
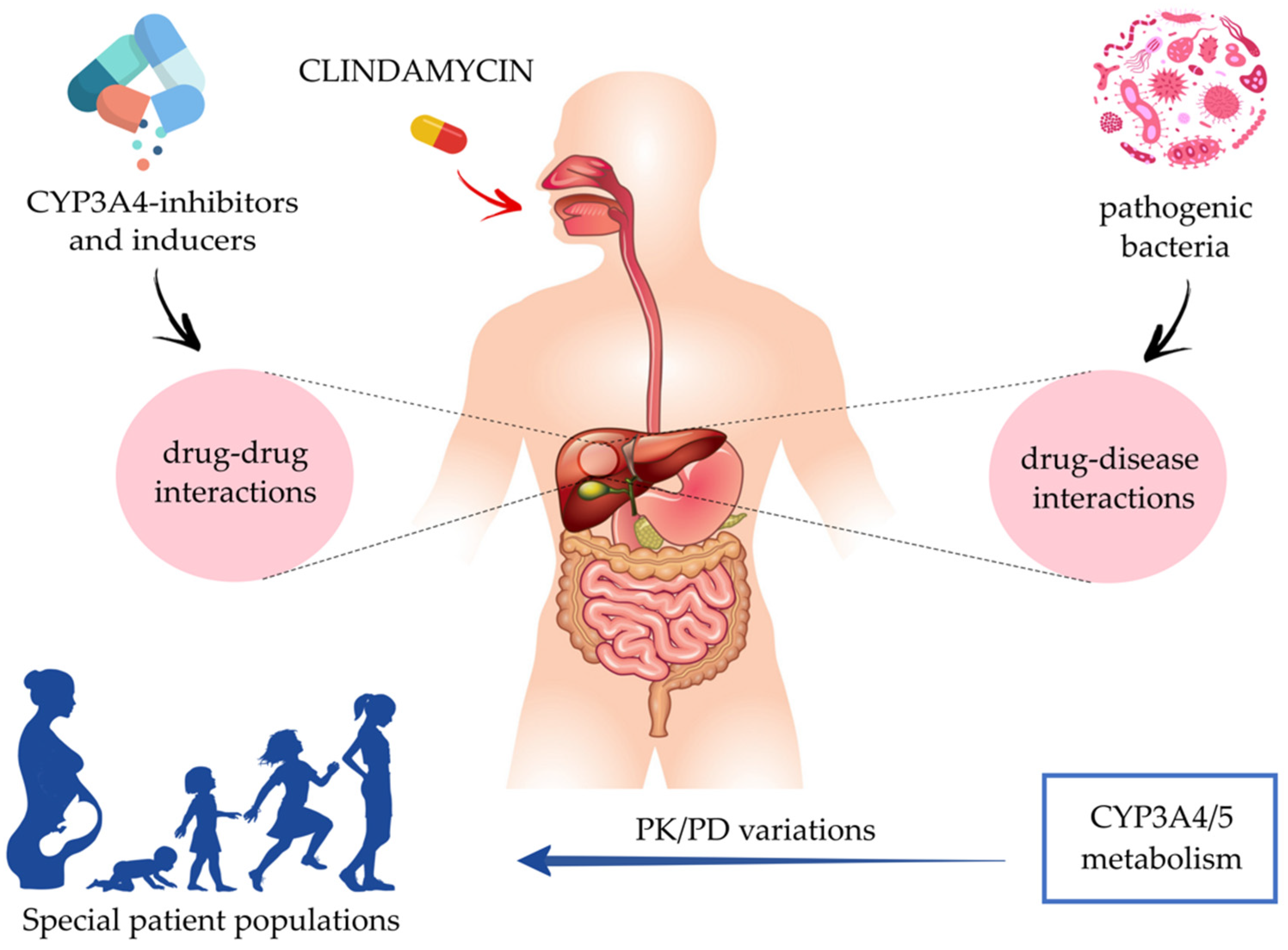
Ways to Improve Insights into Clindamycin Pharmacology and Pharmacokinetics Tailored to Practice

 ,
,  and
and
Abstract
:1. Introduction
2. Pharmacology and Target
3. Pharmacokinetics
3.1. Pharmacokinetics in Reference Population
3.2. Pharmacokinetics in Special Patient Populations
3.2.1. Pediatrics

{kind=link}
{kind=link}
| Pediatrics | Pregnant, Breastfeeding and Postpartum Women | |||||
|---|---|---|---|---|---|---|
| PK Covariates | Neonates (0 to <28 Days) | Infants (28 Days to <2 Years) | Young Children (2 to <6 Years) | Old Children (6 to <12 Years) | Adolescents (12 to 18 Years) | |
| Absorption | ||||||
| Gastric pH 2 | pH ~7: postnatal peak pH ~2.0–2.7: rapid decrease after birth | pH ~2–3 | pH ~2–3 | pH ~2–3 | pH ~2–3 | Pregnant women: increased gastric acid secretion, but no major changes in gastric pH |
| Gastric emptying | Highly variable | Highly variable until ~6 months | More stable | More stable | More stable | Pregnant women: gastric emptying does not appear to be affected |
| GI 1 transit time | Slower than adults | Slower than adults | Adult values | Adult values | Adult values | Pregnant women: GI transit time could be longer in the third trimester when intestinal motility is lower |
| Other factors | NS 1 | Pregnant women: nausea and vomiting also diminish absorption in the early pregnancy | ||||
| Distribution | ||||||
| Protein binding: maturational changes 3 | Low protein binding rate 7 | Low protein binding rate until ~10 months, then adult value rate | Adult value rate (77 mg/mL) | Adult value rate (77 mg/mL) | Adult value rate (77 mg/mL) | Pregnant women: reduction in AAG 1 and albumin fractions over pregnancy trimesters. From ~100% (prepregnant, first trimester) to ~80% (second, third trimesters) [8] |
| Protein binding: nonmaturational changes 4 | Generally increase in serum AAG concentrations | Type of delivery (cesarean or vaginal): increase in AAG serum concentrations, no significant changes in albumin | ||||
| Transplacental distribution | NA 1 | Breastfeeding women: human breast milk concentrations of ~0.7 – 3.8 mcg/mL during lactation | ||||
| Metabolism | ||||||
| CYP3A4 1 enzyme activity 5 | Postnatal increase in microsomal levels 8, 50% of adult levels | 50% of adult levels until 1 year, then adult values are slowly reached | Adult values are slowly reached (between 1 to 5 years) | Adult values | Adult values | Pregnant women: drastic increase in CYP3A4 enzyme activity from prepregnant (~100%) to first, second and third trimester (~210%) [8] |
| Drug CL 1 CYP3A4-substrate (midazolam) 6 | Results from a popPK 1 model quantifying CL changes [34]: 30% decrease in midazolam CL in the presence of increasing inflammation (3-fold), 26% decrease in midazolam CL in the presence of increasing organ failure (from 1 organ to 2) | Pregnant women: ~100-fold increase in sex hormones. Increase of CL [35] for pregnant women (593 ± 237 L/min) compared with postpartum (343 ± 103 L/min) | ||||
3.2.2. Pregnant, Breastfeeding, and Postpartum Women
4. Clinical Practice and Efficacy
4.1. Surgical Prophylaxis in the Event of Beta-Lactam Allergy
4.2. Prophylaxis and Treatment of Pregnancy Infections
4.3. Treatment of Diabetic Foot Infections
4.4. Treatment of Bone and Joint, Fracture-Related, and Periprosthetic Joint Infections
5. CYP3A4-Mediated Drug–Drug Interactions
5.1. CYP3A4-Inhibition: Macrolides and Antiretroviral Drugs—Clindamycin
5.2. CYP3A4-Induction: Rifampicin–Clindamycin
6. Drug–Disease Interactions: Impact of Inflammation on CYP3A4/5 Activity
7. Safety and Adverse Event Profile
8. Discussion
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- European Comission Health Research and Innovation. Antimicrobial Drug Resistance (AMR). Available online: https://ec.europa.eu/info/research-and-innovation/research-area/health-research-and-innovation/antimicrobial-drug-resistance-amr_en (accessed on 10 April 2022).
