
Short, Synthetic Cationic Peptides Have Antibacterial Activity against Mycobacterium smegmatis by Forming Pores in Membrane and Synergizing with Antibiotics


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial and Mammalian Cells
2.2. Peptides
2.3. Bioinformatics
2.4. Bactericidal Assays
2.5. Minimum Inhibitory Concentration (MIC) Measurements
2.6. Checkerboard Assay
2.7. Bacterial Cytoplasmic Membrane Depolarization Assay

2.8. Bacterial Cytoplasmic Membrane Permeation

2.9. In Vitro Intracellular Activity in Infected Macrophages
3. Results
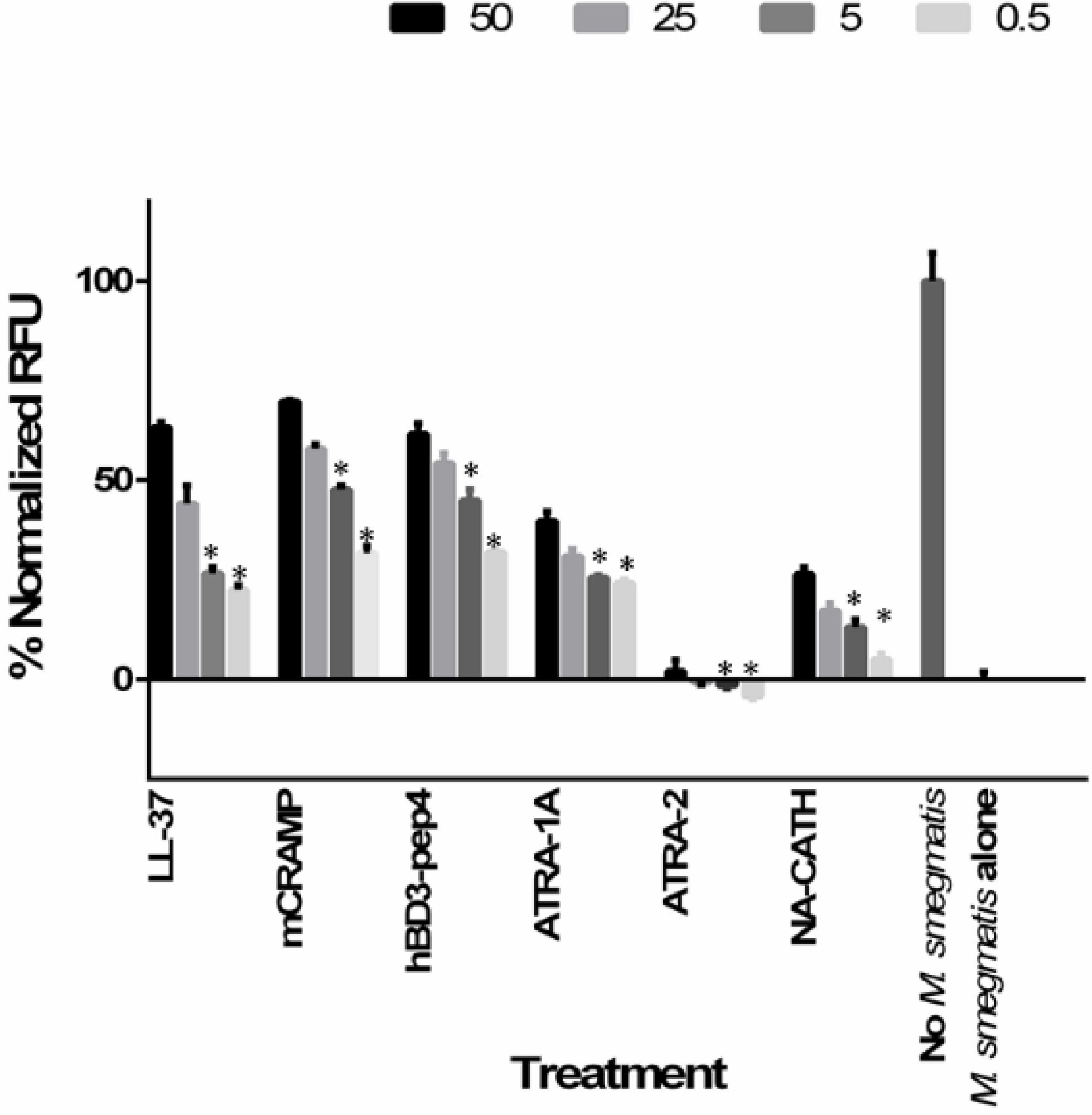
3.1. Synthetic ATRA Peptides Exert Antimycobacterial Effects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Net Charge at pH 7 | Hydro-Phobicity |
|---|---|---|---|
| NA-CATH | KRFKKFFKKLKNSVKKRAKKFFKKPKVIGVTFPF (ATRA-1) (ATRA-2) | +15 | 38% |
| ATRA-1A | KRAKKFFKKLK-NH2 | +8 | 36% |
| ATRA-2 | KRAKKFFKKPK-NH2 | +8 | 27% |
| hBD3-Pep4 | RGRRSSRRKK-NH2 | +7 | 0% |
| mCRAMP | GLLRKGGEKIGEKLKKIGQKIKNFFQKLVPQPEQ | +6 | 29% |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | +6 | 35% |
| Peptide/Antibiotic | Molecular Weight (g/mol) | EC50 | Fold vs. Rifampicin | ||
|---|---|---|---|---|---|
| μg/mL | 95% Confidence Interval | μM | |||
| Rifampicin | 822.94 | 0.13 | 0.061 to 0.276 | 0.159 | 1.0 |
| Polymyxin B | 1301.56 | 0.12 | 0.086 to 0.180 | 0.096 | 1.65 |
| Fosmidomycin | 183.1 | 0.14 | 0.062 to 0.326 | 0.780 | 0.20 |
| NA-CATH | 4175.26 | 1.88 | 0.622 to 5.68 | 0.451 | 0.354 |
| hBD3-Pep4 | 1286.51 | 0.36 | 0.159 to 0.824 | 0.287 | 0.554 |
| LL-37 | 7793.33 | 0.27 | 0.150 to 0.491 | 0.060 | 2.65 |
| mCRAMP | 3878.67 | 0.17 | 0.093 to 0.325 | 0.044 | 3.61 |
| ATRA-1A | 1420.84 | 0.05 | 0.034 to 0.075 | 0.035 | 4.54 |
| ATRA-2 | 1404.80 | >100 | - | - | - |

3.2. Cathelicidin Peptides mCRAMP, NA-CATH, and LL-37 Are Antimycobacterial
3.3. Human β-Defensin Derivative hBD3-Pep4 Exerts Antimycobacterial Activity
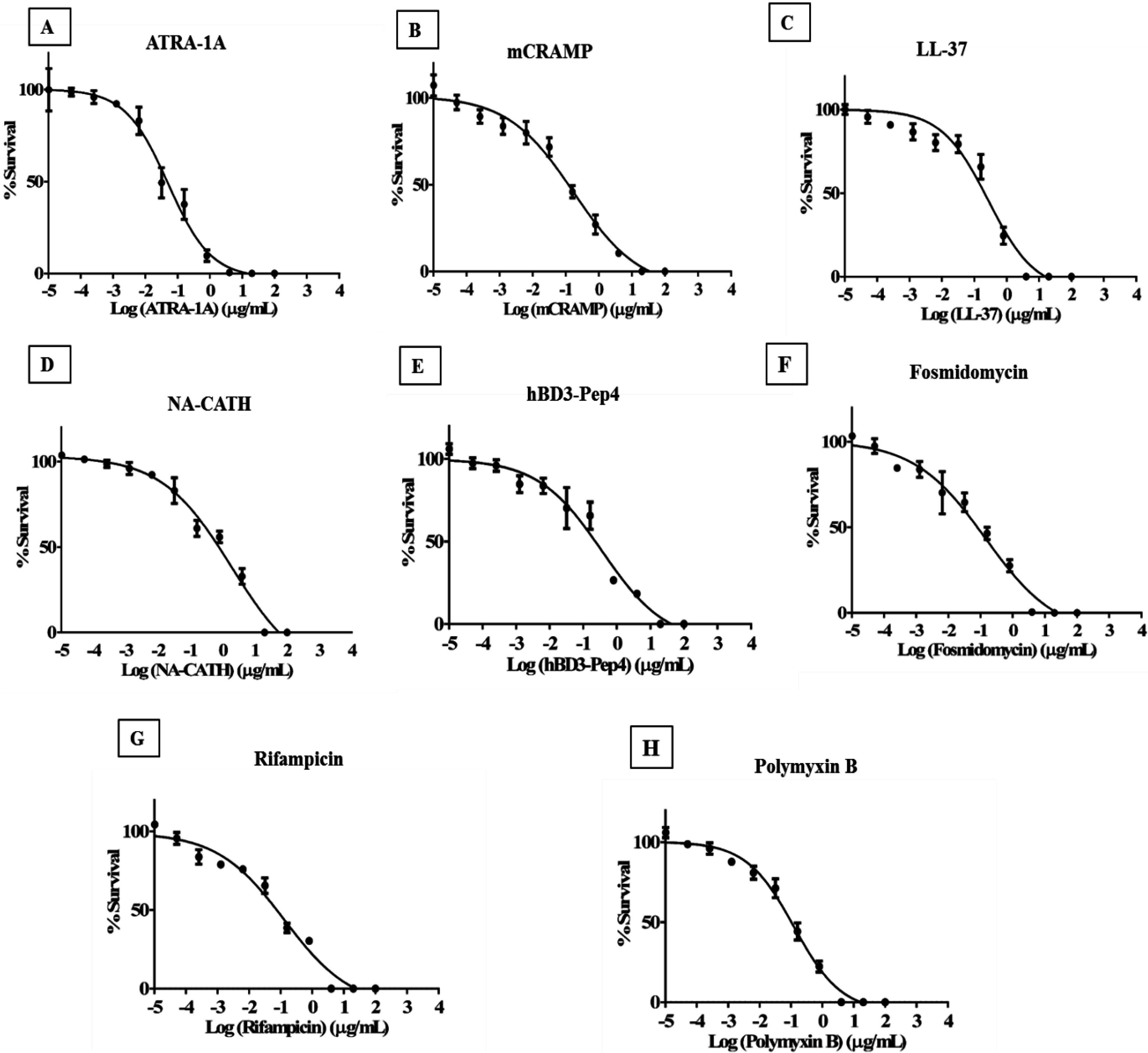
3.4. Minimum Inhibitory Concentration of Peptides to Inhibit Mycobacterial Growth
| Peptide/Antibiotic | MIC (μg/mL) | MIC μM | Fold vs. Rifampicin (μM) |
|---|---|---|---|
| Rifampicin | 3.9 | 4.73 | 1 |
| Fosmidomycin | 7.8 | 1.86 | 0.39 |
| Polymyxin B | 7.8 | 5.99 | 1.26 |
| mCRAMP | 15.6 | 4.02 | 0.85 |
| LL-37 | 31.3 | 4.01 | 0.85 |
