
Synthesis and Antimicrobial Activity of New Heteroaryl(aryl) Thiazole Derivatives Molecular Docking Studies
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Antibacterial Activity
2.2.2. Antifungal Activity
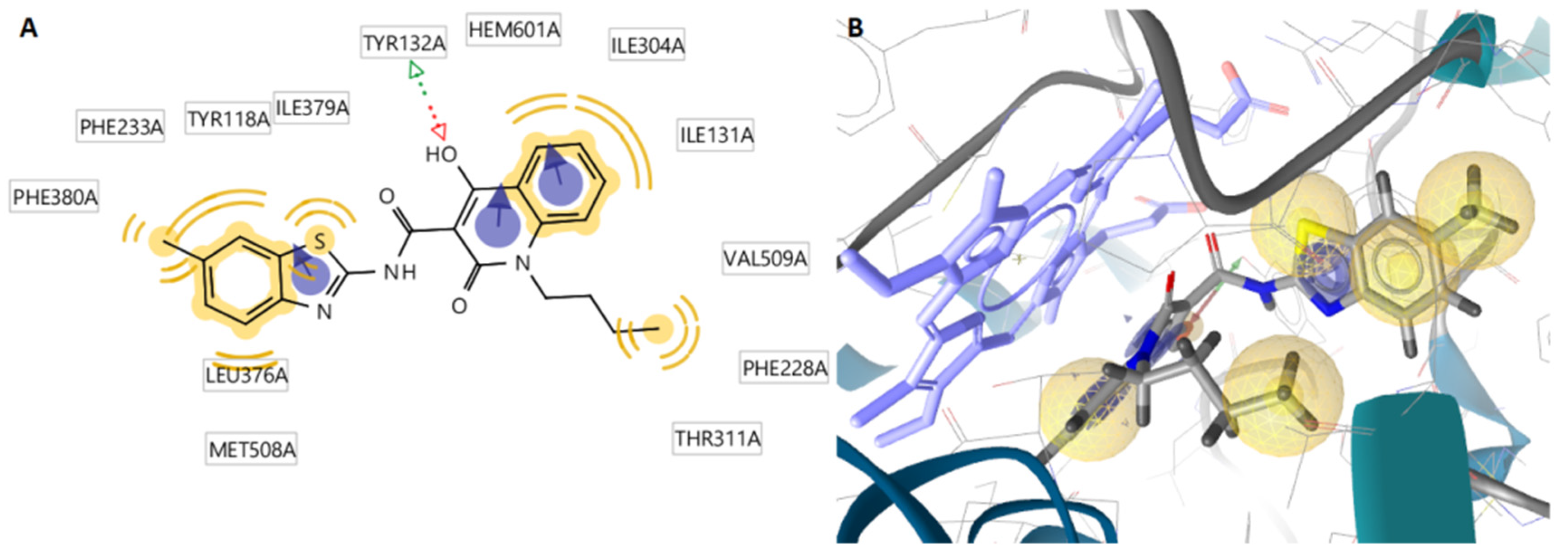
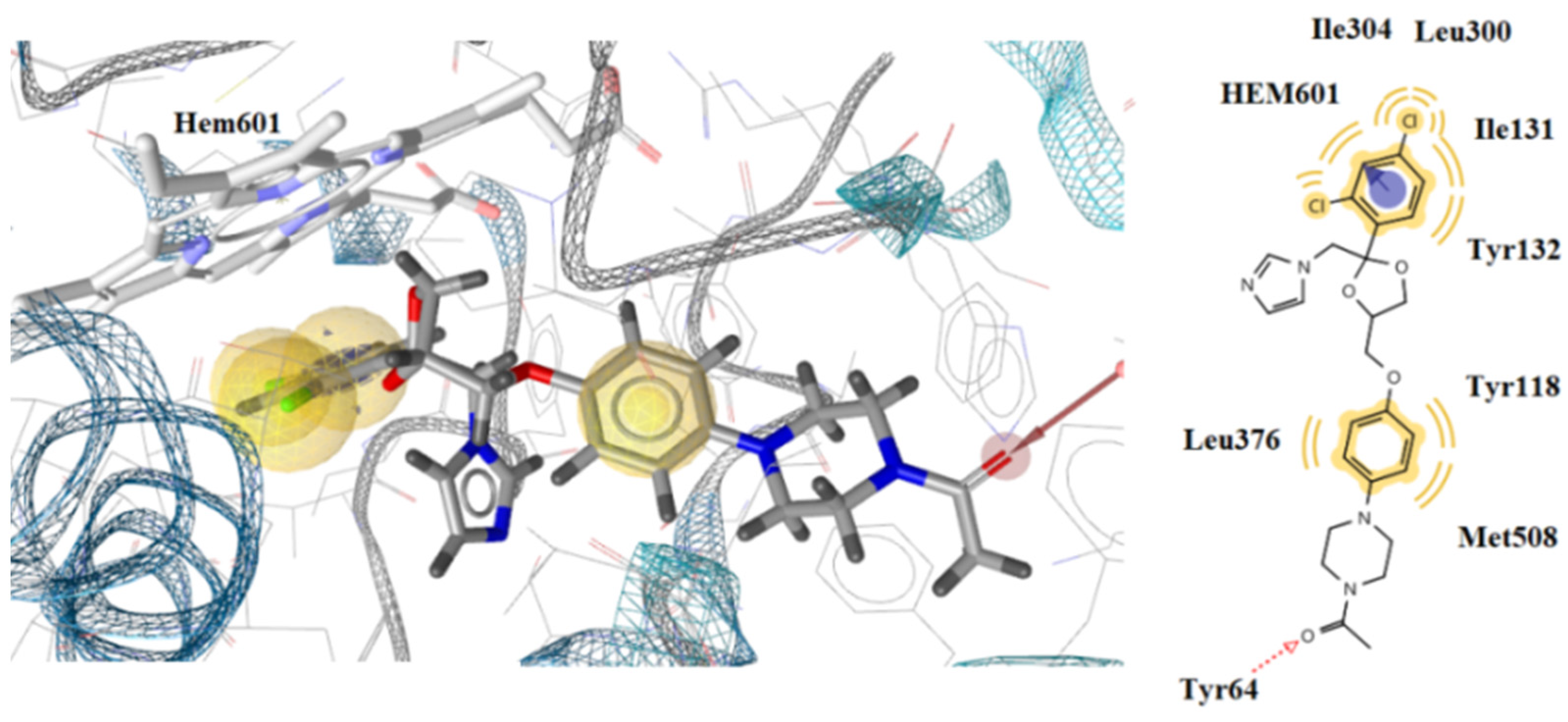
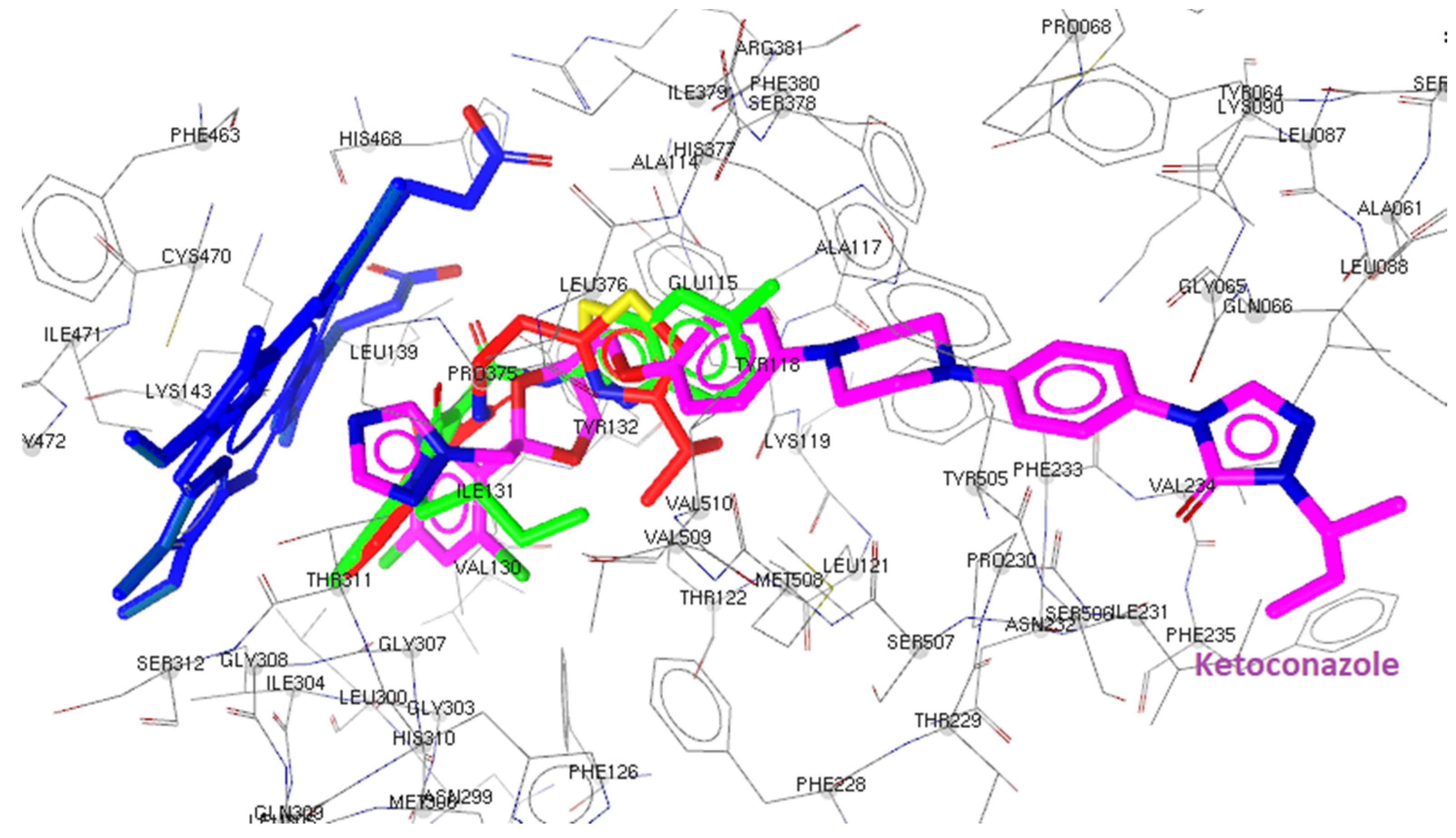
2.3. Docking Studies
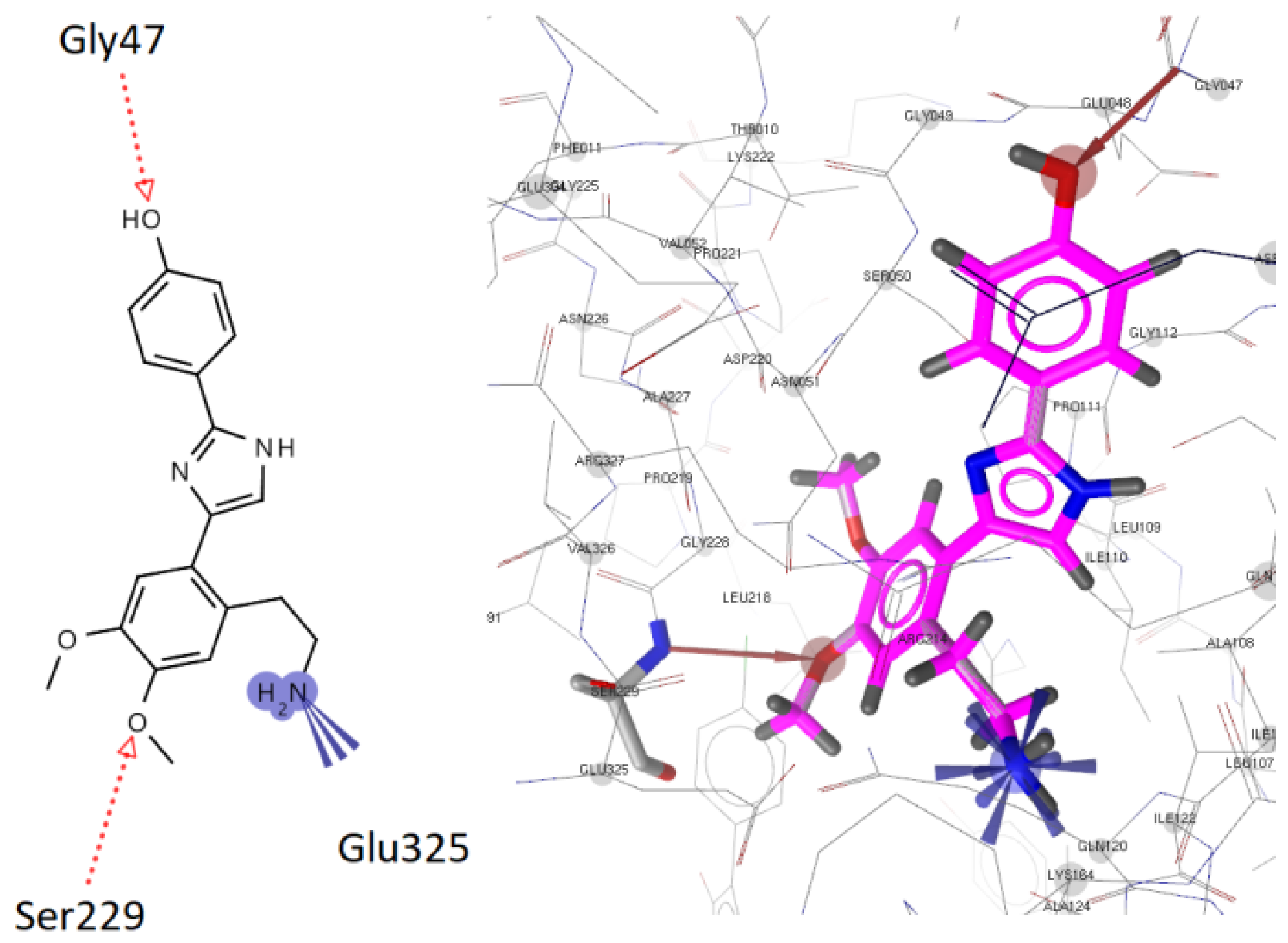
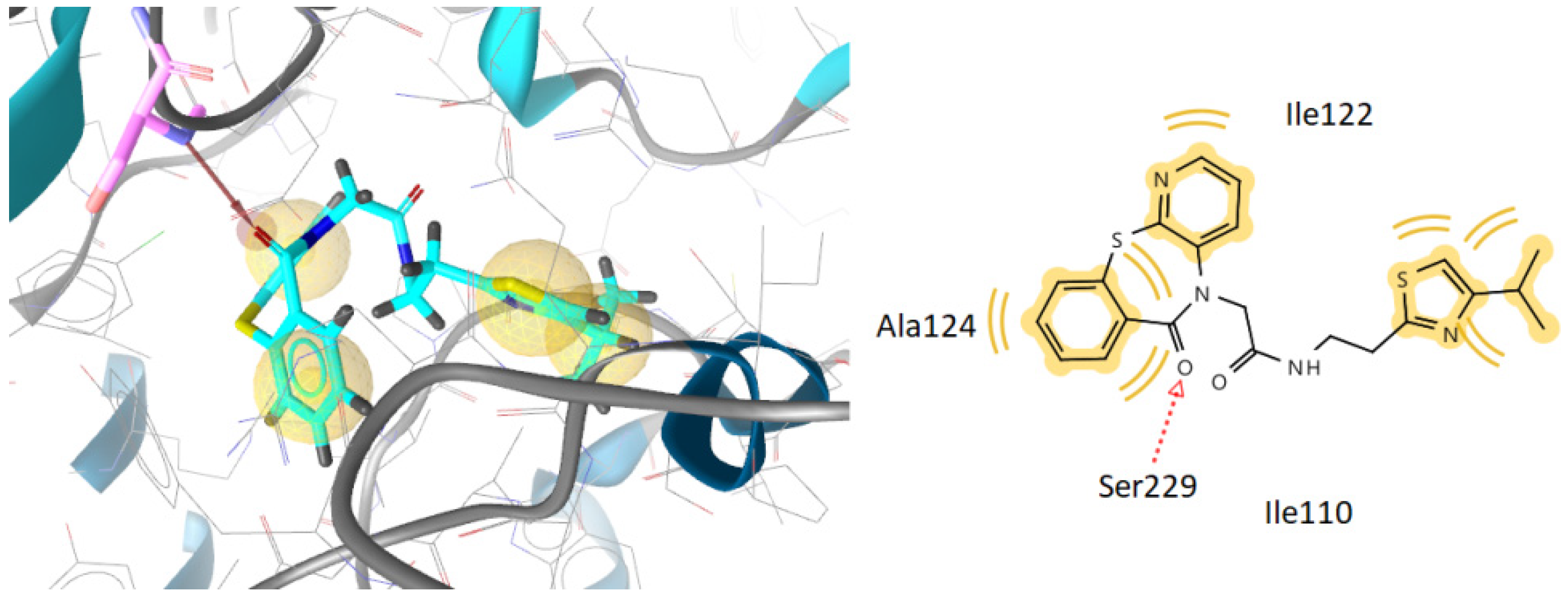
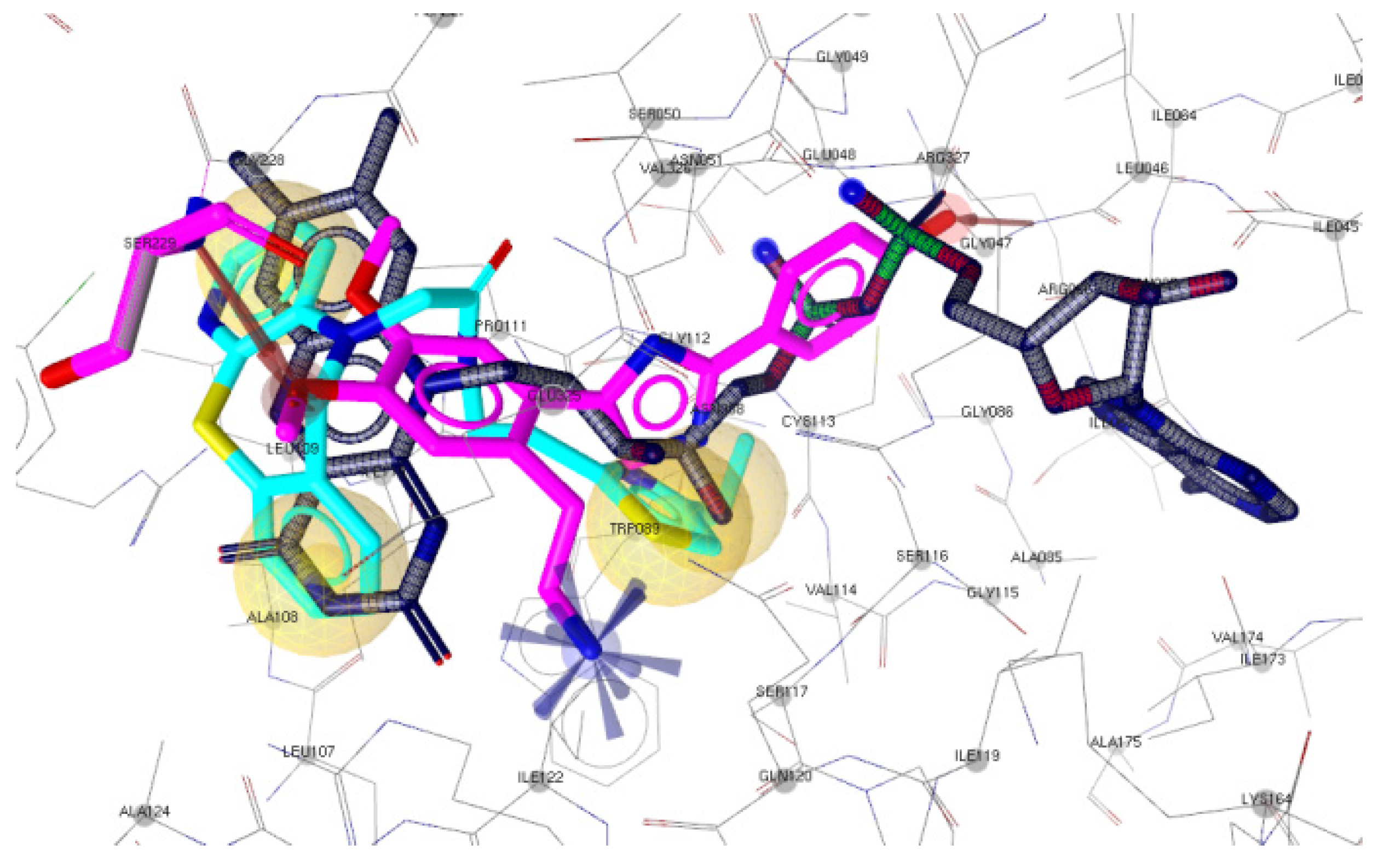
2.3.1. Docking to Antibacterial Targets
2.3.2. Docking to Antifungal Targets
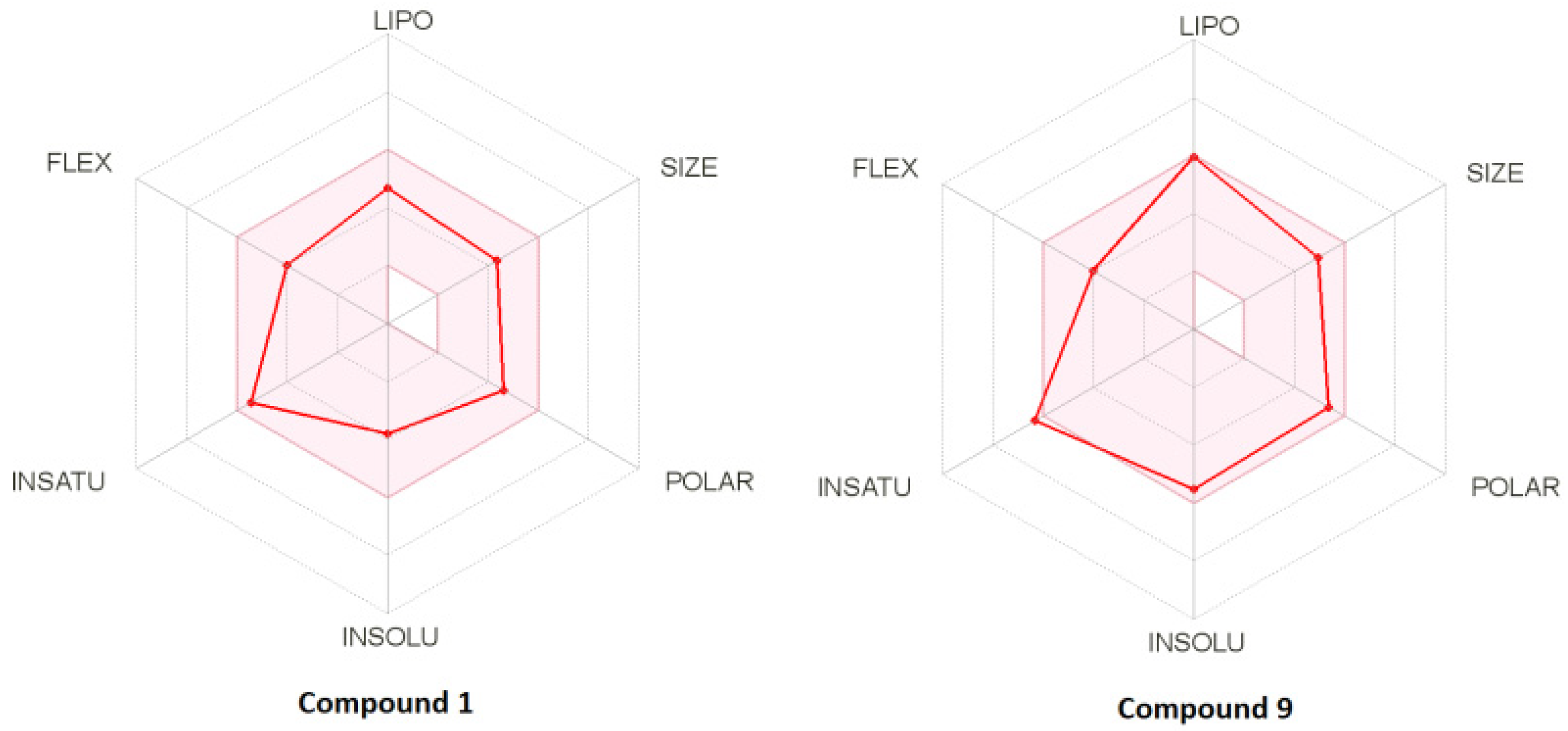
2.4. Drug-Likeness
3. Materials and Methods
3.1. General Information
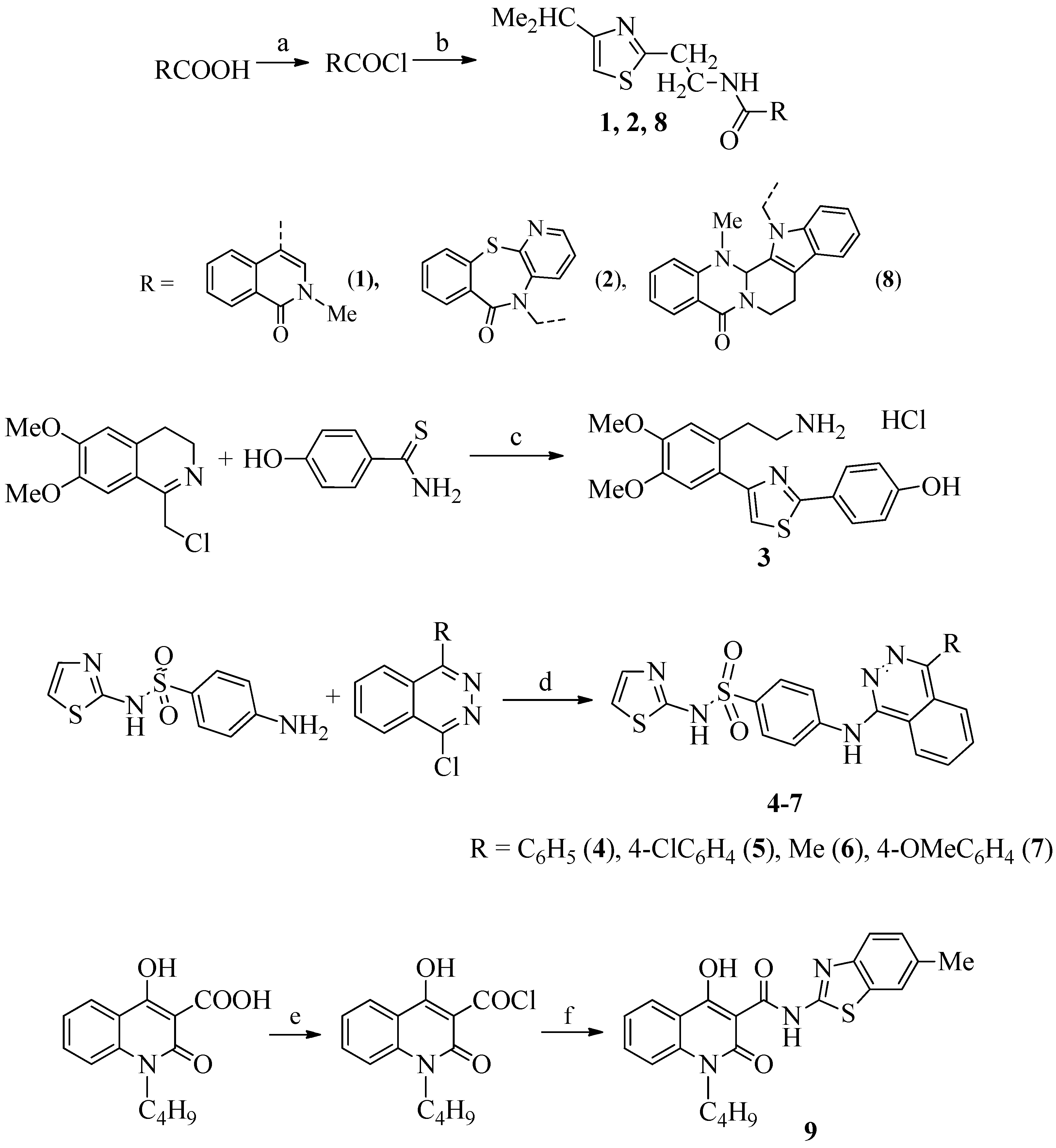
3.1.1. General Procedure for the Synthesis of Compounds 1, 2, and 8
3.1.2. Synthesis of Compound 3 [61]
3.1.3. General Procedure for the Synthesis of Compounds 4–7
3.2. Biological Evaluation
3.2.1. Antibacterial Action
3.2.2. Antifungal Activity
3.2.3. Inhibition of Biofilm Formation
3.3. Molecular Modeling Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tripathi, A.C.; Gupta, S.J.; Fatima, G.N.; Sonar, P.K.; Verma, A.; Saraf, S.K. 4-Thiazolidinones: The advances continue…. Eur. J. Med. Chem. 2014, 72, 52–77. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, D.; Kryshchyshyn, A.; Lesyk, R. 5-Ene-4-thiazolidinones—An efficient tool in medicinal chemistry. Eur. J. Med. Chem. 2017, 140, 542–594. [Google Scholar] [CrossRef] [PubMed]
- Bikobo, D.S.N.; Vodnar, D.C.; Stana, A.; Tiperciuc, B.; Nastasă, C.; Douchet, M.; Oniga, O. Synthesis of 2-phenylamino-thiazole derivatives as antimicrobial agents. J. Saudi Chem. Soc. 2017, 21, 861–868. [Google Scholar] [CrossRef]
- AlThagafi, I.; El-Metwaly, N.; Farghaly, T.A. New Series of Thiazole Derivatives: Synthesis, Structural Elucidation, Antimicrobial Activity, Molecular Modeling and MOE Docking. Molecules 2019, 24, 1741. [Google Scholar] [CrossRef] [PubMed]
- Biernasiuk, A.; Kawczyńska, M.; Berecka-Rycerz, A.; Rosada, B.; Gumieniczek, A.; Malm, A.; Dzitko, K.; Łączkowski, K.Z. Synthesis, antimicrobial activity, and determination of the lipophilicity of ((cyclohex-3-enylmethylene)hydrazinyl)thiazole derivatives. Med. Chem. Res. 2019, 28, 2023–2036. [Google Scholar] [CrossRef]
- Salem, M.A. Synthesis of New Thiazole, Bithiazolidinone and Pyrano[2,3-d]thiazole Derivatives as Potential Antimicrobial Agents. Croat. Chem. Acta 2017, 90, 7–15. [Google Scholar] [CrossRef]
- Bondock, S.; Fouda, A.M. Synthesis and evaluation of some new 5-(hetaryl)thiazoles as potential antimicrobial agents. Synth. Commun. 2018, 48, 561–573. [Google Scholar] [CrossRef]
- Borcea, A.-M.; Ionuț, I.; Crișan, O.; Oniga, O. An Overview of the Synthesis and Antimicrobial, Antiprotozoal, and Antitumor Activity of Thiazole and Bisthiazole Derivatives. Molecules 2021, 26, 624. [Google Scholar] [CrossRef]
- Carbone, A.; Cascioferro, S.; Parrino, B.; Carbone, D.; Pecoraro, C.; Schillaci, D.; Cusimano, M.G.; Cirrincione, G.; Diana, P. Thiazole Analogues of the Marine Alkaloid Nortopsentin as Inhibitors of Bacterial Biofilm Formation. Molecules 2021, 26, 81. [Google Scholar] [CrossRef]