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guay, D. Update on clindamycin in the management of bacterial, fungal and protozoal infections. Expert Opin. Pharmacother. 2007, 8, 2401–2444. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of Methicillin-Resistant Staphylococcus aureus Infections in Adults and Children. Clin. Infect. Dis. 2011, 52, e18–e55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynalda, M.A.; Hutzler, J.M.; Koets, M.D.; Podoll, T.; Wienkers, L.C. In vitro metabolism of clindamycin in human liver and intestinal microsomes. Drug Metab. Dispos. 2003, 31, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Barton, G.; Fischer, A. Pharmacokinetic considerations and dosing strategies of antibiotics in the critically ill patient. J. Intensive Care Soc. 2015, 16, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.E.; Mouton, J.W.; Oostvogel, P.M.; Dörr, P.J.; Voskuyl, R.A.; DeJongh, J.; Steegers, E.A.P.; Danhof, M. Pharmacokinetics of clindamycin in pregnant women in the peripartum period. Antimicrob. Agents Chemother. 2010, 54, 2175–2181. [Google Scholar] [CrossRef] [Green Version]
- Allegaert, K.; Muller, A.E.; Russo, F.; Schoenmakers, S.; Deprest, J.; Koch, B.C.P. Pregnancy-related pharmacokinetics and antimicrobial prophylaxis during fetal surgery, cefazolin and clindamycin as examples. Prenat. Diagn. 2020, 40, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Schreckenberger, P.C.; Ilendo, E.; Ristow, K.L. Incidence of Constitutive and Inducible Clindamycin Resistance in Staphylococcus aureus and Coagulase-Negative Staphylococci in a Community and a Tertiary Care Hospital. J. Clin. Microbiol. 2004, 42, 2777–2779. [Google Scholar] [CrossRef] [Green Version]
- European Committee on Antimicrobial Susceptibility Testing. MIC Distributions and Epidemiological Cut-Off Value (ECOFF) Setting, EUCAST SOP 10. 2022. Available online: https://www.eucast.org/organization/subcommittees/mic_distributions_and_ecoffs/ (accessed on 10 April 2022).
- Spížek, J.; Řezanka, T. Lincosamides: Chemical structure, biosynthesis, mechanism of action, resistance, and applications. Biochem. Pharmacol. 2017, 133, 20–28. [Google Scholar] [CrossRef]
- Levison, M.E.; Levison, J.H. Pharmacokinetics and Pharmacodynamics of Antibacterial Agents. Infect. Dis. Clin. N. Am. 2009, 23, 791–815. [Google Scholar] [CrossRef] [Green Version]
- Wade, K.C.; Benjamin, D.K. Clinical Pharmacology of Anti-Infective Drugs. In Infectious Diseases of the Fetus and Newborn; Elsevier: Amsterdam, The Netherlands, 2011; pp. 1160–1211. [Google Scholar]
- Chavez-Bueno, S.; Bozdogan, B.; Katz, K.; Bowlware, K.L.; Cushion, N.; Cavuoti, D.; Ahmad, N.; McCracken, G.H.; Appelbaum, P.C. Inducible Clindamycin Resistance and Molecular Epidemiologic Trends of Pediatric Community-Acquired Methicillin-Resistant Staphylococcus aureus in Dallas, Texas. Antimicrob. Agents Chemother. 2005, 49, 2283–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, R. Mechanisms of Resistance to Macrolides and Lincosamides: Nature of the Resistance Elements and Their Clinical Implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.S.; Jorgensen, J.H. Inducible Clindamycin Resistance in Staphylococci: Should Clinicians and Microbiologists be Concerned? Clin. Infect. Dis. 2005, 40, 280–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, P.S.; Davis, S.L. Pharmacokinetic and Pharmacodynamic Issues in the Treatment of Bacterial Infectious Diseases. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Bouazza, N.; Pestre, V.; Jullien, V.; Curis, E.; Urien, S.; Salmon, D.; Tréluyer, J.M. Population pharmacokinetics of clindamycin orally and intravenously administered in patients with osteomyelitis. Br. J. Clin. Pharmacol. 2012, 74, 971–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatti, G.; Flaherty, J.; Bubp, J.; White, J.; Borin, M.; Gambertoglio, J. Comparative study of bioavailabilities and pharmacokinetics of clindamycin in healthy volunteers and patients with AIDS. Antimicrob. Agents Chemother. 1993, 37, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.L.; Pharm, D. A Monthly Newsletter for Health Care Professionals. Pediatr. Pharmacother. 2010, 16, 2–5. [Google Scholar]
- Hand, W.L.; King-Thompson, N.L. Membrane transport of clindamycin in alveolar macrophages. Antimicrob. Agents Chemother. 1982, 21, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, T.H. Cellular Transport of Drugs. Clin. Infect. Dis. 1994, 19, 916–921. [Google Scholar] [CrossRef]
- van den Broek, A.K.; Prins, J.M.; Visser, C.E.; van Hest, R.M. Systematic review: The bioavailability of orally administered antibiotics during the initial phase of a systemic infection in non-ICU patients. BMC Infect. Dis. 2021, 21, 285. [Google Scholar] [CrossRef]
- Picardi, J.L.; Lewis, H.P.; Tan, J.S.; Phair, J.P. Clindamycin concentrations in the central nervous system of primates before and after head trauma. J. Neurosurg. 1975, 43, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Castle, S.S. Clindamycin. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–4. [Google Scholar]