| ATRA-1A | 31.3 | 22.01 | 5.11 |
| hBD3-Pep4 | 62.6 | 48.65 | 10.28 |
| NA-CATH | 250 | 59.80 | 12.64 |
| ATRA-2 | >125 | - | - |
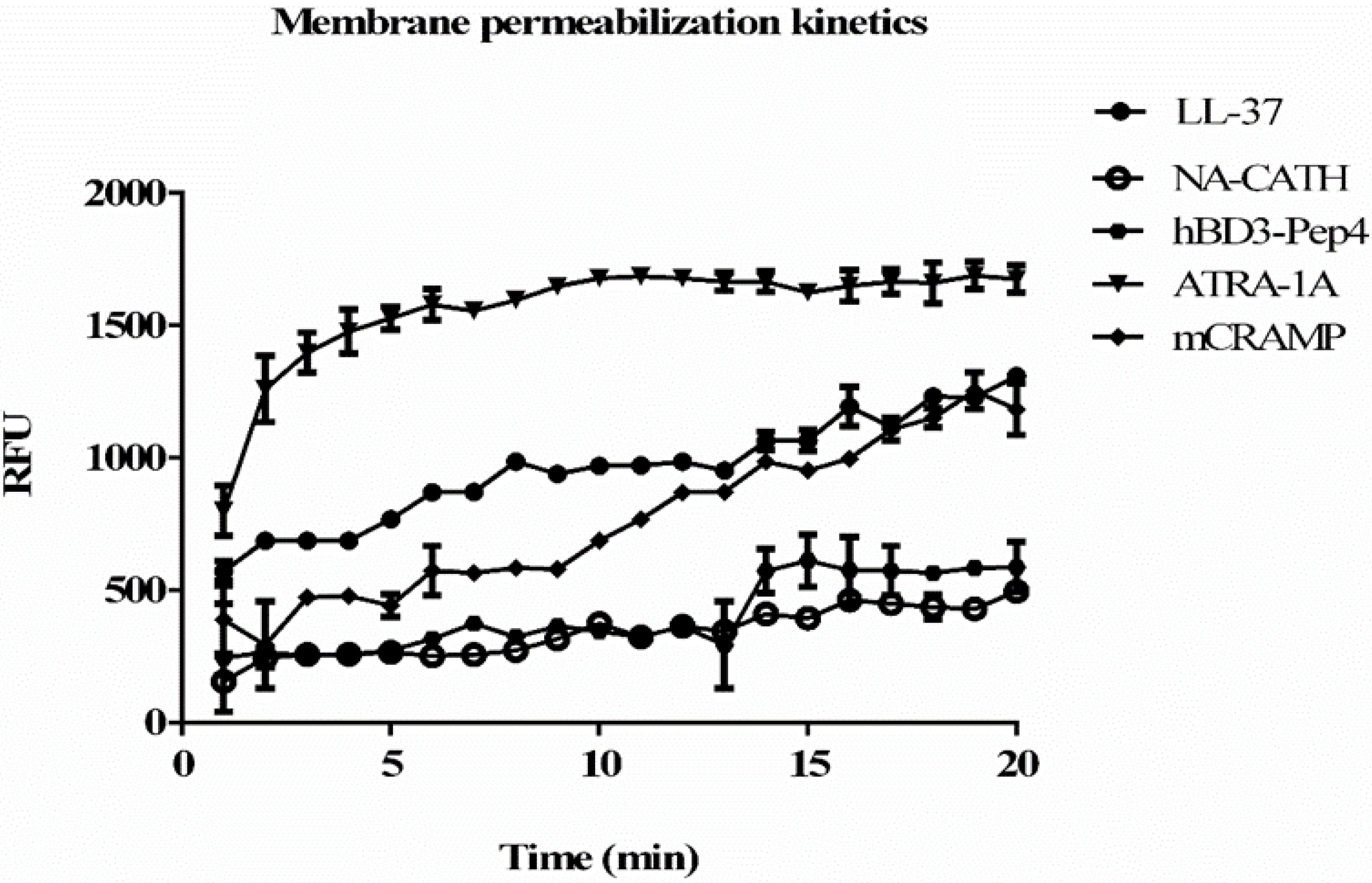
3.5. Membrane Permeablization and Depolarization by Peptides
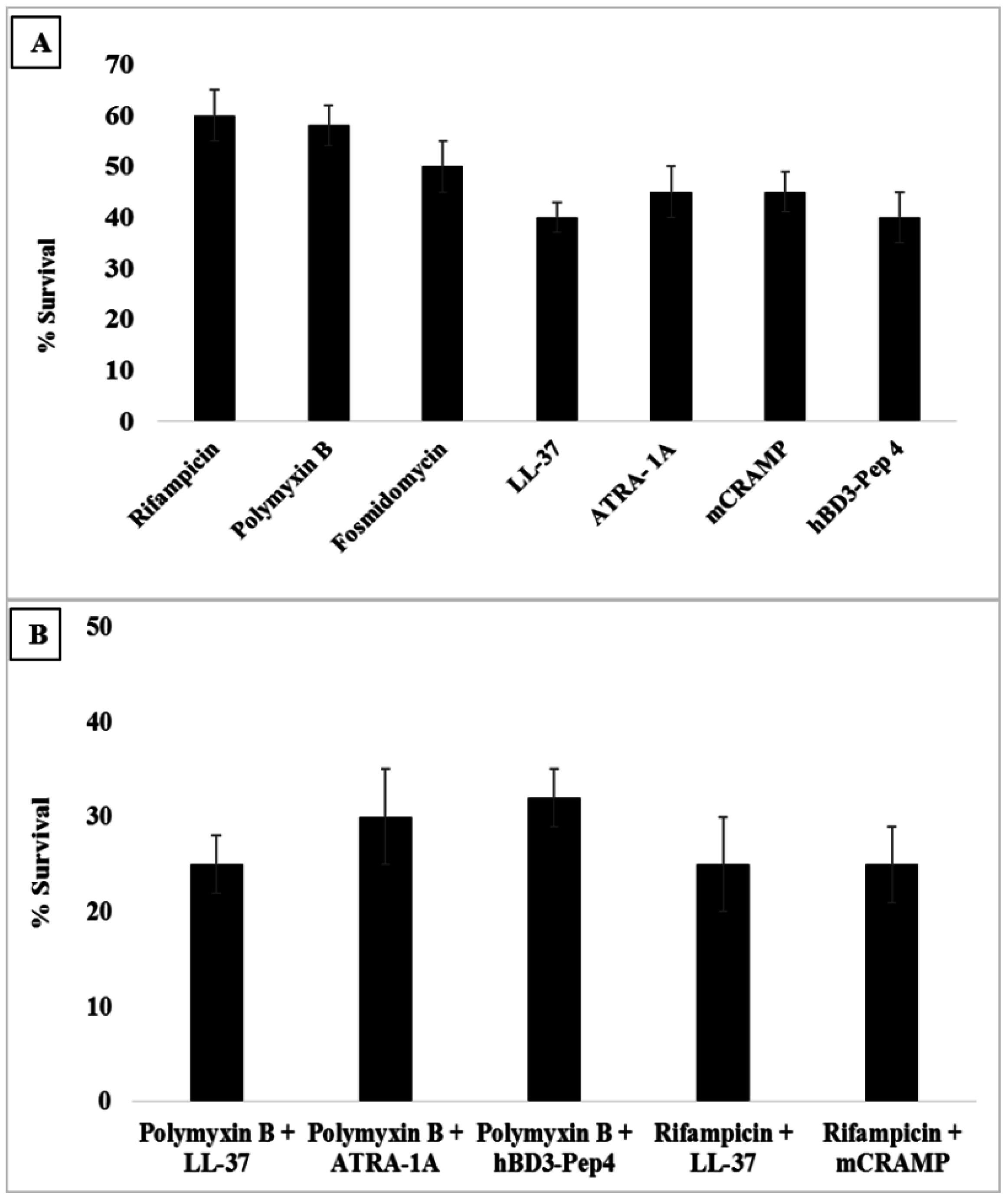
3.6. Synergy with Antibiotics
| Drug Combination | MIC (μg/mL) | FIC | FICI | |
|---|---|---|---|---|
| Alone | Combined | |||
| Rifampicin ATRA-1A | 3.9 | 1.95 | 0.5 | 0.56 |
| 31.3 | 3.9 | 0.06 | ||
| Rifampicin hBD3-Pep4 | 3.9 | 1.95 | 0.5 | 0.56 |
| 62.6 | 3.9 | 0.06 | ||
| Rifampicin LL-37 | 3.9 | 0.97 | 0.25 | 0.32 |
| 31.3 | 3.9 | 0.12 | ||
| Rifampicin mCRAMP | 3.9 | 0.97 | 0.25 | 0.35 |
| 15.6 | 1.95 | 0.12 | ||
| Polymyxin B ATRA-1A | 7.8 | 1.95 | 0.5 | 0.37 |
| 31.3 | 3.9 | 0.12 | ||
| Polymyxin B hBD3-Pep 4 | 7.8 | 1.95 | 0.25 | 0.37 |
| 62.6 | 7.8 | 0.12 | ||
| Polymyxin B LL-37 | 7.8 | 1.95 | 0.25 | 0.37 |
| 31.3 | 3.9 | 0.12 | ||
| Polymyxin B mCRAMP | 7.8 | 1.95 | 0.25 | 0.5 |
| 15.6 | 3.9 | 0.25 | ||
3.7. Intracellular Killing of Mycobacteria Peptide-Treated Macrophages

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Papanastasiou, E.A.; Hua, Q.; Sandouk, A.; Son, U.H.; Christenson, A.J.; van Hoek, M.L.; Bishop, B.M. Role of acetylation and charge in antimicrobial peptides based on human beta-defensin-3. Acta Pathol. Microbiol. Immunol. Scand. 2009, 117, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R. Multidrug and extensively drug-resistant TB (M/XDR-TB): Problems and solutions. Indian J. Tuberc. 2010, 57, 180–191. [Google Scholar] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Ushio, H.; Hara, M.; Yokoi, H.; Tominaga, M.; Takamori, K.; Kajiwara, N.; Saito, H.; Nagaoka, I.; Ogawa, H.; et al. Antimicrobial peptides human beta-defensins and cathelicidin LL-37 induce the secretion of a pruritogenic cytokine IL-31 by human mast cells. J. Immunol. 2010, 184, 3526–3534. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Jo, E.K. Antimicrobial peptides in innate immunity against Mycobacteria. Immune Netw. 2011, 11, 245–252. [Google Scholar] [CrossRef] [PubMed]