- Kamble, R.D.; Meshram, R.J.; Hese, S.V.; More, R.A.; Kamble, S.S.; Gacche, R.N.; Dawane, B.S. Synthesis and in silico investigation of thiazoles bearing pyrazoles derivatives as anti-inflammatory agents. Comput. Biol. Chem. 2016, 61, 86–96. [Google Scholar] [CrossRef]
- Mohareb, R.; Al-Omran, F.; Abdelaziz, M.; Ibrahim, R. Anti-inflammatory and Anti-ulcer Activities of New Fused Thiazole Derivatives Derived from 2-(2-Oxo-2H-chromen-3-yl)thiazol-4(5H)-one. Acta Chim. Slov. 2017, 64, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Gümüş, M.; Yakan, M.; Koca, İ. Recent advances of thiazole hybrids in biological applications. Future Med. Chem. 2019, 11, 1979–1998. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, Z.A.; Masaret, G.S.; Amin, M.; Abdallah, M.A.; Farghaly, T. Anti-inflammatory, Analgesic and Anti-ulcerogenic Activities of Novel bis-thiadiazoles, bis-thiazoles and bis-formazanes. Med. Chem. 2017, 13, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Kryshchyshyn, A.; Roman, O.; Lozynskyi, A.; Lesyk, R. Thiopyrano[2,3-d]Thiazoles as New Efficient Scaffolds in Medicinal Chemistry. Sci. Pharm. 2018, 86, 26. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Moghimi, S.; Foroumadi, A. Thiazole in the targeted anticancer drug discovery. Future Med. Chem. 2019, 11, 1929–1952. [Google Scholar] [CrossRef]
- Carbone, D.; Vestuto, V.; Ferraro, M.R.; Ciaglia, T.; Pecoraro, C.; Sommella, E.; Cascioferro, S.; Salviati, E.; Novi, S.; Tecce, M.F.; et al. Metabolomics-assisted discovery of a new anticancer GLS-1 inhibitor chemotype from a nortopsentin-inspired library: From phenotype screening to target identification. Eur. J. Med. Chem. 2022, 234, 114233. [Google Scholar] [CrossRef]
- Di Franco, S.; Parrino, B.; Gaggianesi, M.; Pantina, V.D.; Bianca, P.; Nicotra, A.; Mangiapane, L.R.; Iacono, M.L.; Ganduscio, G.; Veschi, V.; et al. CHK1 inhibitor sensitizes resistant colorectal cancer stem cells to nortopsentin. iScience 2021, 24, 102664. [Google Scholar] [CrossRef]
- Khatik, G.; Datusalia, A.K.; Ahsan, W.; Kaur, P.; Vyas, M.; Mittal, A.; Nayak, S. A Retrospect Study on Thiazole Derivatives as the Potential Antidiabetic Agents in Drug Discovery and Developments. Curr. Drug Discov. Technol. 2018, 15, 163–177. [Google Scholar] [CrossRef]
- Petrou, A.; Eleftheriou, P.; Geronikaki, A.; Akrivou, M.G.; Vizirianakis, I. Novel Thiazolidin-4-ones as Potential Non-nucleoside Inhibitors of HIV-1 Reverse Transcriptase. Molecules 2019, 24, 3821. [Google Scholar] [CrossRef]
- Xu, Z.; Ba, M.; Zhou, H.; Cao, Y.; Tang, C.; Yang, Y.; He, R.; Liang, Y.; Zhang, X.; Li, Z.; et al. 2,4,5-Trisubstituted thiazole derivatives: A novel and potent class of non-nucleoside inhibitors of wild type and mutant HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2014, 85, 27–42. [Google Scholar] [CrossRef]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [PubMed]
- Jacob, P.J.; Manju, S.L. Identification and development of thiazole leads as COX-2/5-LOX inhibitors through in-vitro and in-vivo biological evaluation for anti-inflammatory activity. Bioorg. Chem. 2020, 100, 103882. [Google Scholar] [CrossRef] [PubMed]
- Grozav, A.; Porumb, I.-D.; Găină, L.I.; Filip, L.; Hanganu, D. Cytotoxicity and Antioxidant Potential of Novel 2-(2-((1H-indol-5yl)methylene)-hydrazinyl)-thiazole Derivatives. Molecules 2017, 22, 260. [Google Scholar] [CrossRef] [PubMed]