- Brandon Bookstaver, P.; Bland, C.M.; Griffin, B.; Stover, K.R.; Eiland, L.S.; McLaughlin, M. A review of antibiotic use in pregnancy. Pharmacotherapy 2015, 35, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration, HHS. International Conference on Harmonisation; guidance on E11 clinical investigation of medicinal products in the pediatric population; availability. Notice. Fed. Regist. 2000, 65, 78493–78494. [Google Scholar]
- Laughon, M.M.; Avant, D.; Tripathi, N.; Hornik, C.P.; Cohen-Wolkowiez, M.; Clark, R.H.; Smith, P.B.; Rodriguez, W. Drug labeling and exposure in neonates. JAMA Pediatr. 2014, 168, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.; Delmore, P.; Bloom, B.T.; Cotten, C.M.; Poindexter, B.B.; McGowan, E.; Shattuck, K.; Bradford, K.K.; Smith, P.B.; Cohen-Wolkowiez, M.; et al. Clindamycin Pharmacokinetics and Safety in Preterm and Term Infants. Antimicrob. Agents Chemother. 2016, 60, 2888–2894. [Google Scholar] [CrossRef] [Green Version]
- Van Den Abeele, J.; Rayyan, M.; Hoffman, I.; Van de Vijver, E.; Zhu, W.; Augustijns, P. Gastric fluid composition in a paediatric population: Age-dependent changes relevant for gastrointestinal drug disposition. Eur. J. Pharm. Sci. 2018, 123, 301–311. [Google Scholar] [CrossRef]
- Mooij, M.G.; de Koning, B.A.; Huijsman, M.L.; de Wildt, S.N. Ontogeny of oral drug absorption processes in children. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1293–1303. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, T. Essentials for starting a pediatric clinical study (1): Pharmacokinetics in children. J. Toxicol. Sci. 2009, 34, SP307–SP312. [Google Scholar] [CrossRef] [Green Version]
- Stillhart, C.; Vučićević, K.; Augustijns, P.; Basit, A.W.; Batchelor, H.; Flanagan, T.R.; Gesquiere, I.; Greupink, R.; Keszthelyi, D.; Koskinen, M.; et al. Impact of gastrointestinal physiology on drug absorption in special populations—An UNGAP review. Eur. J. Pharm. Sci. 2020, 147, 105280. [Google Scholar] [CrossRef]
- Bonner, J.J.; Vajjah, P.; Abduljalil, K.; Jamei, M.; Rostami-Hodjegan, A.; Tucker, G.T.; Johnson, T.N. Does age affect gastric emptying time? A model-based meta-analysis of data from premature neonates through to adults. Biopharm. Drug Dispos. 2015, 36, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Booker, P.D.; Taylor, C.; Saba, G. Perioperative changes in alpha 1-acid glycoprotein concentrations in infants undergoing major surgery. Br. J. Anaesth. 1996, 76, 365–368. [Google Scholar] [CrossRef]
- İpek, İ.Ö.; Saracoglu, M.; Bozaykut, A. α 1-Acid glycoprotein for the early diagnosis of neonatal sepsis. J. Matern. Neonatal Med. 2010, 23, 617–621. [Google Scholar] [CrossRef]
- Tracy, T.S.; Chaudhry, A.S.; Prasad, B.; Thummel, K.E.; Schuetz, E.G.; Zhong, X.-B.; Tien, Y.-C.; Jeong, H.; Pan, X.; Shireman, L.M.; et al. Interindividual Variability in Cytochrome P450-Mediated Drug Metabolism. Drug Metab. Dispos. 2016, 44, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, K.; Wright, I.M.R.; Schneider, J.J.; Jones, A.L.; Martin, J.H. Pharmacokinetics in neonatal prescribing: Evidence base, paradigms and the future. Br. J. Clin. Pharmacol. 2015, 80, 1281–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Groen, B.D.; Nicolaï, J.; Kuik, A.C.; Van Cruchten, S.; van Peer, E.; Smits, A.; Schmidt, S.; de Wildt, S.N.; Allegaert, K.; De Schaepdrijver, L.; et al. Ontogeny of Hepatic Transporters and Drug-Metabolizing Enzymes in Humans and in Nonclinical Species. Pharmacol. Rev. 2021, 73, 597–678. [Google Scholar] [CrossRef]
- Elens, L.; van Gelder, T.; Hesselink, D.A.; Haufroid, V.; van Schaik, R.H. CYP3A4*22: Promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics 2013, 14, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Brussee, J.M.; Vet, N.J.; Krekels, E.H.J.; Valkenburg, A.J.; Jacqz-Aigrain, E.; van Gerven, J.M.A.; Swart, E.L.; van den Anker, J.N.; Tibboel, D.; de Hoog, M.; et al. Predicting CYP3A-mediated midazolam metabolism in critically ill neonates, infants, children and adults with inflammation and organ failure. Br. J. Clin. Pharmacol. 2018, 84, 358–368. [Google Scholar] [CrossRef]
- Barrett, S.; Blake, J.; Bradley, C.; Cleary, B.; Cronin, C.; Delaney, A.; Drew, R.; Duddy, P.; Fitzgerald, D.; Flaherty, E.; et al. Medication Guidelines for Obstetrics and Gynaecology: Antimicrobial safety in pregnancy and lactation. Inst. Obstet. Gynaecol. Health Serv. Exec. 2017, 2, 80. [Google Scholar]
- Wear, C.D.; Towers, C.V.; Brown, M.S.; Weitz, B.; Porter, S.; Wolfe, L. Transplacental passage of clindamycin from mother to neonate. J. Perinatol. 2016, 36, 960–961. [Google Scholar] [CrossRef]
- Costantine, M.M. Physiologic and pharmacokinetic changes in pregnancy. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef]
- Pariente, G.; Leibson, T.; Carls, A.; Adams-Webber, T.; Ito, S.; Koren, G. Pregnancy-Associated Changes in Pharmacokinetics: A Systematic Review. PLoS Med. 2016, 13, e1002160. [Google Scholar] [CrossRef] [PubMed]