- De Smet, K.; Contreras, R. Human antimicrobial peptides: Defensins, cathelicidins and histatins. Biotechnol. Lett. 2005, 27, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Durr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.; Santos, J.C.; Mishra, B.B.; Jena, P.; Progida, C.; Sorensen, O.E.; Gallo, R.; Appelberg, R.; Griffiths, G. Cathelicidin is involved in the intracellular killing of Mycobacteria in macrophages. Cell. Microbiol. 2011, 13, 1601–1617. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T. Defensins of vertebrate animals. Curr. Opin. Immunol. 2002, 14, 96–102. [Google Scholar] [CrossRef]
- Han, S.; Bishop, B.M.; van Hoek, M.L. Antimicrobial activity of human beta-defensins and induction by Francisella. Biochem. Biophys. Res. Commun. 2008, 371, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Driessen, N.N.; Appelmelk, B.J.; Besra, G.S. Lipoarabinomannan and related glycoconjugates: Structure, biogenesis and role in Mycobacterium tuberculosis physiology and host-pathogen interaction. FEMS Microbiol. Rev. 2011, 35, 1126–1157. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.A.; Papanastasiou, E.A.; Juba, M.; Bishop, B. Covalent modification of a ten-residue cationic antimicrobial peptide with levofloxacin. Front. Chem. 2014, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Wong, E.B.; Cohen, K.A.; Bishai, W.R. Rising to the challenge: New therapies for tuberculosis. Trends Microbiol. 2013, 21, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Fattorini, L.; Gennaro, R.; Zanetti, M.; Tan, D.; Brunori, L.; Giannoni, F.; Pardini, M.; Orefici, G. In vitro activity of protegrin-1 and beta-defensin-1, alone and in combination with isoniazid, against Mycobacterium tuberculosis. Peptides 2004, 25, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Verma, I.; Khuller, G.K. Antibacterial activity of human neutrophil peptide-1 against Mycobacterium tuberculosis H37Rv: In vitro and ex vivo study. Eur. Respir. J. 2000, 16, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.M. The potential of human neutrophil peptides in tuberculosis therapy. Int. J. Tuberc. Lung Dis. 2003, 7, 1027–1032. [Google Scholar] [PubMed]
