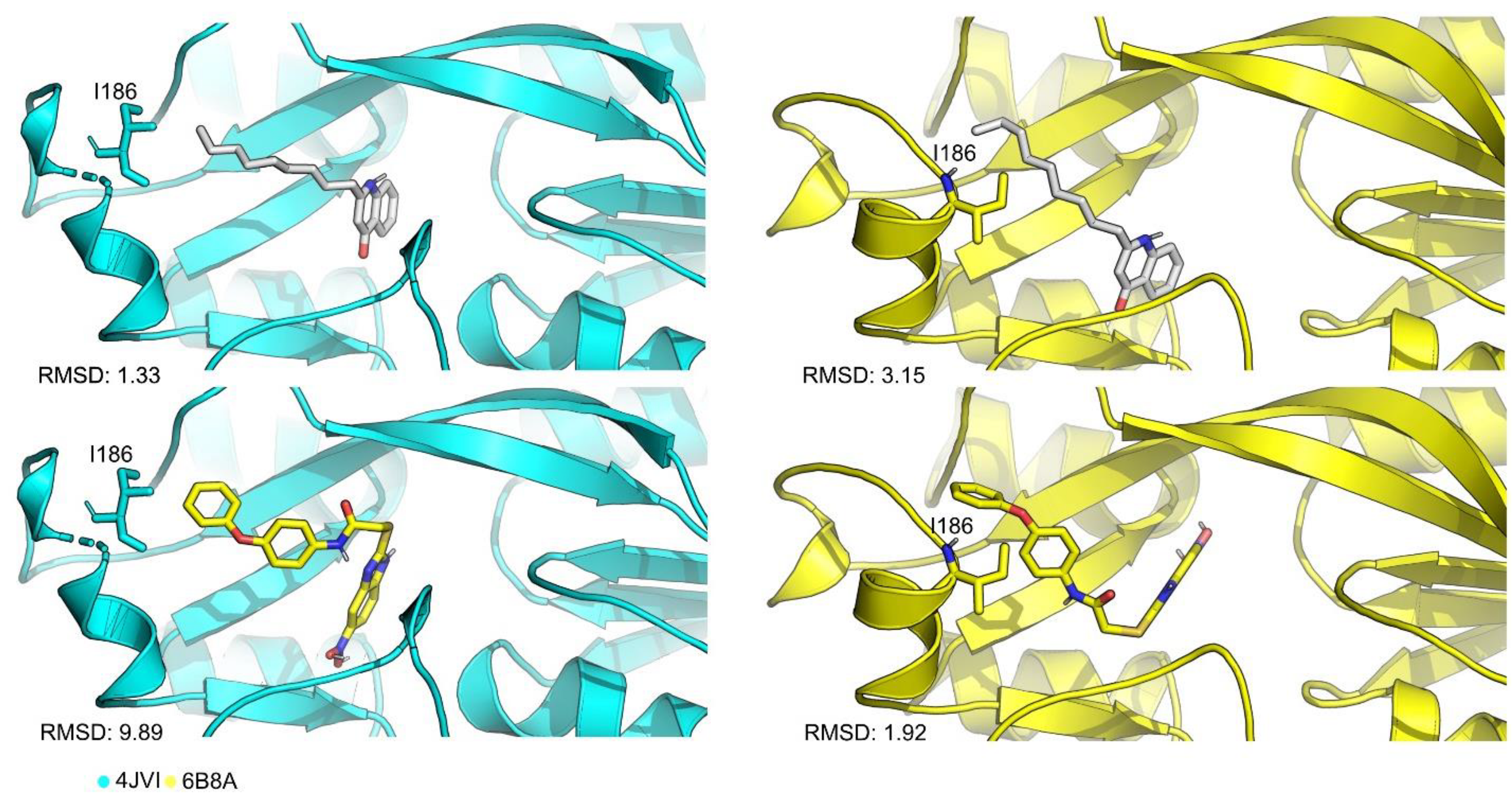
3.2. Protein–Ligand Docking Protocol Validation
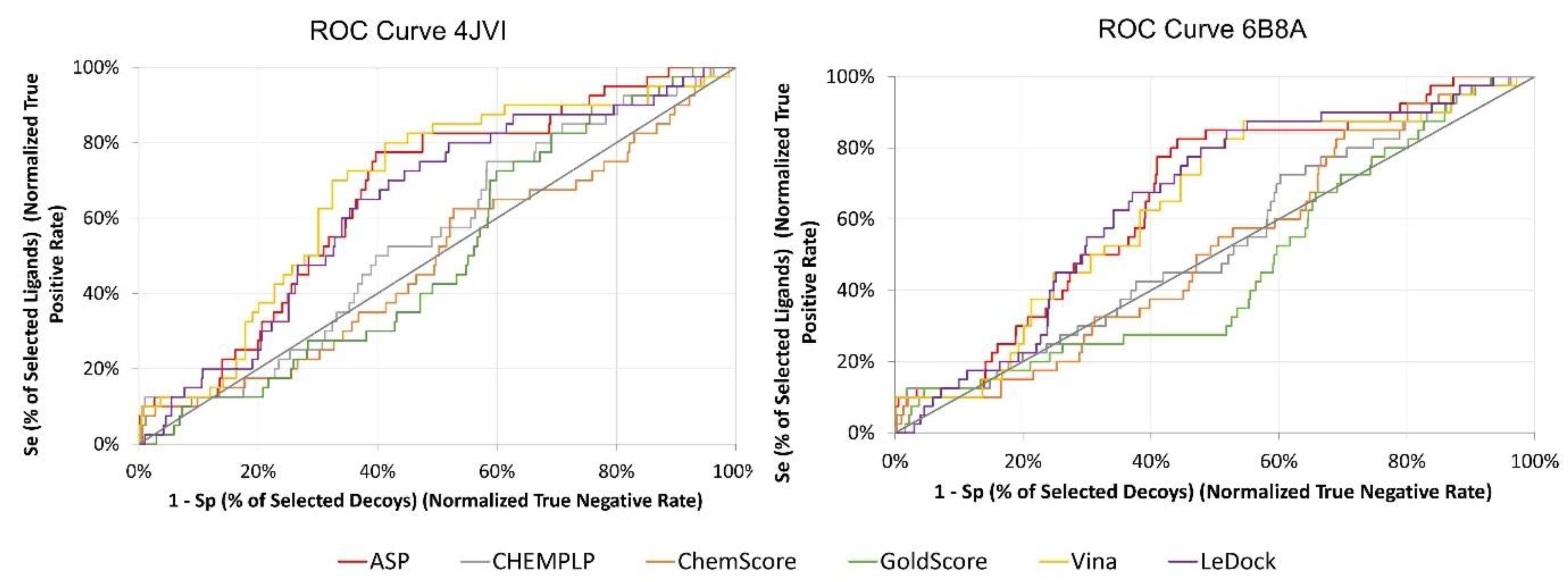
3.3. Virtual Screening Protocol Optimization
3.4. Virtual Screening for Drug Repurposing
3.5. Molecular Dynamics Simulations
3.6. Free-Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allegretta, G.; Maurer, C.K.; Eberhard, J.; Maura, D.; Hartmann, R.W.; Rahme, L.; Empting, M. In-depth Profiling of MvfR-Regulated Small Molecules in Pseudomonas aeruginosa after Quorum Sensing Inhibitor Treatment. Front. Microbiol. 2017, 24, 924. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Cámara, M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: A tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009, 12, 182–191. [Google Scholar] [CrossRef]
- Kamaruzzaman, N.F.; Tan, L.P.; Mat Yazid, K.A.; Saeed, S.I.; Hamdan, R.H.; Choong, S.S.; Wong, W.K.; Chivu, A.; Gibson, A.J. Targeting the Bacterial Protective Armour; Challenges and Novel Strategies in the Treatment of Microbial Biofilm. Materials 2018, 11, 1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, M.; Tasneem, U.; Hussain, T.; Andleeb, S. Bacterial Biofilm: Its Composition, Formation and Role in Human Infections. J. Microbiol. Biotechnol. 2015, 4, 1–14. [Google Scholar]
- Dufour, D.; Leung, V.; Lévesque, C.M. Bacterial biofilm: Structure, function, and antimicrobial resistance. Endod. Top. 2010, 22, 2–16. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, S.; Sen, S.K.; Pattanaik, S.; Raut, S. Review on bacterial biofilm: An universal cause of contamination. Biocatal. Agric. Biotechnol. 2016, 7, 56–66. [Google Scholar] [CrossRef]
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell. Infect. Microbiol. 2017, 7, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Das, A.; Mallik, S. Inhibition of Quorum Sensing in Pseudomonas aeruginosa: A Review. Indian J. Pharm. Sci. 2019, 81, 797–806. [Google Scholar] [CrossRef]
- Li, S.; Chen, S.; Fan, J.; Cao, Z.; Ouyang, W.; Tong, N.; Hu, X.; Hu, J.; Li, P.; Feng, Z.; et al. Anti-biofilm effect of novel thiazole acid analogs against Pseudomonas aeruginosa through IQS pathways. Eur. J. Med. Chem. 2018, 145, 64–73. [Google Scholar] [CrossRef]
- Abelyan, N.; Grabski, H.; Tiratsuyan, S. In silico Screening of Flavones and its Derivatives as Potential Inhibitors of Quorum-Sensing Regulator LasR of Pseudomonas aeruginosa. Mol. Biol. 2020, 54, 153–163. [Google Scholar] [CrossRef]
- Lazdunski, A.M.; Ventre, I.; Sturgis, J.N. Regulatory circuits and communication in gram-negative bacteria. Nat. Rev. Microbiol. 2004, 2, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Maura, D.; Hazan, R.; Kitao, T.; Ballok, A.E.; Rahme, L.G. Evidence for Direct Control of Virulence and Defense Gene Circuits by the Pseudomonas aeruginosa Quorum Sensing Regulator, MvfR. Sci. Rep. 2016, 6, 34083. [Google Scholar] [CrossRef]
