New Chloramphenicol Derivatives from the Viewpoint of Anticancer and Antimicrobial Activity

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
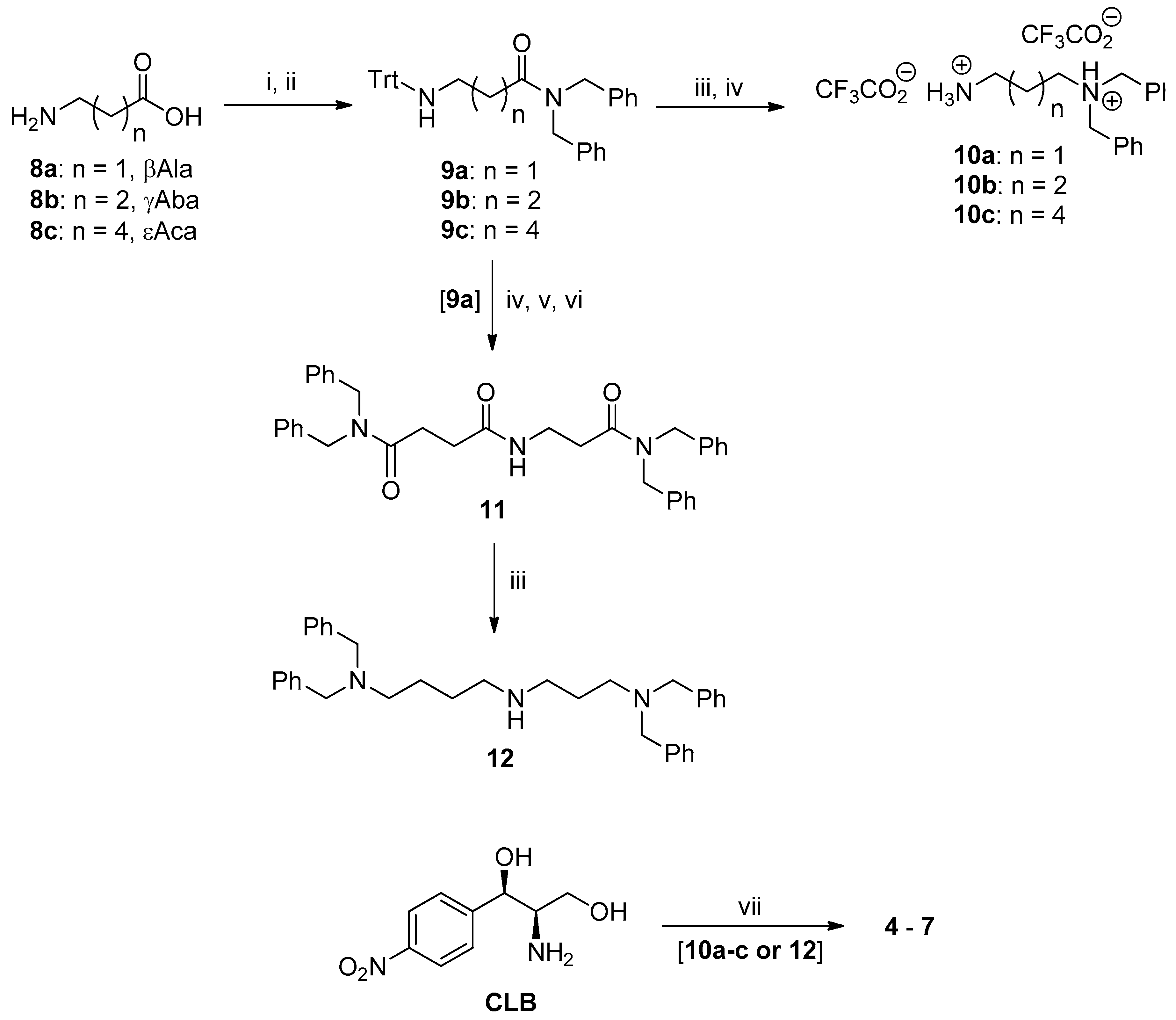
2.1. Synthesis of PA–CAM Conjugates
2.2. Biological Evaluation
2.2.1. Bacterial Strains and Cell Lines
2.2.2. In Vivo Antibacterial Activity