- Chiloiro, M.; Darconza, G.; Piccioli, E.; De Carne, M.; Clemente, C.; Riezzo, G. Gastric emptying and orocecal transit time in pregnancy. J. Gastroenterol. 2001, 36, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Larijani, G.E.; Norris, M.C.; Ala-Kokko, T.I.; Leighton, B.A.; Desimone, C. Serum Concentration of Alpha,-Acid Glycoprotein and Albumin following Cesarean Section and Vaginal Delivery. DICP 1990, 24, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Abduljalil, K.; Furness, P.; Johnson, T.N.; Rostami-Hodjegan, A.; Soltani, H. Anatomical, Physiological and Metabolic Changes with Gestational Age during Normal Pregnancy. Clin. Pharmacokinet. 2012, 51, 365–396. [Google Scholar] [CrossRef]
- Hebert, M.; Easterling, T.; Kirby, B.; Carr, D.; Buchanan, M.; Rutherford, T.; Thummel, K.; Fishbein, D.; Unadkat, J. Effects of Pregnancy on CYP3A and P-glycoprotein Activities as Measured by Disposition of Midazolam and Digoxin: A University of Washington Specialized Center of Research Study. Clin. Pharmacol. Ther. 2008, 84, 248–253. [Google Scholar] [CrossRef]
- Papageorgiou, I.; Grepper, S.; Unadkat, J.D. Induction of Hepatic CYP3A Enzymes by Pregnancy-Related Hormones: Studies in Human Hepatocytes and Hepatic Cell Lines. Drug Metab. Dispos. 2013, 41, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.B.; Bistas, K.G.; Le, J.K. Clindamycin. [Updated 10 April 2022]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK519574/ (accessed on 10 April 2022).
- Depypere, M.; Kuehl, R.; Metsemakers, W.-J.; Senneville, E.; McNally, M.A.; Obremskey, W.T.; Zimmerli, W.; Atkins, B.L.; Trampuz, A. Fracture-Related Infection (FRI) Consensus Group Recommendations for Systemic Antimicrobial Therapy in Fracture-Related Infection: A Consensus From an International Expert Group. J. Orthop. Trauma 2020, 34, 30–41. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Surveillance of Surgical Site Infections and Prevention Indicators in European Hospitals: HAI-Net SSI protocol, version 2.2; European Centre for Disease Prevention and Control: Solna, Sweden, 2017. [Google Scholar]
- Young, P.Y.; Khadaroo, R.G.; FRCSC. Surgical Site Infections. Surg. Clin. N. Am. 2014, 94, 1245–1264. [Google Scholar] [CrossRef]
- Mundhada, A.; Tenpe, S. A study of organisms causing surgical site infections and their antimicrobial susceptibility in a tertiary care Government Hospital. Indian J. Pathol. Microbiol. 2015, 58, 195. [Google Scholar] [CrossRef]
- Bratzler, D.W.; Dellinger, E.P.; Olsen, K.M.; Perl, T.M.; Auwaerter, P.G.; Bolon, M.K.; Fish, D.N.; Napolitano, L.M.; Sawyer, R.G.; Slain, D.; et al. Clinical Practice Guidelines for Antimicrobial Prophylaxis in Surgery. Surg. Infect. (Larchmt). 2013, 14, 73–156. [Google Scholar] [CrossRef]
- Antibiotic Prophylaxis for Inpatient and Outpatient Surgery. Available online: https://www.smh.com/Portals/0/Documents/Services/Surgery%20Forms/Antibiotic%20Surgical%20Prophylaxis%20Protocol%20SMH%20October%202018%20Update.pdf (accessed on 10 April 2022).
- Lepetic, A.; Vujacich, C.; Calmaggi, A.; Guerrini, G.M.; Arzac, M.d.R.G. Surgical antibiotic prophylaxis. Morb. Obes. 2009, 173–194. [Google Scholar] [CrossRef]
- Heesen, M.; Klöhr, S.; Rossaint, R.; Allegeaert, K.; Deprest, J.; Van de Velde, M.; Straube, S. Concerning the timing of antibiotic administration in women undergoing caesarean section: A systematic review and meta-analysis. BMJ Open 2013, 3, e002028. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, T.; Landy, H.J. Surgical site infections after cesarean delivery: Epidemiology, prevention and treatment. Matern. Health Neonatol. Perinatol. 2017, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Verani, J.R.; McGee, L.; Schrag, S.J. Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention (CDC) Prevention of perinatal group B streptococcal disease--revised guidelines from CDC, 2010. MMWR Recomm. Rep. 2010, 59, 1–36. [Google Scholar] [PubMed]
- ACOG Committee Opinion No. 485: Prevention of early-onset group B streptococcal disease in newborns. Obstet. Gynecol. 2011, 117, 1019–1027. [CrossRef]
- Melin, P. Neonatal group B streptococcal disease: From pathogenesis to preventive strategies. Clin. Microbiol. Infect. 2011, 17, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharkey, M.; Hu, S.S. Diabetic Foot Infections. In Emergency Management of Infectious Diseases; Chin, R.L., Ed.; Cambridge University Press: Cambridge, UK, 2007; pp. 143–146. [Google Scholar]
- Lipsky, B.A.; Berendt, A.R.; Cornia, P.B.; Pile, J.C.; Peters, E.J.G.; Armstrong, D.G.; Deery, H.G.; Embil, J.M.; Joseph, W.S.; Karchmer, A.W.; et al. 2012 Infectious Diseases Society of America Clinical Practice Guideline for the Diagnosis and Treatment of Diabetic Foot Infections. Clin. Infect. Dis. 2012, 54, e132–e173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, M.G.; Ekkelenkamp, M.B.; Peters, E.J.G. Are clindamycin and ciprofloxacin appropriate for the empirical treatment of diabetic foot infections? Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 453–456. [Google Scholar] [CrossRef]
- Lipsky, B.A. Medical Treatment of Diabetic Foot Infections. Clin. Infect. Dis. 2004, 39, S104–S114. [Google Scholar] [CrossRef]
- Government of South Australia. Diabetic Foot Infections: Antibiotic Management Clinical Guideline. Gov. South Aust. 2019. [Google Scholar]