- De Latour, F.A.; Amer, L.S.; Papanstasiou, E.A.; Bishop, B.M.; van Hoek, M.L. Antimicrobial activity of the Naja atra cathelicidin and related small peptides. Biochem. Biophys. Res. Commun. 2010, 396, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Amer, L.S.; Bishop, B.M.; van Hoek, M.L. Antimicrobial and antibiofilm activity of cathelicidins and short, synthetic peptides against Francisella. Biochem. Biophys. Res. Commun. 2010, 396, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.N.; Bishop, B.M.; van Hoek, M.L. Susceptibility of Pseudomonas aeruginosa biofilm to alpha-helical peptides: D-enantiomer of LL-37. Front. Microbiol. 2011, 2, e128. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.N.; Bishop, B.M.; van Hoek, M.L. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 2011, 11, e114. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. Apd2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.S.; Namjoshi, O.A.; Verma, R.; Polanowski, R.; Krueger, S.M.; Sherman, D.; Rott, M.A.; Schwan, W.R.; Monte, A.; Cook, J.M. A new class of potential anti-tuberculosis agents: Synthesis and preliminary evaluation of novel acrylic acid ethyl ester derivatives. Bioorganic Med. Chem. 2010, 18, 4178–4186. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.H.; Houck, H.J.; Brown, P.; Bennett, D. Reproducibility of the microdilution checkerboard method for antibiotic synergy. Antimicrob. Agents Chemother. 1993, 37, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Tseng, W.; Guina, T.; Miller, S.I.; Nikaido, H. PhoPQ-mediated regulation produces a more robust permeability barrier in the outer membrane of Salmonella enterica serovar typhimurium. J. Bacteriol. 2007, 189, 7213–7222. [Google Scholar] [CrossRef] [PubMed]
- Khara, J.S.; Wang, Y.; Ke, X.Y.; Liu, S.; Newton, S.M.; Langford, P.R.; Yang, Y.Y.; Ee, P.L. Anti-mycobacterial activities of synthetic cationic alpha-helical peptides and their synergism with rifampicin. Biomaterials 2014, 35, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Silva-Gomes, S.; Silva, J.P.; Gama, M.; Rosa, G.; Gallo, R.L.; Appelberg, R. Endogenous cathelicidin production limits inflammation and protective immunity to Mycobacterium avium in mice. Immun. Inflamm. Dis. 2014, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ramon-Garcia, S.; Mikut, R.; Ng, C.; Ruden, S.; Volkmer, R.; Reischl, M.; Hilpert, K.; Thompson, C.J. Targeting Mycobacterium tuberculosis and other microbial pathogens using improved synthetic antibacterial peptides. Antimicrob. Agents Chemother. 