- Djukic, M.; Fesatidou, M.; Xenikakis, I.; Geronikaki, A.; Angelova, V.T.; Savic, V.; Pasic, M.; Krilovic, B.; Djukic, D.; Gobeljic, B.; et al. In vitro antioxidant activity of thiazolidinone derivatives of 1,3-thiazole and 1,3,4-thiadiazole. Chem. Interact. 2018, 286, 119–131. [Google Scholar] [CrossRef]
- Brito, C.C.B.; Silva, H.V.C.D.; Brondani, D.J.; Faria, A.R.; Ximenes, R.M.; Silva, I.M.D.; Albuquerque, J.F.C.; Castilho, M.S. Synthesis and biological evaluation of thiazole derivatives as LbSOD inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.A.; dos Santos, P.F.; da Costa, M.O.L.; Pavani, T.; Xander, P.; Geraldo, M.M.; Mengarda, A.C.A.; de Moraes, J.; Rando, D.G.G. 4-Phenyl-1,3-thiazole-2-amines as scaffolds for new antileishmanial agents. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 26. [Google Scholar] [CrossRef]
- Sowjanya, C.H.; Swamy, S.S.; Gomathi, S.; Babu, A.K. Synthesis, Chemistry and Anti-Hypertensive Activity of Some New Thiazole-Thiadiazole Derivatives. Int. J. Adv. Res. Med. Pharm. Sci. 2016, 1, 6–10. [Google Scholar]
- Salar, U.; Khan, K.M.; Chigurupati, S.; Taha, M.; Wadood, A.; Vijayabalan, S.; Ghufran, M.; Perveen, S. New Hybrid Hydrazinyl Thiazole Substituted Chromones: As Potential α-Amylase Inhibitors and Radical (DPPH & ABTS) Scavengers. Sci. Rep. 2017, 7, 16980. [Google Scholar] [CrossRef]
- Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold. J. Enzym. Inhib. Med. Chem. 2011, 26, 1–21. [Google Scholar] [CrossRef]
- Kenchappa, R.; Bodke, Y.D.; Telkar, S.; Sindhe, M.A. Antifungal and anthelmintic activity of novel benzofuran derivatives containing thiazolo benzimidazole nucleus: An in vitro evaluation. J. Chem. Biol. 2016, 10, 11–23. [Google Scholar] [CrossRef]
- White, C.A., Jr. Nitazoxanide: A new broad spectrum antiparasitic agent. Expert Rev. Anti. Infect. Ther. 2004, 2, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rouf, A.; Tanyeli, C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Kardos, N.; Demain, A.L. Penicillin: The medicine with the greatest impact on therapeutic outcomes. Appl. Microbiol. Biotechnol. 2011, 92, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Pasqualotto, A.C.; O Thiele, K.; Goldani, L.Z. Novel triazole antifungal drugs: Focus on isavuconazole, ravuconazole and albaconazole. Curr. Opin. Investig. drugs 2010, 11, 165–174. [Google Scholar] [PubMed]
- Thierbach, G.; Reichenbach, H. Myxothiazol, a new antibiotic interfering with respiration. Antimicrob. Agents Chemother. 1981, 19, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Borelli, C.; Schaller, M.; Niewerth, M.; Nocker, K.; Baasner, B.; Berg, D.; Tiemann, R.; Tietjen, K.; Fugmann, B.; Lang-Fugmann, S.; et al. Modes of Action of the New Arylguanidine Abafungin beyond Interference with Ergosterol Biosynthesis and in vitro Activity against Medically Important Fungi. Chemotherapy 2008, 54, 245–259. [Google Scholar] [CrossRef]
- Li, X.-H.; Yang, X.-L.; Ling, Y.; Fan, Z.-J.; Liang, X.-M.; Wang, D.-Q.; Chen, A.F.-H.; Li, Z.-M. Synthesis and Fungicidal Activity of Novel 2-Oxocycloalkylsulfonylureas. J. Agric. Food Chem. 2005, 53, 2202–2206. [Google Scholar] [CrossRef]