- Schütz, C.; Empting, M. Targeting the Pseudomonas quinolone signal quorum sensing system for the discovery of novel anti-infective pathoblockers. Beilstein J. Org. Chem. 2018, 14, 2627–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturi, V. Regulation of quorum sensing in Pseudomonas. FEMS Microbiol. Rev. 2006, 30, 274–291. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.B.; Bassler, B.L. Quorum Sensing in Bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [Green Version]
- DIckey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Mühlen, S.; Dersch, P. Anti-virulence strategies to target bacterial infections. In Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2015; Volume 398, pp. 147–183. [Google Scholar]
- Thomann, A.; Brengel, C.; Börger, C.; Kail, D.; Steinbach, A.; Empting, M.; Hartmann, R.W. Structure-Activity Relationships of 2-Sufonylpyrimidines as Quorum-Sensing Inhibitors to Tackle Biofilm Formation and eDNA Release of Pseudomonas aeruginosa. ChemMedChem 2016, 11, 2522–2533. [Google Scholar] [CrossRef]
- Ilangovan, A.; Fletcher, M.; Rampioni, G.; Pustelny, C.; Rumbaugh, K.; Heeb, S.; Cámara, M.; Truman, A.; Chhabra, S.R.; Emsley, J.; et al. Structural Basis for Native Agonist and Synthetic Inhibitor Recognition by the Pseudomonas aeruginosa Quorum Sensing Regulator PqsR (MvfR). PLoS Pathog. 2013, 9, e1003508. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Cheng, J.; Wang, Y.; Shen, X. The Pseudomonas Quinolone Signal (PQS): Not Just for Quorum Sensing Anymore. Front. Cell. Infect. Microbiol. 2018, 8, 230. [Google Scholar] [CrossRef]
- Zender, M.; Witzgall, F.; Kiefer, A.; Kirsch, B.; Maurer, C.K.; Kany, A.M.; Xu, N.; Schmelz, S.; Börger, C.; Blankenfeldt, W.; et al. Flexible Fragment Growing Boosts Potency of Quorum-Sensing Inhibitors against Pseudomonas aeruginosa Virulence. ChemMedChem 2019, 14, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Kitao, T.; Lepine, F.; Babloudi, S.; Walte, F.; Steinbacher, S.; Maskos, K.; Blaesse, M.; Negri, M.; Pucci, M.; Zahler, B.; et al. Molecular Insights into Function and Competitive Inhibition of Pseudomonas aeruginosa Multiple Virulence Factor Regulator. mBio 2018, 9, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zender, M.; Klein, T.; Henn, C.; Kirsch, B.; Maurer, C.K.; Kail, D.; Ritter, C.; Dolezal, O.; Steinbach, A.; Hartmann, R.W. Discovery and Biophysical Characterization of 2-Amino-oxadiazoles as Novel Antagonists of PqsR, an Important Regulator of Pseudomonas aeruginosa Virulence. J. Med. Chem. 2013, 56, 6761–6774. [Google Scholar] [CrossRef] [PubMed]