2013, 57, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Rivas Santiago, C.E.; Castaneda-Delgado, J.E.; Leon-Contreras, J.C.; Hancock, R.E.; Hernandez-Pando, R. Activity of LL-37, CRAMP and antimicrobial peptide-derived compounds E2, E6 and CP26 against Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2013, 41, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Lee, D.G.; Yang, S.T.; Kim, Y.; Kim, J.I.; Hahm, K.S.; Shin, S.Y. CRAMP analog having potent antibiotic activity without hemolytic activity. Protein Pept. Lett. 2002, 9, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Sanchez, J.I.; Garcia-Perez, B.E.; Munoz-Duarte, A.R.; Baltierra-Uribe, S.L.; Mejia-Lopez, H.; Lopez-Lopez, C.; Bautista-De Lucio, V.M.; Robles-Contreras, A.; Luna-Herrera, J. Defensin production by human limbo-corneal Fibroblasts infected with Mycobacteria. Pathogens 2013, 2, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Corrales-Garcia, L.; Ortiz, E.; Castaneda-Delgado, J.; Rivas-Santiago, B.; Corzo, G. Bacterial expression and antibiotic activities of recombinant variants of human beta-defensins on pathogenic bacteria and M. tuberculosis. Protein Expr. Purif. 2013, 89, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Aoki, W.; Ueda, M. Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals 2013, 6, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- McKenney, E.S.; Sargent, M.; Khan, H.; Uh, E.; Jackson, E.R.; San Jose, G.; Couch, R.D.; Dowd, C.S.; van Hoek, M.L. Lipophilic prodrugs of FR900098 are antimicrobial against Francisella novicida in vivo and in vitro and show GlpT independent efficacy. PLoS ONE 2012, 7, e38167. [Google Scholar] [CrossRef] [PubMed]
- Mackie, R.S.; McKenney, E.S.; van Hoek, M.L. Resistance of Francisella novicida to fosmidomycin associated with mutations in the glycerol-3-phosphate transporter. Front. Microbiol. 2012, 3, e226. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, M.; Lindh, M.; Bjorkelid, C.; Suresh, S.; Wieckowska, A.; Iyer, H.; Karlen, A.; Larhed, M. Substitution of the phosphonic acid and hydroxamic acid functionalities of the DXR inhibitor FR900098: An attempt to improve the activity against Mycobacterium tuberculosis. Bioorganic Med. Chem. Lett. 2011, 21, 5403–5407. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Zhao, D.; Lyu, Y.; Tian, L.; Yin, X.; Yang, L.; Teng, K.; Zhou, X. Antimycobacterial activity of pichia pastoris-derived mature bovine neutrophil beta-defensins 5. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, Y.; Ratnakar, P.; Rao, A.G.; Costello, M.L.; Mathieu-Costello, O.; Lehrer, R.I.; Catanzaro, A. In vitro activity of the antimicrobial peptides human and rabbit defensins and porcine leukocyte protegrin against Mycobacterium tuberculosis. Infect. Immun. 1996, 64, 926–932. [Google Scholar] [PubMed]
- Anantharaman, A.; Rizvi, M.S.; Sahal, D. Synergy with rifampin and kanamycin enhances potency, kill kinetics, and selectivity of de novo-designed antimicrobial peptides. Antimicrob. Agents Chemother. 2010, 54, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Barriere, S.L. Bacterial resistance to beta-lactams, and its prevention with combination antimicrobial therapy. Pharmacotherapy 1992, 12, 397–402. [Google Scholar] [PubMed]
- Cirioni, O.; Silvestri, C.; Ghiselli, R.; Orlando, F.; Riva, A.; Mocchegiani, F.; Chiodi, L.; Castelletti, S.; Gabrielli, E.; Saba, V.; et al. Protective effects of the combination of alpha-helical antimicrobial peptides and rifampicin in three rat models of Pseudomonas aeruginosa infection. J. Antimicrob. Chemother. 2008, 62, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Kalita, A.; Verma, I.; Khuller, G.K. Role of human neutrophil peptide-1 as a possible adjunct to antituberculosis chemotherapy. J. Infect. Dis. 2004, 190, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Jordao, L.; Bleck, C.K.; Mayorga, L.; Griffiths, G.; Anes, E. On the killing of Mycobacteria by macrophages. Cell. Microbiol. 2008, 10, 529–548. [Google Scholar] [CrossRef] [PubMed]
- Yamshchikov, A.V.; Kurbatova, E.V.; Kumari, M.; Blumberg, H.M.; Ziegler, T.R.; Ray, S.M.; Tangpricha, V. Vitamin D status and antimicrobial peptide cathelicidin (LL-37) concentrations in patients with active pulmonary tuberculosis. Am. J. Clin. Nutr. 2010, 92, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Rehn, A.; Rahman, J.; Andersson, J.; Svensson, M.; Brighenti, S. Pulmonary tuberculosis patients with a vitamin D deficiency demonstrate low local expression of the antimicrobial peptide LL-37 but enhanced FoxP3+ regulatory T cells and IgG-secreting cells. Clin. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Larcombe, L.; Orr, P.; Turner-Brannen, E.; Slivinski, C.R.; Nickerson, P.W.; Mookherjee, N. Effect of vitamin D supplementation on Mycobacterium tuberculosis-induced innate immune responses in a canadian dene first nations cohort. PLoS ONE 2012, 7, e40692. [Google Scholar] [CrossRef] [PubMed]