- Abbasi, M.; Nazifi, S.M.R.; Nazifi, Z.S.; Massah, A.R. Synthesis, characterization and in vitro antibacterial activity of novel phthalazine sulfonamide derivatives. J. Chem. Sci. 2017, 129, 1257–1266. [Google Scholar] [CrossRef]
- Sławiński, J.; Pogorzelska, A.; Żołnowska, B.; Kedzia, A.; Ziółkowska-Klinkosz, M.; Kwapisz, E. Synthesis and Anti-Yeast Evaluation of Novel 2-Alkylthio-4-chloro-5-methyl-N-[imino-(1-oxo-(1H)-phthalazin-2-yl)methyl]benzenesulfonamide Derivatives. Molecules 2014, 19, 13704–13723. [Google Scholar] [CrossRef]
- Mourad, A.K.; Makhlouf, A.A.; Soliman, A.Y.; Mohamed, S.A. Phthalazines and phthalazine hybrids as antimicrobial agents: Synthesis and biological evaluation. J. Chem. Res. 2020, 44, 31–41. [Google Scholar] [CrossRef]
- Hussein, E.M.; Al-Rooqi, M.M.; Abd El-Galil, S.M.; Ahmed, S.A. Design, synthesis, and biological evaluation of novel N4-substituted sulfonamides: Acetamides derivatives as dihydrofolate reductase (DHFR) inhibitors. BMC Chem. 2019, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Kachaeva, M.V.; Hodyna, D.M.; Semenyuta, I.V.; Pilyo, S.G.; Prokopenko, V.M.; Kovalishyn, V.V.; Metelytsia, L.O.; Brovarets, V.S. Design, synthesis and evaluation of novel sulfonamides as potential anticancer agents. Comput. Biol. Chem. 2018, 74, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Rubab, L.; Rehman, M.U.; Anjum, R.; Ullah, S.; Marjana, M.; Qadeer, S.; Sana, S. Coumarin sulfonamide derivatives: An emerging class of therapeutic agents. Heterocycl. Commun. 2020, 26, 46–59. [Google Scholar] [CrossRef]
- Bonardi, A.; Nocentini, A.; Bua, S.; Combs, J.; Lomelino, C.; Andring, J.; Lucarini, L.; Sgambellone, S.; Masini, E.; McKenna, R.; et al. Sulfonamide Inhibitors of Human Carbonic Anhydrases Designed through a Three-Tails Approach: Improving Ligand/Isoform Matching and Selectivity of Action. J. Med. Chem. 2020, 63, 7422–7444. [Google Scholar] [CrossRef] [PubMed]
- Gokcen, T.; Gulcin, I.; Ozturk, T.; Goren, A.C. A class of sulfonamides as carbonic anhydrase I and II inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31 (Suppl. 2), 180–188. [Google Scholar] [CrossRef] [PubMed]
- Esirden, I.; Tanç, M.; Supuran, C.T.; Kaya, M. Microwave assisted synthesis of novel tetrazole/sulfonamide derivatives based on octahydroacridine, xanthene and chromene skeletons as inhibitors of the carbonic anhydrases isoforms I, II, IV and VII. Bioorg. Med. Chem. Lett. 2016, 27, 86–89. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, Y.; Li, G.; Chen, L.; Nie, M.; Wang, Z.; Ji, H. Coumarin Sulfonamides and Amides Derivatives: Design, Synthesis, and Antitumor Activity In Vitro. Molecules 2021, 26, 786. [Google Scholar] [CrossRef]
- Azzam, R.A.; Elboshi, H.A.; Elgemeie, G.H. Novel Synthesis and Antiviral Evaluation of New Benzothiazole-Bearing N-Sulfonamide 2-Pyridone Derivatives as USP7 Enzyme Inhibitors. ACS Omega 2020, 5, 30023–30036. [Google Scholar] [CrossRef]
- Dash, R.N.; Moharana, A.K.; Subudhi, B.B. Sulfonamides: Antiviral Strategy for Neglected Tropical Disease Virus. Curr. Org. Chem. 2020, 24, 1018–1041. [Google Scholar] [CrossRef]
- Qadir, M.A.; Ahmed, M.; Iqbal, M. Synthesis, Characterization, and Antibacterial Activities of Novel Sulfonamides Derived through Condensation of Amino Group Containing Drugs, Amino Acids, and Their Analogs. BioMed Res. Int. 2015, 2015, 938486. [Google Scholar] [CrossRef]