- Mellini, M.; Di Muzio, E.; D’Angelo, F.; Baldelli, V.; Ferrillo, S.; Visca, P.; Leoni, L.; Polticelli, F.; Rampioni, G. In silico Selection and Experimental Validation of FDA-Approved Drugs as Anti-quorum Sensing Agents. Front. Microbiol. 2019, 10, 2355. [Google Scholar] [CrossRef] [PubMed]
- Soheili, V.; Tajani, A.S.; Ghodsi, R.; Bazzaz, B.S.F. Anti-PqsR compounds as next-generation antibacterial agents against Pseudomonas aeruginosa: A review. Eur. J. Med. Chem. 2019, 172, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Henn, C.; De Jong, J.C.; Zimmer, C.; Kirsch, B.; Maurer, C.K.; Pistorius, D.; Müller, R.; Steinbach, A.; Hartmann, R.W. Identification of small-molecule antagonists of the Pseudomonas aeruginosa transcriptional regulator PqsR: Biophysically guided hit discovery and optimization. ACS Chem. Biol. 2012, 7, 1496–1501. [Google Scholar] [CrossRef]
- Starkey, M.; Lepine, F.; Maura, D.; Bandyopadhaya, A.; Lesic, B.; He, J.; Kitao, T.; Righi, V.; Milot, S.; Tzika, A.; et al. Identification of Anti-virulence Compounds That Disrupt Quorum-Sensing Regulated Acute and Persistent Pathogenicity. PLoS Pathog. 2014, 10, e1004321. [Google Scholar] [CrossRef]
- Lu, C.; Kirsch, B.; Zimmer, C.; de Jong, J.C.; Henn, C.; Maurer, C.K.; Müsken, M.; Häussler, S.; Steinbach, A.; Hartmann, R.W. Discovery of antagonists of PqsR, a key player in 2-alkyl-4-quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem. Biol. 2012, 19, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Soukarieh, F.; Vico Oton, E.; Dubern, J.F.; Gomes, J.; Halliday, N.; de Pilar Crespo, M.; Ramírez-Prada, J.; Insuasty, B.; Abonia, R.; Quiroga, J.; et al. In Silico and in Vitro-Guided Identification of Inhibitors of Alkylquinolone-Dependent Quorum Sensing in Pseudomonas aeruginosa. Molecules 2018, 23, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, N.K.; Teng, C.; Frei, C.R. Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus on Drug Repurposing. Front. Cell. Infect. Microbiol. 2021, 11, 442. [Google Scholar] [CrossRef]
- Farha, M.A.; Brown, E.D. Drug repurposing for antimicrobial discovery. Nat. Microbiol. 2019, 4, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Choulis, N.H. Miscellaneous drugs, materials, medical devices, and techniques. In Side Effects of Drugs Annual; Elsevier: Amsterdam, The Netherlands, 2009; pp. 757–769. [Google Scholar]
- Alonso, R.; Cuevas, A.; Mata, P. Lomitapide: A review of its clinical use, efficacy, and tolerability. Core Evid. 2019, 14, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Opdam, F.L.; Guchelaar, H.; Beijnen, J.H.; Schellens, J.H.M. Lapatinib for Advanced or Metastatic Breast Cancer. Oncologist 2012, 17, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.T.J.; Jones, R.L.; Huang, P.H. Pazopanib in advanced soft tissue sarcomas. Signal Transduct. Target Ther. 2019, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Longo, N.; Diaz, G.A.; Lichter-Konecki, U.; Schulze, A.; Inbar-Feigenberg, M.; Conway, R.L.; Bannick, A.A.; McCandless, S.E.; Zori, R.; Hainline, B.; et al. Glycerol phenylbutyrate efficacy and safety from an open label study in pediatric patients under 2 months of age with urea cycle disorders. Mol. Genet. Metab. 