- Colston, J.; Atkins, B. Bone and joint infection. Clin. Med. 2018, 18, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheson, E.M.; Bragg, S.W.; Blackwelder, R.S. Diabetes-Related Foot Infections: Diagnosis and Treatment. Am. Fam. Physician 2021, 104, 386–394. [Google Scholar] [PubMed]
- Mader, J.T.; Adams, K.; Morrison, L. Comparative evaluation of cefazolin and clindamycin in the treatment of experimental Staphylococcus aureus osteomyelitis in rabbits. Antimicrob. Agents Chemother. 1989, 33, 1760–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depypere, M.; Morgenstern, M.; Kuehl, R.; Senneville, E.; Moriarty, T.F.; Obremskey, W.T.; Zimmerli, W.; Trampuz, A.; Lagrou, K.; Metsemakers, W.-J. Pathogenesis and management of fracture-related infection. Clin. Microbiol. Infect. 2020, 26, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, M.D.; Hischebeth, G.T.R.; Randau, T.M.; Gathen, M.; Schildberg, F.A.; Fröschen, F.S.; Kohlhof, H.; Gravius, S. Difficult-to-treat pathogens significantly reduce infection resolution in periprosthetic joint infections. Diagn. Microbiol. Infect. Dis. 2020, 98, 115114. [Google Scholar] [CrossRef]
- Metsemakers, W.-J.; Morgenstern, M.; Senneville, E.; Borens, O.; Govaert, G.A.M.; Onsea, J.; Depypere, M.; Richards, R.G.; Trampuz, A.; Verhofstad, M.H.J.; et al. General treatment principles for fracture-related infection: Recommendations from an international expert group. Arch. Orthop. Trauma Surg. 2020, 140, 1013–1027. [Google Scholar] [CrossRef] [Green Version]
- Seng, P.; Vernier, M.; Gay, A.; Pinelli, P.-O.; Legré, R.; Stein, A. Clinical features and outcome of bone and joint infections with streptococcal involvement: 5-year experience of interregional reference centres in the south of France. New Microbes New Infect. 2016, 12, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-K.; Rombach, I.; Zambellas, R.; Walker, A.S.; McNally, M.A.; Atkins, B.L.; Lipsky, B.A.; Hughes, H.C.; Bose, D.; Kümin, M.; et al. Oral versus Intravenous Antibiotics for Bone and Joint Infection. N. Engl. J. Med. 2019, 380, 425–436. [Google Scholar] [CrossRef]
- Czekaj, J.; Dinh, A.; Moldovan, A.; Vaudaux, P.; Gras, G.; Hoffmeyer, P.; Lew, D.; Bernard, L.; Uçkay, I. Efficacy of a combined oral clindamycin-rifampicin regimen for therapy of staphylococcal osteoarticular infections. Scand. J. Infect. Dis. 2011, 43, 962–967. [Google Scholar] [CrossRef]
- Zeller, V.; Dzeing-Ella, A.; Kitzis, M.-D.; Ziza, J.-M.; Mamoudy, P.; Desplaces, N. Continuous Clindamycin Infusion, an Innovative Approach to Treating Bone and Joint Infections. Antimicrob. Agents Chemother. 2010, 54, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Curis, E.; Pestre, V.; Jullien, V.; Eyrolle, L.; Archambeau, D.; Morand, P.; Gatin, L.; Karoubi, M.; Pinar, N.; Dumaine, V.; et al. Pharmacokinetic variability of clindamycin and influence of rifampicin on clindamycin concentration in patients with bone and joint infections. Infection 2015, 43, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Trampuz, A.; Zimmerli, W. Antimicrobial Agents in Orthopaedic Surgery. Drugs 2006, 66, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Leijtens, B.; Elbers, J.B.W.; Sturm, P.D.; Kullberg, B.J.; Schreurs, B.W. Clindamycin-rifampin combination therapy for staphylococcal periprosthetic joint infections: A retrospective observational study. BMC Infect. Dis. 2017, 17, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berbari, E.F.; Kanj, S.S.; Kowalski, T.J.; Darouiche, R.O.; Widmer, A.F.; Schmitt, S.K.; Hendershot, E.F.; Holtom, P.D.; Huddleston, P.M.; Petermann, G.W.; et al. 2015 Infectious Diseases Society of America (IDSA) Clinical Practice Guidelines for the Diagnosis and Treatment of Native Vertebral Osteomyelitis in Adultsa. Clin. Infect. Dis. 2015, 61, e26–e46. [Google Scholar] [CrossRef] [Green Version]
- Spilf, O. Recommendations for bone and joint prosthetic device infections in clinical practice (prosthesis, implants, osteosynthesis). Méd. Mal. Infect. 2010, 40, 185–211. [Google Scholar] [CrossRef]
- Pea, F. Penetration of Antibacterials into Bone. Clin. Pharmacokinet. 2009, 48, 125–127. [Google Scholar] [CrossRef]
- Zeller, V.; Magreault, S.; Heym, B.; Salmon, D.; Kitzis, M.D.; Billaud, E.; Marmor, S.; Jannot, A.S.; Salomon, L.; Jullien, V. Influence of the clindamycin administration route on the magnitude of clindamycin–rifampicin interaction: A prospective pharmacokinetic study. Clin. Microbiol. Infect. 2021, 27, 1857.e1–1857.e7. [Google Scholar] [CrossRef]
- Boselli, E.; Allaouchiche, B. Bone tissue diffusion of antibiotics [Diffusion osseuse des antibiotiques]. Press. Med. 1999, 28, 2265–2276. [Google Scholar]
- Thabit, A.K.; Fatani, D.F.; Bamakhrama, M.S.; Barnawi, O.A.; Basudan, L.O.; Alhejaili, S.F. Antibiotic penetration into bone and joints: An updated review. Int. J. Infect. Dis. 2019, 81, 128–136. [Google Scholar] [CrossRef] [Green Version]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 12.0. 2022. Available online: http://www.eucast.org (accessed on 10 April 2022).