- Beheshtimaal, K.; Khazaeili, T.; Asakere, N.; Mousavi, F.; Massah, A.R.; Assakere, N. Synthesis of Some Novel Sulfonamide-imines as Potential Antimicrobial Agents. Lett. Org. Chem. 2018, 15, 111–117. [Google Scholar] [CrossRef]
- Gao, H.-D.; Liu, P.; Yang, Y.; Gao, F. Sulfonamide-1,3,5-triazine–thiazoles: Discovery of a novel class of antidiabetic agents via inhibition of DPP-4. RSC Adv. 2016, 6, 83438–83447. [Google Scholar] [CrossRef]
- Zajdel, P.; Marciniec, K.; Maślankiewicz, A.; Grychowska, K.; Satała, G.; Duszyńska, B.; Lenda, T.; Siwek, A.; Nowak, G.; Partyka, A.; et al. Antidepressant and antipsychotic activity of new quinoline- and isoquinoline-sulfonamide analogs of aripiprazole targeting serotonin 5-HT₁A/5-HT₂A/5-HT₇ and dopamine D₂/D₃ receptors. Eur. J. Med. Chem. 2013, 60, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.; Hussein, B. Synthesis and Anti-Oxidant Evaluation of Some Novel Sulfa Drugs. Lett. Drug Des. Dis. 2017, 14, 1425–1432. [Google Scholar] [CrossRef]
- Ivasiv, V.; Albertini, C.; Gonçalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E.J.; Fraga, C.A.M. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef]
- Horishny, V.; Kartsev, V.; Matiychuk, V.; Geronikaki, A.; Anthi, P.; Pogodin, P.; Poroikov, V.; Ivanov, M.; Kostic, M.; Soković, M.D.; et al. 3-Amino-5-(indol-3-yl)methylene-4-oxo-2-thioxothiazolidine Derivatives as Antimicrobial Agents: Synthesis, Computational and Biological Evaluation. Pharmaceuticals 2020, 13, 229. [Google Scholar] [CrossRef]
- Horishny, V.; Kartsev, V.; Geronikaki, A.; Matiychuk, V.; Petrou, A.; Glamoclija, J.; Ciric, A.; Sokovic, M. 5-(1H-Indol-3-ylmethylene)-4-oxo-2-thioxothiazolidin-3-yl)alkancarboxylic Acids as Antimicrobial Agents: Synthesis, Biological Evaluation, and Molecular Docking Studies. Molecules 2020, 25, 1964. [Google Scholar] [CrossRef]
- Rotilie, C.A.; Fass, R.J.; Prior, R.B.; Perkins, R.L. Microdilution technique for antimicrobial susceptibility testing of anaerobic bacteria. Antimicro. Agents Chemother. 1975, 7, 311–315. [Google Scholar] [CrossRef]
- Benson, T.E.; Walsh, C.T.; Massey, V. Kinetic Characterization of Wild-Type and S229A Mutant MurB: Evidence for the Role of Ser 229 as a General Acid. Biochemistry 1997, 36, 796–805. [Google Scholar] [CrossRef]
- Zubenko, A.A.; Morkovnik, A.S.; Divaeva, L.N.; Demidov, O.P.; Kartsev, V.G.; Sochnev, V.S.; Klimenko, A.I.; Dobaeva, N.M.; Borodkin, G.S.; Bodryakov, A.N.; et al. Thiourea assisted recyclization of 1-(chloromethyl)dihydroisoquinolines: A convenient route to β-(o-thiazolylaryl)ethylamines. Mendeleev Commun. 2021, 31, 125–127. [Google Scholar] [CrossRef]
- Simakov, S.; Kartsev, V.; Petrou, A.; Nicolaou, I.; Geronikaki, A.; Ivanov, M.; Kostic, M.; Glamočlija, J.; Soković, M.; Talea, D.; et al. 4-(Indol-3-yl)thiazole-2-amines and 4-ιndol-3-yl)thiazole Acylamines as Νovel Antimicrobial Agents: Synthesis, In Silico and In Vitro Evaluation. Pharmaceuticals 2021, 14, 1096. [Google Scholar] [CrossRef] [PubMed]