2021, 132, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, A.; Vaishampayan, U. Cabozantinib for the treatment of kidney cancer. Expert Rev. Anticancer Ther. 2017, 17, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, L.; Shen, K.; Jin, H.; Li, H.; Cheng, Y.; Ma, X. Efficacy and Safety of Bcl-2 Inhibitor Venetoclax in Hematological Malignancy: A Systematic Review and Meta-Analysis of Clinical Trials. Front. Pharmacol. 2019, 10, 697. [Google Scholar] [CrossRef]
- McManus, D.S. Antifungal drugs. In Side Effects of Drugs Annual; Elsevier: Amsterdam, The Netherlands, 2016; pp. 243–253. [Google Scholar]
- Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.R. Cefsulodin and ceftazidime, two antipseudomonal cephalosporins. Clin. Pharm. 1984, 3, 373–385. [Google Scholar] [CrossRef]
- Paggiaro, P.; Bacci, E. Montelukast in asthma: A review of its efficacy and place in therapy. Ther. Adv. Chronic Dis. 2011, 2, 47–58. [Google Scholar] [CrossRef]
- Castle, S.S. Cefoperazone. In XPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–5. [Google Scholar]
- Berman, H.M.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhães, R.P.; Vieira, T.F.; Fernandes, H.S.; Melo, A.; Simões, M.; Sousa, S.F. The Biofilms Structural Database. Trends Biotechnol. 2020, 38, 937–940. [Google Scholar] [CrossRef]
- Soukarieh, F.; Liu, R.; Romero, M.; Roberston, S.N.; Richardson, W.; Lucanto, S.; Oton, E.V.; Qudus, N.R.; Mashabi, A.; Grossman, S.; et al. Hit Identification of New Potent PqsR Antagonists as Inhibitors of Quorum Sensing in Planktonic and Biofilm Grown Pseudomonas aeruginosa. Front. Chem. 2020, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.; Soukarieh, F.; Richardson, W.; Liu, R.; Mashabi, A.; Emsley, J.; Williams, P.; Cámara, M.; Stocks, M.J. Novel quinazolinone inhibitors of the Pseudomonas aeruginosa quorum sensing transcriptional regulator PqsR. Eur. J. Med. Chem. 2020, 208, 112778. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 757–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef] [Green Version]
- Vieira, T.F.; Magalhães, R.P.; Sousa, S.F. Tailoring Specialized Scoring Functions for More Efficient Virtual Screening. Front. Drug Chem. Clin. Res. 2019, 2, 1–4. [Google Scholar]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerqueira, N.M.F.S.A.; Gesto, D.; Oliveira, E.F.; Santos-Martins, D.; Brás, N.F.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys. 2015, 582, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Kirsch, B.; Maurer, C.K.; de Jong, J.C.; Braunshausen, A.; Steinbach, A.; Hartmann, R.W. Optimization of anti-virulence PqsR antagonists regarding aqueous solubility and biological properties resulting in new insights in structure–activity relationships. Eur. J. Med. Chem. 2014, 79, 173–183. [Google Scholar] [CrossRef]