- Afsal, K.; Harishankar, M.; Banurekha, V.V.; Meenakshi, N.; Parthasarathy, R.T.; Selvaraj, P. Effect of 1,25-dihydroxy vitamin D on cathelicidin expression in patients with and without cavitary tuberculosis. Tuberculosis 2014, 94, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Reyrat, J.M. Laboratory maintenance of Mycobacterium smegmatis. Curr. Protoc. Microbiol. 2009. [Google Scholar] [CrossRef]
- Bhatt, A.; Molle, V.; Besra, G.S.; Jacobs, W.R., Jr.; Kremer, L. The Mycobacterium tuberculosis FAS-IIcondensing enzymes: Their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 2007, 64, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Smeulders, M.J.; Keer, J.; Speight, R.A.; Williams, H.D. Adaptation of Mycobacterium smegmatis to stationary phase. J. Bacteriol. 1999, 181, 270–283. [Google Scholar] [PubMed]
- Baloni, P.; Padiadpu, J.; Singh, A.; Gupta, K.R.; Chandra, N. Identifying feasible metabolic routes in Mycobacterium smegmatis and possible alterations under diverse nutrient conditions. BMC Microbiol. 2014, 14, e276. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bhakta, S. An integrated surrogate model for screening of drugs against Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012, 67, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.N.; Daniels, L. Characterization of the MSMEG_2631 gene (mmp) encoding a multidrug and toxic compound extrusion (MATE) family protein in Mycobacterium smegmatis and exploration of its polyspecific nature using biolog phenotype microarray. J. Bacteriol. 2013, 195, 1610–1621. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Weiss, J. Antimicrobial peptides of phagocytes and epithelia. Semin. Hematol. 1997, 34, 343–354. [Google Scholar] [PubMed]
- Bals, R. Epithelial antimicrobial peptides in host defense against infection. Respir. Res. 2000, 1, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duplantier, A.J.; van Hoek, M.L. The human cathelicidin antimicrobial peptide LL-37 as a potential treatment for polymicrobial infected wounds. Front. Immunol. 2013, 4, e143. [Google Scholar] [CrossRef] [PubMed]