- Victor, K.; Boris, L.; Athina, G.; Anthi, P.; Marija, S.; Marina, K.; Oliver, R.; Marina, S. Design, synthesis and antimicrobial activity of usnic acid derivatives. MedChemComm 2018, 9, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Kostić, M.; Smiljković, M.; Petrović, J.; Glamočlija, J.; Barros, L.; Ferreira, I.C.F.R.; Ćirić, A.; Soković, M. Chemical, nutritive composition and a wide range of bioactive properties of honey mushroom Armillaria mellea (Vahl: Fr.) Kummer. Food Funct. 2017, 8, 3239–3249. [Google Scholar] [CrossRef]
- Kritsi, E.; Matsoukas, M.-T.; Potamitis, C.; Detsi, A.; Ivanov, M.; Sokovic, M.; Zoumpoulakis, P. Novel Hit Compounds as Putative Antifungals: The Case of Aspergillus fumigatus. Molecules 2019, 24, 3853. [Google Scholar] [CrossRef] [PubMed]
- Aleksić, M.; Stanisavljević, D.; Smiljković, M.; Vasiljević, P.; Stevanović, M.; Soković, M.; Stojković, D. Pyrimethanil: Between efficient fungicide against Aspergillus rot on cherry tomato and cytotoxic agent on human cell lines. Ann. Appl. Biol. 2019, 175, 228–235. [Google Scholar] [CrossRef]
- Cady, N.C.; McKean, K.A.; Behnke, J.; Kubec, R.; Mosier, A.P.; Kasper, S.H.; Burz, D.S.; Musah, R.A. Inhibition of Biofilm Formation, Quorum Sensing and Infection in Pseudomonas aeruginosa by Natural Products-Inspired Organosulfur Compounds. PLoS ONE 2012, 7, e38492. [Google Scholar] [CrossRef]
- Wolber, G.; Lange, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Compounds | S.a. | B.c. | L.m. | E.c. | S.T. | En.cl. | |
|---|---|---|---|---|---|---|---|---|
| 1 |  | MIC | 0.70 ± 0.19 | 0.35 ± 0.09 | 0.35 ± 0.09 | >3.75 | >3.75 | 0.23 ± 0.00 |
| MBC | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | >3.75 | >3.75 | 0.47 ± 0.00 | ||
| 2 |  | MIC | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.70 ± 0.19 | 0.35 ± 0.09 | 0.35 ± 0.09 | 0.35 ± 0.09 |
| MBC | 1.88 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | ||
| 3 |  | MIC | 0.70 ± 0.19 | 0.23 ± 0.00 | 0.70 ± 0.19 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.70 ± 0.19 |
| MBC | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | ||
| 4 |  | MIC | 1.41 ± 0.38 | 0.70 ± 0.19 | 0.70 ± 0.19 | 0.17 ± 0.00 | 0.70 ± 0.19 | 0.70 ± 0.19 |
| MBC | 1.88 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | 0.23 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | ||
| 5 |  | MIC | 1.41 ± 0.38 | 0.70 ± 0.19 | 0.70 ± 0.19 | 0.23 ± 0.00 | 0.47 ± 0.00 | 0.70 ± 0.19 |
| MBC | 1.88 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | ||
| 6 |  | MIC | 2.31 ± 0.76 | 1.41 ± 0.38 | 0.70 ± 0.19 | >3.75 | 0.70 ± 0.19 | 0.70 ± 0.19 |
| MBC | 3.75 ± 0.00 | 1.88 ± 0.00 | 0.94 ± 0.00 | >3.75 | 0.94 ± 0.00 | 0.94 ± 0.00 | ||
| 7 |  | MIC | 1.41 ± 0.00 | 0.70 ± 0.19 | 0.70 ± 0.19 | 0.35 ± 0.09 | 0.70 ± 0.19 | 0.70 ± 0.19 |
| MBC | 1.88 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | ||
| 8 |  | MIC | 1.41 ± 0.38 | 0.47 ± 0.00 | 0.35 ± 0.09 | >3.75 | 0.35 ± 0.09 | 0.23 ± 0.00 |
| MBC | 1.88 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | >3.75 | 0.47 ± 0.00 | 0.47 ± 0.00 | ||
| 9 |  | MIC | 0.94 ± 0.00 | 0.17 ± 0.00 | 0.35 ± 0.09 | 0.70 ± 0.19 | 0.17 ± 0.19 | 0.70 ± 0.19 |
| MBC | 1.88 ± 0.00 | 0.23 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | 0.23 ± 0.00 | 0.94 ± 0.00 | ||
| Streptomycin | MIC | 0.10 ± 0.00 | 0.02 ± 0.00 | 0.15 ± 0.00 | 0.10 ± 0.00 | 0.10 ± 0.00 | 0.02 ± 0.00 | |
| MBC | 0.20 ± 0.01 | 0.05 ± 0.00 | 0.30 ± 0.01 | 0.20 ± 0.00 | 0.20 ± 0.01 | 0.05 ± 0.00 | ||
| Ampicillin | MIC | 0.10 ± 0.00 | 0.10 ± 0.00 | 0.15 ± 0.00 | 0.15 ± 0.00 | 0.10 ± 0.00 | 0.10 ± 0.00 | |