- Hossain, M.A.; Sattenapally, N.; Parikh, H.I.; Li, W.; Rumbaugh, K.P.; German, N.A. Design, synthesis, and evaluation of compounds capable of reducing Pseudomonas aeruginosa virulence. Eur. J. Med. Chem. 2020, 185, 111800. [Google Scholar] [CrossRef]
- Lesic, B.; Lépine, F.; Déziel, E.; Zhang, J.; Zhang, Q.; Padfield, K.; Castonguay, M.H.; Milot, S.; Stachel, S.; Tzika, A.A.; et al. Inhibitors of Pathogen Intercellular Signals as Selective Anti-Infective Compounds. PLoS Pathog. 2007, 3, e126. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Empereur-Mot, C.; Zagury, J.F.; Montes, M. Screening Explorer–An Interactive Tool for the Analysis of Screening Results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Aminpour, M.; Montemagno, C.; Tuszynski, J.A. An Overview of Molecular Modeling for Drug Discovery with Specific Illustrative Examples of Applications. Molecules 2019, 24, 1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Published Online 2016; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comp. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Martins, F.G.; Melo, A.; Sousa, S.F. Identification of New Potential Inhibitors of Quorum Sensing through a Specialized Multi-Level Computational Approach. Molecules 2021, 26, 2600. [Google Scholar] [CrossRef]
- Vieira, T.F.; Martins, F.G.; Moreira, J.P.; Barbosa, T.; Sousa, S.F. In Silico Identification of Possible Inhibitors for Protein Kinase B (PknB) of Mycobacterium tuberculosis. Molecules 2021, 26, 6162. [Google Scholar] [CrossRef]
- Pereira, R.B.; Pinto, N.F.S.; Fernandes, M.J.G.; Vieira, T.F.; Rodrigues, A.R.O.; Pereira, D.M.; Sousa, S.F.; Castanheira, E.M.S.; Fortes, A.G.; Gonçalves, M.S.T. Amino Alcohols from Eugenol as Potential Semisynthetic Insecticides: Chemical, Biological, and Computational Insights. Molecules 2021, 26, 6616. [Google Scholar] [CrossRef]
- Natal, C.M.; Fernandes, M.J.G.; Pinto, N.F.S.; Pereira, R.B.; Vieira, T.F.; Rodrigues, A.R.O.; Pereira, D.M.; Sousa, S.F.; Fortes, A.G.; Castanheira, E.M.S.; et al. New carvacrol and thymol derivatives as potential insecticides: Synthesis, biological activity, computational studies and nanoencapsulation. RSC Adv. 2021, 11, 34024–34035. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [PubMed]