- Fahy, R.J.; Wewers, M.D. Pulmonary defense and the human cathelicidin hCAP-18/LL-37. Immunol. Res. 2005, 31, 75–89. [Google Scholar] [CrossRef]
- Jiang, Z.; Higgins, M.P.; Whitehurst, J.; Kisich, K.O.; Voskuil, M.I.; Hodges, R.S. Anti-tuberculosis activity of alpha-helical antimicrobial peptides: De novo designed l- and d-enantiomers versus L- and d-LL-37. Protein Pept. Lett. 2011, 18, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.M.; Wu, Z.; Tucker, K.; Lu, W.; Lubkowski, J. Antimicrobial characterization of human beta-defensin 3 derivatives. Antimicrob. Agents Chemother. 2003, 47, 2804–2809. [Google Scholar] [CrossRef] [PubMed]
- Schutte, B.C.; Mitros, J.P.; Bartlett, J.A.; Walters, J.D.; Jia, H.P.; Welsh, M.J.; Casavant, T.L.; McCray, P.B., Jr. Discovery of five conserved beta-defensin gene clusters using a computational search strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Schutte, B.C.; Schudy, A.; Linzmeier, R.; Guthmiller, J.M.; Johnson, G.K.; Tack, B.F.; Mitros, J.P.; Rosenthal, A.; Ganz, T.; et al. Discovery of new human beta-defensins using a genomics-based approach. Gene 2001, 263, 211–218. [Google Scholar] [PubMed]
- Yang, D.; Biragyn, A.; Kwak, L.W.; Oppenheim, J.J. Mammalian defensins in immunity: More than just microbicidal. Trends Immunol. 2002, 23, 291–296. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, K.; Singh, S.; Van Hoek, M.L. Short, Synthetic Cationic Peptides Have Antibacterial Activity against Mycobacterium smegmatis by Forming Pores in Membrane and Synergizing with Antibiotics. Antibiotics 2015, 4, 358-378. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics4030358
Gupta K, Singh S, Van Hoek ML. Short, Synthetic Cationic Peptides Have Antibacterial Activity against Mycobacterium smegmatis by Forming Pores in Membrane and Synergizing with Antibiotics. Antibiotics. 2015; 4(3):358-378. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics4030358
Chicago/Turabian StyleGupta, Kajal, Sameer Singh, and Monique L. Van Hoek. 2015. "Short, Synthetic Cationic Peptides Have Antibacterial Activity against Mycobacterium smegmatis by Forming Pores in Membrane and Synergizing with Antibiotics" Antibiotics 4, no. 3: 358-378. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics4030358