| MBC | 0.15 ± 0.00 | 0.15 ± 0.00 | 0.30 ± 0.02 | 0.20 ± 0.01 | 0.20 ± 0.00 | 0.15 ± 0.01 | ||
| Compound | MRSA | P.a. | E.c. | MIC | 0% MIC | |
|---|---|---|---|---|---|---|
| 2 | MIC | 0.94 ± 0.00 | 0.23 ± 0.00 | 0.94 ± 0.00 | 14.59 | 7.08 |
| MBC | 1.88 ± 0.00 | 0.47 ± 0.00 | 1.88 ± 0.00 | |||
| 3 | MIC | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.47 ± 0.00 | 19.97 | 8.84 |
| MBC | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | |||
| 4 | MIC | 0.94 ± 0.00 | 0.23 ± 0.00 | 0.94 ± 0.00 | 4.31 | NE |
| MBC | 1.88 ± 0.00 | 0.47 ± 0.00 | 1.88 ± 0.00 | |||
| Streptomycin | MIC | 0.10 ± 0.00 | 0.05 ± 0.00 | 0.10 ± 0.00 | 71.94 | 55.42 |
| MBC | / | 0.10 ± 0.00 | 0.20 ± 0.00 | |||
| Ampicillin | MIC | / | 0.20 ± 0.01 | 0.20 ± 0.00 | 67.36 | 30.35 |
| MBC | / | / | / |
| Compounds | A.f. | A.n. | A.v. | P.f. | T.v. | P.v.c. | |
|---|---|---|---|---|---|---|---|
| 1 | MIC | 0.35 ± 0.08 | 0.08 ± 0.00 | 0.23 ± 0.00 | 0.17 ± 0.00 | 0.11 ± 0.00 | 0.17 ± 0.00 |
| MFC | 0.47 ± 0.00 | 0.11 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | |
| 2 | MIC | 0.35 ± 0.08 | 0.23 ± 0.00 | 0.35 ± 0.08 | 0.23 ± 0.00 | 0.17 ± 0.00 | 0.35 ± 0.08 |
| MFC | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.47 ± 0.00 | |
| 3 | MIC | 0.17 ± 0.00 | 0.11 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.11 ± 0.00 | 0.35 ± 0.08 |
| MFC | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.47 ± 0.00 | |
| 4 | MIC | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.35 ± 0.08 | 0.23 ± 0.00 | 0.23 ± 0.00 |
| MFC | 0.94 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | |
| 5 | MIC | 0.23 ± 0.00 | 0.06 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.17 ± 0.00 | 0.17 ± 0.00 |
| MFC | 0.47 ± 0.00 | 0.11 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | |
| 6 | MIC | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.17 ± 0.00 | 0.47 ± 0.00 |
| MFC | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.94 ± 0.00 | |
| 7 | MIC | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.23 ± 0.00 |
| MFC | 0.94 ± 0.00 | 0.47 ± 0.00 | 0.47 ± 0.00 | 0.94 ± 0.00 | 0.94 ± 0.00 | 0.47 ± 0.00 | |
| 8 | MIC | 0.23 ± 0.00 | 0.11 ± 0.00 | 0.11 ± 0.00 | 0.17 ± 0.00 | 0.08 ± 0.00 | 0.11 ± 0.00 |
| MFC | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.11 ± 0.00 | 0.23 ± 0.00 | |
| 9 | MIC | 0.23 ± 0.00 | 0.17 ± 0.00 | 0.17 ± 0.00 | 0.17 ± 0.00 | 0.06 ± 0.00 | 0.17 ± 0.00 |
| MFC | 0.47 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.23 ± 0.00 | 0.11 ± 0.00 | 0.23 ± 0.00 | |
| Bifonazole | MIC | 0.15 ± 0.00 | 0.15 ± 0.00 | 0.10 ± 0.00 | 0.20 ± 0.00 | 0.15 ± 0.00 | 0.10 ± 0.00 |
| MFC | 0.20 ± 0.00 | 0.20 ± 0.00 | 0.20 ± 0.00 | 0.25 ± 0.00 | 0.20 ± 0.00 | 0.20 ± 0.00 | |
| Ketoconazole | MIC | 0.20 ± 0.00 | 0.20 ± 0.00 | 0.20 ± 0.00 | 0.20 ± 0.00 | 1.00 ± 0.01 | 0.20 ± 0.00 |
| MFC | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.50 ± 0.00 | 0.50 ± 0.00 | 1.50 ± 0.00 | 0.30 ± 0.010 |
| Comp. | Est. Binding Energy (kcal/mol) | I-H E. coli MurB | Residues E. coli MurB | ||||
|---|---|---|---|---|---|---|---|
| E. coli Gyrase 1KZN | Thymidylate Kinase 4QGG | E. coli Primase 1DDE | E. coli MurA JV4T | E. coli MurB 2Q85 | |||
| 1 | −3.46 | - | - | −3.85 | −7.02 | 1 | Arg158 |