- Sousa, S.F.; Coimbra, J.T.S.; Paramos, D.; Pinto, R.; Guimarães, R.S.; Teixeira, V.; Fernandes, P.A.; Ramos, M.J. Molecular dynamics analysis of a series of 22 potential farnesyltransferase substrates containing a CaaX-motif. J. Mol. Model. 2013, 19, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Molecular dynamics simulations on the critical states of the farnesyltransferase enzyme. Bioorg. Med. Chem. 2009, 17, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.A.; Perez, M.A.S.; Moreira, I.S.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Computational Alanine Scanning Mutagenesis: MM-PBSA vs TI. J. Chem. Theory Comput. 2013, 9, 1311–1319. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Redocking RMSD (Å) | ||||||||
|---|---|---|---|---|---|---|---|---|
| PDB Code | Ligand | Vina | LeDock | CHEMPLP | GoldScore | ChemScore | ASP | Average per Target |
| 4JVD | NNQ | 6.67 | 3.51 | 2.16 | 3.20 | 1.18 | 2.68 | 3.23 |
| 4JVI | QZN | 1.59 | 3.07 | 1.33 | 2.93 | 3.18 | 1.85 | 2.33 |
| 6B8A | M64 | 0.34 | 0.58 | 0.46 | 0.63 | 3.48 | 1.92 | 1.24 |
| 6Q7U | HLH | 7.26 | 5.80 | 3.71 | 3.14 | 2.64 | 2.11 | 4.11 |
| 6Q7V | C11 | 5.77 | 6.16 | 3.49 | 5.73 | 3.76 | 1.77 | 4.98 |
| 6Q7W | C20 | 3.54 | 4.50 | 1.99 | 5.15 | 3.17 | 1.71 | 3.34 |
| 6TPR | NV5 | 9.15 | 5.23 | 1.54 | 1.16 | 4.59 | 1.35 | 3.84 |
| 6Z07 | Q4E | 1.22 | 1.41 | 0.92 | 0.87 | 1.31 | 1.32 | 1.18 |
| 6Z17 | Q4W | 1.18 | 7.25 | 1.59 | 4.08 | 2.32 | 1.96 | 3.06 |
| 6Z5K | QAE | 1.43 | 4.46 | 1.17 | 8.13 | 3.95 | 1.60 | 3.46 |
| 6YZ3 | Q25 | 1.11 | 7.67 | 1.25 | 0.99 | 3.46 | 1.56 | 2.67 |
| Average by SF | 3.57 | 4.51 | 1.78 | 3.27 | 3.00 | 1.81 | ||
| 4JVI | 6B8A | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EF 1% | AUC% | TG | RIE | BEDROC | EF 1% | AUC | TG | RIE | BEDROC | |
| Vina | 10.23 | 0.68 | 0.21 | 2.34 | 0.14 | 10.35 | 0.63 | 0.19 | 2.13 | 0.13 |
| CHEMPLP | 10.40 | 0.55 | 0.08 | 2.22 | 0.13 | 5.20 | 0.54 | 0.07 | 2.07 | 0.12 |
| ChemScore | 5.20 | 0.49 | 0.005 | 1.68 | 0.10 | 2.60 | 0.52 | 0.02 | 1.65 | 0.10 |
| ASP | 10.40 | 0.66 | 0.21 | 2.39 | 0.14 | 10.39 | 0.66 | 0.25 | 2.33 | 0.14 |
| Drug Name & Code | Description | Structure | ASP Score |
|---|---|---|---|
| Nilotinib | Bcr-Abl tyrosine kinase inhibitor (TKI) used in the treatment of chronic myelogenous leukemia (CML) |  | 54.96 |
| Indocyanine Green | Dye used in medical diagnosis. It has been used to measure cardiac output, liver function, and in ophthalmic angiography [33]. |  | 50.55 |
| Lomitapide | Used to treat patients with Homozygous familial hypercholesterolaemia (HoFH). It is an inhibitor of MTP, an enzyme responsible for the synthesis of low-density lipoproteins in the liver [34]. |  | 50.01 |
| Valrubicin | Chemotherapy drug used to treat carcinoma in situ bladder tumors. |  | 49.89 |
| Lapatinib | Inhibitor of tyrosine kinase domains of epidermal growth factor receptor and human epidermal growth factor receptor (HER)-2. Used to treat metastatic HER-2 + breast cancer [35]. |  | 49.84 |