- Bernard, A.; Kermarrec, G.; Parize, P.; Caruba, T.; Bouvet, A.; Mainardi, J.L.; Sabatier, B.; Nich, C. Dramatic reduction of clindamycin serum concentration in staphylococcal osteoarticular infection patients treated with the oral clindamycin-rifampicin combination. J. Infect. 2015, 71, 200–206. [Google Scholar] [CrossRef]
- Zhou, S.; Chan, E.; Li, X.; Huang, M. Clinical outcomes and management of mechanism-based inhibition of cytochrome P450 3A4. Ther. Clin. Risk Manag. 2005, 1, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Rahmioglu, N.; Heaton, J.; Clement, G.; Gill, R.; Surdulescu, G.; Zlobecka, K.; Hodgkiss, D.; Ma, Y.; Hider, R.C.; Smith, N.W.; et al. Genetic epidemiology of induced CYP3A4 activity. Pharmacogenet. Genom. 2011, 21, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lu, A.Y. Interindividual Variability in Inhibition and Induction of Cytochrome P450 Enzymes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 535–567. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H. CYP induction-mediated drug interactions: In vitro assessment and clinical implications. Pharm. Res. 2006, 23, 1089–1116. [Google Scholar] [CrossRef] [PubMed]
- Molenaar-Kuijsten, L.; Van Balen, D.E.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. A Review of CYP3A Drug-Drug Interaction Studies: Practical Guidelines for Patients Using Targeted Oral Anticancer Drugs. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Agarwal, S.; Agarwal, S.K. Lopinavir-Ritonavir in SARS-CoV-2 Infection and Drug-Drug Interactions with Cardioactive Medications. Cardiovasc. Drugs Ther. 2021, 35, 427–440. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on the investigation of drug interactions. Eur. Med. Agency 2012, 44, 59. [Google Scholar]
- Pfizer. Fact Sheet for Patients, Parents, and Caregivers Emergency Use Authorization (EUA) of Paxolavid for Coronavirus Disease 2019 (COVID-19); Pfizer: New York, NY, USA, 2022; pp. 1–6. [Google Scholar]
- Komorowski, A.S.; Tseng, A.; Vandersluis, S.; Leung, E.; Ciccotelli, W.; Langford, B.J.; Andany, N.; Razak, F.; Wadhwa, W.; Jüni, P.; et al. Evidence-based recommendations on the use of nirmatrelvir/ritonavir (Paxlovid) for adults in Ontario; Ontario COVID-19 Science Advisory Table: Toronto, ON, Canada, 2022; Volume 3, p. 57. [Google Scholar] [CrossRef]
- Tompkins, L.M.; Wallace, A.D. Mechanisms of cytochrome P450 induction. J. Biochem. Mol. Toxicol. 2007, 21, 176–181. [Google Scholar] [CrossRef]
- Huwyler, J.; Wright, M.; Gutmann, H.; Drewe, J. Induction of Cytochrome P450 3A4 and P-Glycoprotein by the Isoxazolyl- Penicillin Antibiotic Flucloxacillin. Curr. Drug Metab. 2006, 7, 119–126. [Google Scholar] [CrossRef]
- Comuth, W.J.; Comuth, J.A.; Hauer, R.N.W.; Malingré, M.M. Interaction of flucloxacillin and quinidine. Eur. J. Clin. Pharmacol. 2012, 68, 891–893. [Google Scholar] [CrossRef]
- Muilwijk, E.W.; Dekkers, B.G.J.; Henriet, S.S.V.; Verweij, P.E.; Witjes, B.; Lashof, A.M.L.O.; Groeneveld, G.H.; van der Hoeven, J.; Alffenaar, J.W.C.; Russel, F.G.M.; et al. Flucloxacillin results in suboptimal plasma voriconazole concentrations. Antimicrob. Agents Chemother. 2017, 61, e00915-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenhof, H.; Schouw, H.M.; Besouw, M.T.P.; Touw, D.J.; Gracchi, V. Flucloxacillin decreases tacrolimus blood trough levels: A single-center retrospective cohort study. Eur. J. Clin. Pharmacol. 2020, 76, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, R.; Wauters, J.; De Cock, P.; Buyle, F.; Leys, J.; Van Brantegem, P.; Gijsen, M.; Annaert, P.; Debaveye, Y.; Lagrou, K.; et al. Concomitant Treatment with Voriconazole and Flucloxacillin: A Combination to Avoid. Antibiotics 2021, 10, 1112. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, R.; Wauters, J.; Vandenbriele, C.; Lagrou, K.; Vos, R.; Debaveye, Y.; Spriet, I. Interaction between flucloxacillin and azoles: Is isavuconazole next? Mycoses 2021, 64, 1508–1511. [Google Scholar] [CrossRef]
- Niemi, M.; Backman, J.T.; Fromm, M.F.; Neuvonen, P.J.; Kivistö, K.T. Pharmacokinetic Interactions with Rifampicin. Clin. Pharmacokinet. 2003, 42, 819–850. [Google Scholar] [CrossRef]