| 2 | −4.52 | −3.18 | −2.94 | −5.03 | −9.16 | 1 | Ser229 |
| 3 | −4.88 | −2.71 | - | −5.14 | −9.96 | 2 | Gly47, Ser229 |
| 4 | −3.82 | −3.11 | - | −4.69 | −8.70 | 1 | Ser229 |
| 5 | −3.96 | - | −2.91 | −3.67 | −7.53 | 1 | Arg213 |
| 6 | −4.62 | −2.54 | −3.47 | −5.75 | −8.65 | 2 | Gly122, Arg213 |
| 7 | −2.66 | - | - | −4.12 | −7.05 | 1 | Arg213 |
| 8 | −2.15 | −3.54 | −1.28 | −4.63 | −8.51 | 2 | Arg213, Ser229 |
| 9 | −2.74 | - | - | −3.79 | −8.52 | 1 | Ser229 |
| Est. Binding Energy (kcal/mol) | |||||
|---|---|---|---|---|---|
| N/N | DNA TopoIV 1S16 | CYP51 of C. albicans 5V5Z | I-H | Residues CYP51 of C. albicans | Interactions with HEM601 |
| 1 | −2.17 | −9.15 | 1 | Tyr132 | Hydrophobic, Aromatic |
| 2 | −3.10 | −7.95 | 1 | Tyr132 | Hydrophobic |
| 3 | - | −8.11 | 1 | Tyr118 | Hydrophobic |
| 4 | - | −7.52 | 1 | Tyr118 | Hydrophobic |
| 5 | −1.42 | −7.50 | 1 | Tyr118 | Hydrophobic |
| 6 | −1.56 | −8.64 | 1 | Tyr64 | Hydrophobic |
| 7 | −2.25 | −7.12 | - | - | Hydrophobic, Aromatic |
| 8 | - | −7.03 | - | - | Hydrophobic |
| 9 | −2.71 | −9.21 | 1 | Tyr132 | Hydrophobic, Aromatic |
| Ketoconazole | - | -8.23 | 1 | Tyr64 | Hydrophobic, Aromatic |
| No | MW | Number of HBA a | Number of HBD b | Log Po/w (iLOGP) c | Log S d | TPSA e | BBB Permeant f | Lipinski, Ghose, Veber, Egan, and Muegge Violations | Bioavailability Score | Drug-Likeness Model Score |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 355.45 | 3 | 1 | 2.8 | Poorly soluble | 92.23 | No | 0 | 0.55 | 1.03 |
| 2 | 438.12 | 4 | 1 | 3.37 | Poorly soluble | 128.73 | No | 0 | 0.55 | 1.09 |
| 3 | 339.16 | 5 | 3 | 2.33 | Poorly soluble | 93.39 | No | 0 | 0.55 | 0.01 |
| 4 | 459.54 | 5 | 2 | 3.00 | Poorly soluble | 133.49 | No | 0 | 0.55 | −0.12 |
| 5 | 493.99 | 5 | 2 | 3.20 | Poorly soluble | 133.49 | No | 3 * | 0.55 | −0.13 |
| 6 | 396.49 | 5 | 1 | 2.26 | Poorly soluble | 121.46 | No | 0 | 0.55 | 0.27 |
| 7 | 489.57 | 6 | 2 | 3.07 | Poorly soluble | 142.72 | No | 3 * | 0.55 | 0.27 |
| 8 | 513.65 | 3 | 1 | 3.73 | Poorly soluble | 98.71 | No | 1 ** | 0.55 | 1.37 |
| 9 | 407.49 | 4 | 2 | 2.86 | Poorly soluble | 112.46 | No | 0 | 0.55 | 0.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kartsev, V.; Geronikaki, A.; Zubenko, A.; Petrou, A.; Ivanov, M.; Glamočlija, J.; Sokovic, M.; Divaeva, L.; Morkovnik, A.; Klimenko, A. Synthesis and Antimicrobial Activity of New Heteroaryl(aryl) Thiazole Derivatives Molecular Docking Studies. Antibiotics 2022, 11, 1337. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11101337
Kartsev V, Geronikaki A, Zubenko A, Petrou A, Ivanov M, Glamočlija J, Sokovic M, Divaeva L, Morkovnik A, Klimenko A. Synthesis and Antimicrobial Activity of New Heteroaryl(aryl) Thiazole Derivatives Molecular Docking Studies. Antibiotics. 2022; 11(10):1337. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11101337
Chicago/Turabian StyleKartsev, Victor, Athina Geronikaki, Alexander Zubenko, Anthi Petrou, Marija Ivanov, Jasmina Glamočlija, Marina Sokovic, Lyudmila Divaeva, Anatolii Morkovnik, and Alexander Klimenko. 2022. "Synthesis and Antimicrobial Activity of New Heteroaryl(aryl) Thiazole Derivatives Molecular Docking Studies" Antibiotics 11, no. 10: 1337. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11101337