| Pazopanib | Multitarget tyrosine kinase inhibitor approved for the treatment of multiple histological subtypes of soft tissue sarcoma (STS) [36]. |  | 49.69 |
| Ravicti | Used for the treatment of patients with urea cycle disorders (UCDs) [37]. |  | 48.49 |
| Cabozantinib | Tyrosine kinase inhibitor that targets pathways that have been linked to tumor growth. Used for the treatment of metastatic renal cell carcinoma [38]. |  | 48.32 |
| Venetoclax | Inhibitor of B-cell leukemia/lymphoma-2 protein [39]. |  | 48.05 |
| Isavuconazonium | Prodrug used as antifungal for the treatment of invasive aspergillosis and invasive mucormycosis [40]. |  | 48.05 |
| Emend | NK1 antagonist to prevent chemotherapy-induced nausea and vomiting. |  | 46.77 |
| Amethopterin or Methotrexate | Analog and antagonist of folic acid, is commonly used in the treatment of a wide range of malignant and non-malignant diseases [41]. |  | 46.10 |
| Cefsulodin | Broad-spectrum beta-lactamase stable cephalosporin with excellent activity against gram-negative bacilli, including P. aeruginosa [42]. |  | 46.01 |
| Montelukast | Leukotriene receptor antagonist (LTRAs) used for asthma treatment [43]. |  | 45.27 |
| Cefoperazone | Is a parenteral, third-generation cephalosporin that can be given intravenously or intramuscularly [44]. |  | 45.05 |
| ID | Average Protein RMSd (Å) | Average Ligand RMSD | SASA (Å2) | Percentage of Potential Ligand SASA Buried (%) | Average Number H Bonds | ΔGbind (kcal/mol) | Main Contributors (kcal/mol) |
|---|---|---|---|---|---|---|---|
| Venetoclax | 2.3 ± 0.2 | 2.7 ± 0.4 | 344.8 ± 27.0 | 70.4 ± 0.02 | 0.5 ± 0.6 | −70.1 ± 0.3 | TYR258 (−4.9), ILE186 (−4.3), ILE236 (−2.9) |
| Indocyanine Green | 2.2 ± 0.2 | 2.3 ± 0.2 | 298.9 ± 28.3 | 72.3 ± 0.03 | 1.1 ± 0.8 | −58.6 ± 0.3 | ILE186 (−4.2), ILE236 (−3.1), TYR258 (−3.1) |
| Nilotinib | 2.0 ± 0.2 | 1.7 ± 0.4 | 105.2 ± 23.2 | 86.8 ± 0.03 | 0.1 ± 0.3 | −48.1 ± 0.2 | ILE186 (−2.4), LEU208 (−2.1), ILE236 (−3.4) |
| Cabozantinib | 2.3 ± 0.3 | 1.6 ± 0.4 | 135.3 ± 40.5 | 82.3 ± 0.1 | 0.03 ± 0.2 | −44.6 ± 0.2 | ILE236 (−3.2), ILE186 (−2.4), LEU208 (−2.3) |
| Montelukast | 2.2 ± 0.2 | 2.2 ± 0.4 | 228.8 ± 35.6 | 73.8 ± 0.04 | 0.04 ± 0.2 | −43.2 ± 0.2 | ILE236 (−2.8), LEU207 (−2.4), ILE263 (−2.0) |
| Cefoperazone | 2.6 ± 0,3 | 2.9 ± 1.2 | 289.5 ± 41.4 | 66.7 ± 0.04 | 1.0 ± 0.9 | −42.4 ± 0.4 | ARG209 (−3.2), ILE236 (−2.5), LEU208 (−2.3) |
| Valrubicin | 2.4 ± 0.4 | 3.3 ± 0.3 | 276.7 ± 31.4 | 70.6 ± 0.03 | 0.04 ± 0.2 | −41.1 ± 0.2 | LEU207 (−4.2), ILE236 (−2.9), TYR258 (−3.9) |
| Lomitapide | 2.3 ± 0.3 | 3.8 ± 0.4 | 323.4 ± 81.6 | 65.6 ± 0.1 | 0.1 ± 0.3 | −40.6 ± 0.3 | ILE186 (−2.3), LEU208 (−1.9), ILE236 (−2.5) |
| M64 (antagonist) | 2.2 ± 0.2 | 1.2 ± 0.2 | 90.4 ± 19.3 | 86.3 ± 0.03 | 0.1 ± 0.2 | −39.0 ± 0.1 | IlE236 (−3.2), ILE186 (−1.7), TYR258 (−1.7) |