- de Jong, L.M.; Jiskoot, W.; Swen, J.J.; Manson, M.L. Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes 2020, 11, 1509. [Google Scholar] [CrossRef]
- Lenoir, C.; Rodieux, F.; Desmeules, J.A.; Rollason, V.; Samer, C.F. Impact of Inflammation on Cytochromes P450 Activity in Pediatrics: A Systematic Review. Clin. Pharmacokinet. 2021, 60, 1537–1555. [Google Scholar] [CrossRef]
- White, C.M. Inflammation Suppresses Patients’ Ability to Metabolize Cytochrome P450 Substrate Drugs. Ann. Pharmacother. 2021, 56, 809–819. [Google Scholar] [CrossRef]
- Trenholme, G.M.; Williams, R.L.; Rieckmann, K.H.; Frischer, H.; Carson, P.E. Quinine disposition during malaria and during induced fever. Clin. Pharmacol. Ther. 1976, 19, 459–467. [Google Scholar] [CrossRef]
- Sabchareon, A.; Chongsuphajaisiddhi, T.; Attanath, P. Serum quinine concentrations following the initial dose in children with falciparum malaria. Southeast Asian J. Trop. Med. Public Health 1982, 13, 556–562. [Google Scholar]
- White, N.J.; Looareesuwan, S.; Warrell, D.A.; Warrell, M.J.; Bunnag, D.; Harinasuta, T. Quinine pharmacokinetics and toxicity in cerebral and uncomplicated falciparum malaria. Am. J. Med. 1982, 73, 564–572. [Google Scholar] [CrossRef]
- Supanaranond, W.; Davis, T.M.E.; Pukrittayakamee, S.; Silamut, K.; Karbwang, J.; Molunto, P.; Chanond, L.; White, N.J. Disposition of oral quinine in acute falciparum malaria. Eur. J. Clin. Pharmacol. 1991, 40, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Pukrittayakamee, S.; Looareesuwan, S.; Keeratithakul, D.; Davis, T.M.E.; Teja-Isavadharm, P.; Nagachinta, B.; Weber, A.; Smith, A.L.; Kyle, D.; White, N.J. A study of the factors affecting the metabolic clearance of quinine in malaria. Eur. J. Clin. Pharmacol. 1997, 52, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Babalola, C.P.; Bolaji, O.O.; Ogunbona, F.A.; Sowunmi, A.; Walker, O. Pharmacokinetics of quinine in African patients with acute falciparum malaria. Pharm. World Sci. 1998, 20, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Raveh, D.; Rabinowitz, B.; Breuer, G.S.; Rudensky, B.; Yinnon, A.M. Risk factors for Clostridium difficile toxin-positive nosocomial diarrhoea. Int. J. Antimicrob. Agents 2006, 28, 231–237. [Google Scholar] [CrossRef]
- Osborne, N.G.; Ahluwalia, B. Pseudomembranous Colitis. J. Gynecol. Surg. 2009, 25, 129–131. [Google Scholar] [CrossRef]
- Brook, I. Pseudomembranous colitis in children. J. Gastroenterol. Hepatol. 2005, 20, 182–186. [Google Scholar] [CrossRef]
- Garey, K.W.; Jiang, Z.-D.; Yadav, Y.; Mullins, B.; Wong, K.; Dupont, H.L. Peripartum Clostridium difficile infection: Case series and review of the literature. Am. J. Obstet. Gynecol. 2008, 199, 332–337. [Google Scholar] [CrossRef]
- Slimings, C.; Riley, T.V. Antibiotics and healthcare facility-associated Clostridioides difficile infection: Systematic review and meta-analysis 2020 update. J. Antimicrob. Chemother. 2021, 76, 1676–1688. [Google Scholar] [CrossRef]


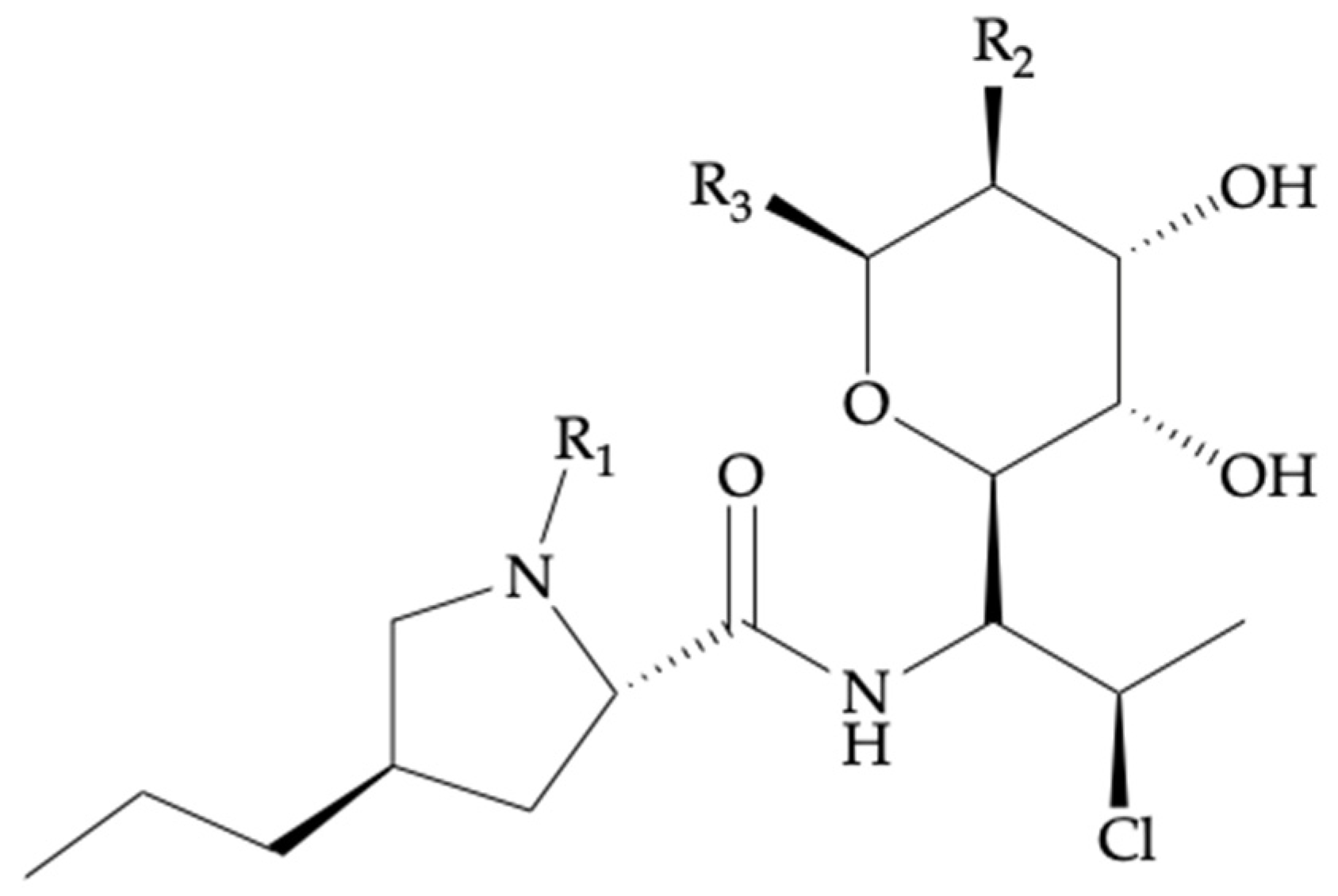
| Compound Name | Biotransformation | R1 | R2 | R3 |
|---|---|---|---|---|
| Clindamycin | - | -CH3 | -OH | -SCH3 |
| Clindamycin phosphate | - | -CH3 | -OPO3H2 | -SCH3 |
| Clindamycin palmitate | - | -CH3 | -OCOC15H31 | -SCH3 |
| Clindamycin sulfoxide 1 | S-Oxidation | -H | -OH | -SCH3 |
| N-demethyl clindamycin 1 | N-Dealkylation | -CH3 | -OH | -SOCH3 |
| Clindamycin | |
|---|---|
| Chemical nomenclature | 7-chloro-7-deoxy-lincomycin |
| Chemical structure | See Figure 2, Table 1 |
| Pharmacotherapeutic group | Lincosamides |
| Indications 1 | 1. Surgical prophylaxis in the event of 2. Beta-lactam allergy; 3. Prophylaxis and treatment of pregnancy infections; 4. Treatment of diabetic foot infections; 5. Treatment of bone and joint, fracture-related, and periprosthetic joint infections |
| Mode of action | Bacterial protein synthesis inhibitor. Binds to 50S ribosome and inhibits peptidyl transferase and translocation |
| Route of administration | PO 2 (CLI HCl, palmitate HCl); IV 2 (CLI phosphate) |
| Formulations | PO (capsules, solution); IV (injection solution) |
| Bacteria Type | Pathogen | Clinical Breakpoints 1 |
|---|---|---|
| Gram-positive aerobes | Staphylococcus spp. | 0.25 |
| Streptococcus spp. 2 | 0.5 | |
| Scheme 0. | 0.5 | |
| Anaerobes | Fusobacterium necrophorum | 0.25 |
| Prevotella spp. | 0.25 | |
| Bacteroides spp. | 4 3 |
| Indications | Predominant Causative Pathogens 1 | Type of Treatment | Admin. Route | Adult Dosing | Followed Guidelines |
|---|---|---|---|---|---|
| Surgical prophylaxis (SSIs 2) in the event of beta-lactam allergy | Clean procedures: S. aureus, CoNS 2 Clean-contaminated procedures: GN 2 spp., Enterococcus spp. 3 | Second line. Monotherapy or combined treatment | IV 2 | 600 mg.q6h (<70 kg) 900 mg.q6h (≥70 kg) | ASHP 2, IDSA 2, SIS 2, SHEA 2, |
| Prophylaxis and treatment of pregnancy infections | S. aureus, CoNS, group B Streptococcus | Second line. Maternal allergy to penicillins | IV | 900 mg.q8h until delivery 4 | CDC 2, American College of Obstetricians and Gynecologists |
| Treatment of DFIs 2 | S. aureus, beta-hemolytic streptococci, GN spp. | Second line. Severe beta-lactam allergy. Combined treatment 5 in the case of IV | Mild DFI: PO 2,5. Moderate or severe DFI: IV 5 | PO: 300–450 mg.q8h IV: 600 mg.q8h | IDSA |
| Treatment of BJIs 2, FRIs 2, and PJIs 2 | Staphylococcus spp., CoNS, Streptococcus spp., Enterococcus spp., Pseudomonas aeruginosa, anaerobic bacteria | Combined treatment with rifampicin. See Section 4.4 T | ~6 weeks IV + ~12 weeks PO 6 | IV or PO: 600 mg.q8h | Consensus from an International Expert Group [52] |
| Pharmacokinetic Studies * | Posology and Route of Administration | Theoretical Target Plasma Concentration 1 | Measured Plasma Concentration 1 | Measurement Technique 1 | |
|---|---|---|---|---|---|
| [Reference] | CLI 4 | RIF 4 | Monotherapy vs. Combined | ||
| Curis et al. [79] | 600 mg.q8h, PO/IV 4 bolus | NS, PO/IV bolus | Cmin 4 = 1.7 | Cmin 2,4 = 1.36 vs. 0.29 Cmax 2,4 = 7.48 vs. 4.46 | HPLC-UV 4 |
| Bernard et al. [89] | 600 mg.q8h, PO 4 | 600 mg.q12h, PO | Cmin = [2,3,4] Cmax 4 = [5,6,7,8] | Cmin 3,4 = 4.7 vs. 0.79 Cmax 3,4 = 10.2 vs. 3.48 | NS 4 |
| Zeller et al. [85] | 2400 mg/day, IV 4 infusion; 750 mg.q8h, PO | 600 mg.q12h, PO | Css 4 = [5,6,7,8] | Cmin 2,4 = 2.09 vs. 0.18 Cmax 2,4 = 7.95 vs. 1.53 | LC-MS/MS 4 |
| Type of CYP3A4- Mediated DDI 1 | Drug | Drug Class | DDI 2 Mechanism | DDI 2 Potency | Indication | Type of Combined Treatment 3 | Admin. Route | Adult Dosing of Drug |
|---|---|---|---|---|---|---|---|---|
| CYP3A4- inhibition | Erythromycin | Macrolide antibiotic | Mechanism-based inhibition | Moderate inhibition | Gastroprokinetic: control acid reflux | Combined in low doses | PO 1 | 125–250 mq.q12h |
| Ritonavir | Antiretroviral: protease inhibitor HIV-1 | Competitive and noncompetitive, irreversible inhibition | Potent inhibition | Mild to moderate COVID-19 1 caused by the severe SARS-CoV-2 1 virus | Paxolavid ® (nirmaltrevir/ ritonavir) | PO | Paxolavid ® (300 mg/100 mg).q12h for 5 days | |
| CYP3A4- induction | Rifampicin | Rifamycin antibiotic | Transcriptional PXR 1 agonism | Potent inhibition | Treatment of BJIs 1 | See Table 6 | PO or IV 1 | See Table 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armengol Álvarez, L.; Van de Sijpe, G.; Desmet, S.; Metsemakers, W.-J.; Spriet, I.; Allegaert, K.; Rozenski, J. Ways to Improve Insights into Clindamycin Pharmacology and Pharmacokinetics Tailored to Practice. Antibiotics 2022, 11, 701. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11050701
Armengol Álvarez L, Van de Sijpe G, Desmet S, Metsemakers W-J, Spriet I, Allegaert K, Rozenski J. Ways to Improve Insights into Clindamycin Pharmacology and Pharmacokinetics Tailored to Practice. Antibiotics. 2022; 11(5):701. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11050701
Chicago/Turabian StyleArmengol Álvarez, Laura, Greet Van de Sijpe, Stefanie Desmet, Willem-Jan Metsemakers, Isabel Spriet, Karel Allegaert, and Jef Rozenski. 2022. "Ways to Improve Insights into Clindamycin Pharmacology and Pharmacokinetics Tailored to Practice" Antibiotics 11, no. 5: 701. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11050701