| Emend | 2.3 ± 0.4 | 1.5 ± 0.2 | 130.2 ± 25.8 | 80.7 ± 0.04 | 0.4 ± 0.5 | −38.9 ± 0.2 | ILE236 (−3.5), LEU207 (−109), TYR258 (−1.7) |
| Pazopanib | 2.3 ± 0.2 | 1.9 ± 0.5 | 203.6 ± 40.5 | 69.4 ± 0.1 | 0.7 ± 0.8 | −37.7 ± 0.3 | LEU208 (−3.7), ILE236 (−3.1), SER196 (−2.0) |
| lapatinib | 2.5 ± 0.4 | 2.8 ± 0.9 | 249.0 ± 56.7 | 69.6 ± 0.1 | 0.5 ± 0.7 | −35.4 ± 0.3 | LEU207 (−2.1), ILE236 (−2.9), ILE263 (−1.8) |
| Ravicti | 2.1 ± 0.2 | 3.3 ± 0.7 | 263.8 ± 50.0 | 69.3 ± 0.1 | 0.03 ± 0.2 | −34.5 ± 0.3 | LEU207 (−2.6), ILE236 (−2.6), ILE263 (−1.7) |
| Cefsulodin | 2.3 ± 0.2 | 1.6 ± 0.4 | 235.4 ± 39.9 | 66.5 ± 0.1 | 0.9 ± 0.8 | −27.4 ± 0.3 | LEU207 (−3.5), ILE236 (−2.2), LEU208 (−2.1) |
| NNQ (natural inducer) | 2.3 ± 0.3 | 1.6 ± 0.4 | 154.8 ± 53.7 | 71.6 ± 0.1 | 0.1 ± 0.3 | −26.1 ± 0.3 | LEU208 (−1.7), ILE236 (−1.5) |
| Isavuconazonium | 2.4 ± 0.2 | 2.7 ± 0.5 | 522.5 ± 47.4 | 42.7 ± 0.1 | 0.04 ± 0.2 | −25.0 ± 0.1 | TYR258 (−3.4), ILE186 (−2.6), LEU189 (−1.1) |
| Methotrexate | 2.8 ± 0.4 | 2.4 ± 0.4 | 312.6 ± 69.2 | 53.8 ± 0.1 | 0.7 ± 0.8 | −22.8 ± 0.3 | ILE186 (−4.1), TYR258 (−3.4), ARG209 (−2.7) |
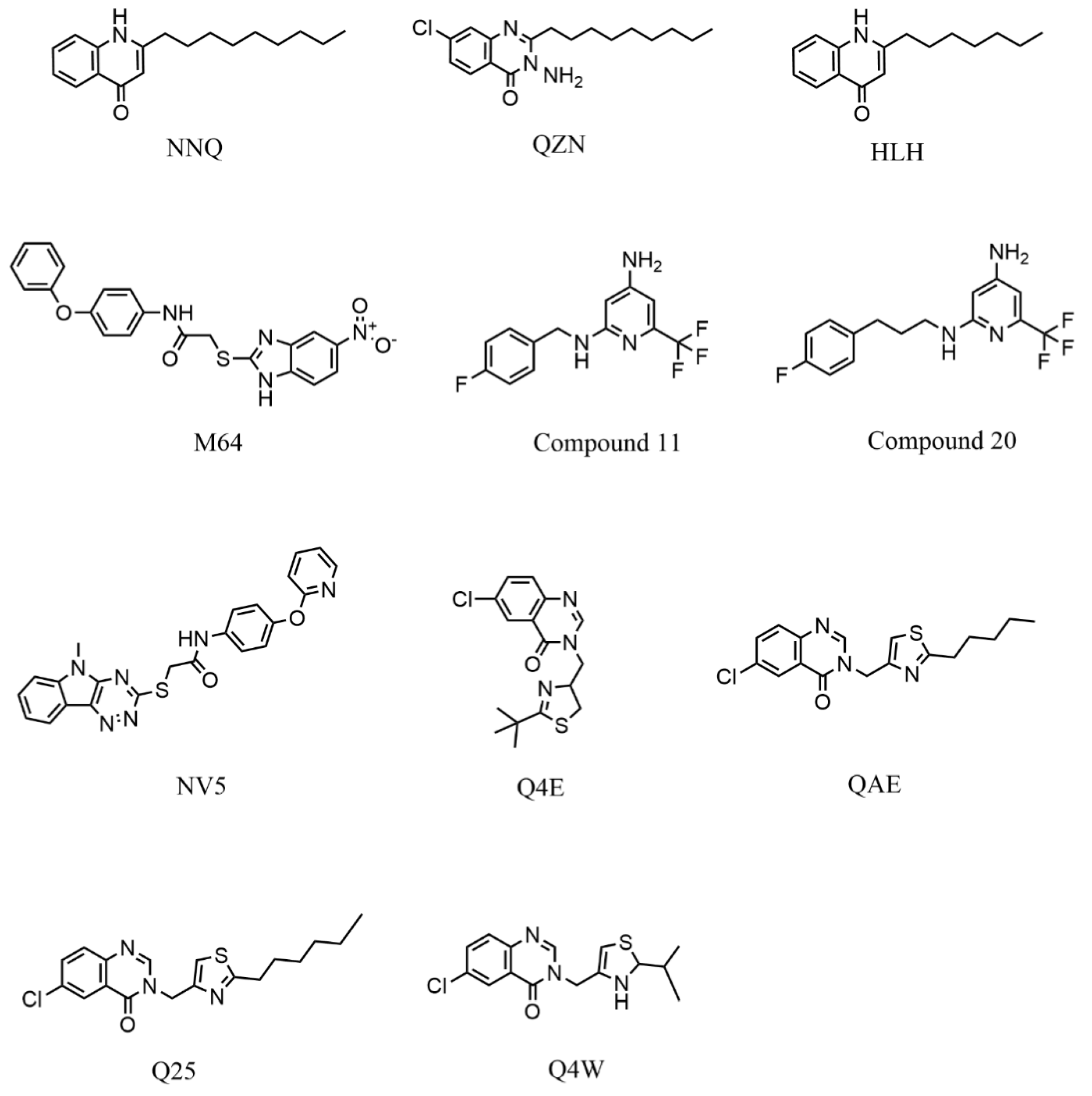
| PDB Code | Protein | Resolution (Å) | Ligand | Strain | References |
|---|---|---|---|---|---|
| 4JVC | Ligand-Binding Domain | 2.50 | UCBPP-PA14 | [20] | |
| 4JVD | Ligand-Binding Domain | 2.95 | NNQ | ||
| 4JVI | Ligand-Binding Domain | 2.90 | QZN | ||
| 6B8A | Ligand-Binding Domain | 2.65 | M64 | PAO1 | [23] |
| 6Q7U | Ligand-Binding Domain | 3.15 | HLH | PAO1 | [22] |
| 6Q7V | Ligand-Binding Domain | 2.56 | HLK | ||
| 6Q7W | Ligand-Binding Domain | 2.82 | HLQ | ||
| 6TPR | Ligand-Binding Domain | 3.20 | NV5 | UCBPP-PA14 | [47] |
| 6Z07 | Ligand-Binding Domain | 2.95 | Q4E | UCBPP-PA14 | [48] |
| 6Z17 | Ligand-Binding Domain | 3.15 | Q4W | ||
| 6Z5K | Ligand-Binding Domain | 3.20 | QAE | ||
| 6YZ3 | Ligand-Binding Domain | 3.00 | Q25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira, T.F.; Magalhães, R.P.; Simões, M.; Sousa, S.F. Drug Repurposing Targeting Pseudomonas aeruginosa MvfR Using Docking, Virtual Screening, Molecular Dynamics, and Free-Energy Calculations. Antibiotics 2022, 11, 185. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11020185
Vieira TF, Magalhães RP, Simões M, Sousa SF. Drug Repurposing Targeting Pseudomonas aeruginosa MvfR Using Docking, Virtual Screening, Molecular Dynamics, and Free-Energy Calculations. Antibiotics. 2022; 11(2):185. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11020185
Chicago/Turabian StyleVieira, Tatiana F., Rita P. Magalhães, Manuel Simões, and Sérgio F. Sousa. 2022. "Drug Repurposing Targeting Pseudomonas aeruginosa MvfR Using Docking, Virtual Screening, Molecular Dynamics, and Free-Energy Calculations" Antibiotics 11, no. 2: 185. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11020185