2.2.3. Affinity Measurement of PA–CAM Conjugates for the E. coli 70S Ribosome
2.2.4. Evaluation of the Anticancer Activity
2.2.5. Immunoblotting
2.2.6. Quantification of the Intracellular Levels of Conjugates 3 and 4 Plus Polyamines
2.2.7. System Modeling and Molecular Dynamics Simulations
2.2.8. Statistics
3. Results
3.1. Antibacterial Activity of Novel PA–CAM Conjugates
3.2. The New PA–CAM Conjugates Compete with CAM to Bind on the Bacterial Ribosome
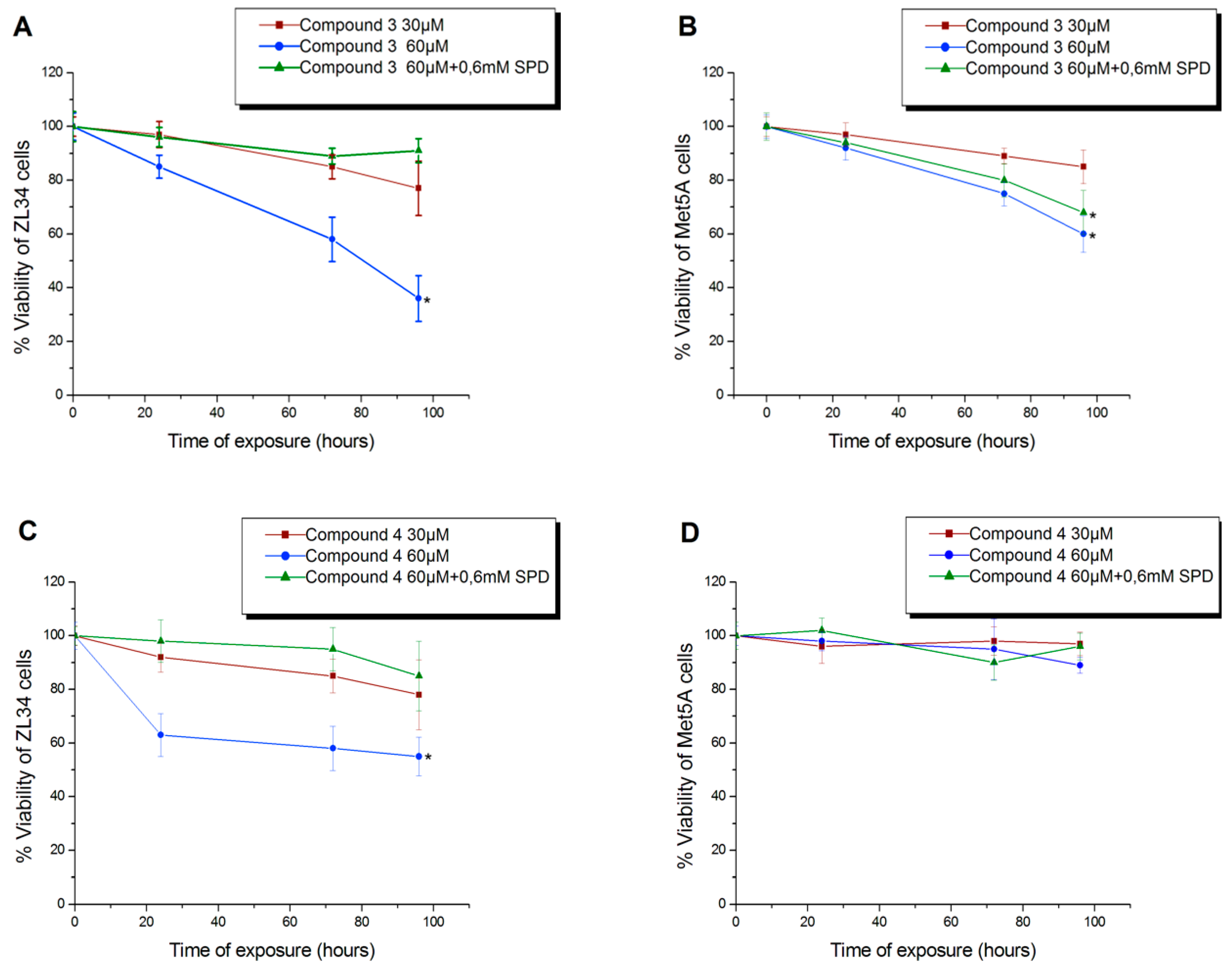
3.3. Antiproliferative Activity of Compound 4
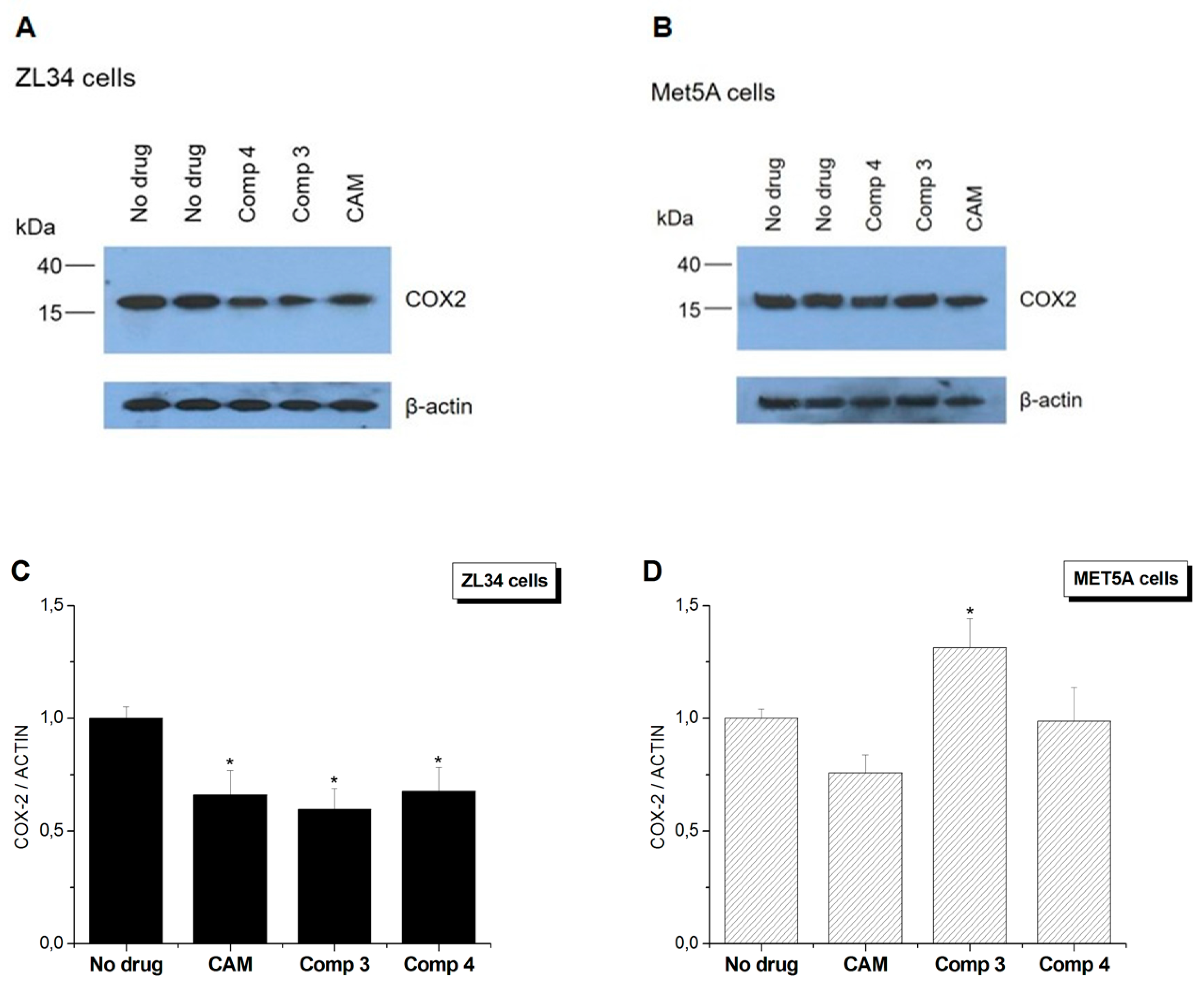
3.4. Conjugates 3 and 4 Are Inhibitors of Mitochondrial Protein Synthesis
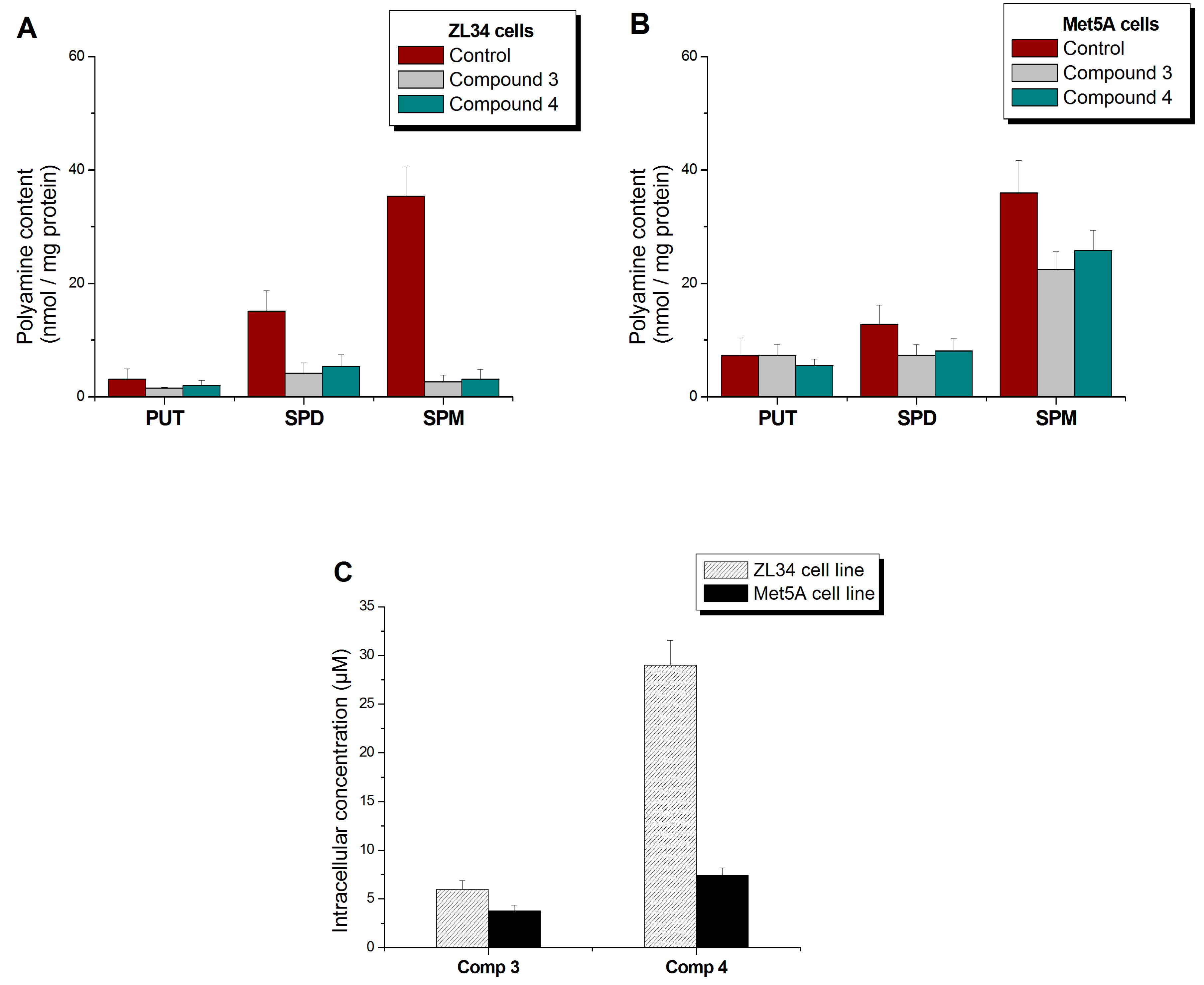
3.5. Derivatives’ and Polyamines’ Intracellular Concentrations
3.6. System Modeling and Molecular Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dinos, G.; Athanassopoulos, C.; Missiri, D.; Giannopoulou, P.; Vlachogiannis, I.; Papadopoulos, G.; Papaioannou, D.; Kalpaxis, D. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef]
- Yuan, Z.R.; Shi, Y. Chloramphenicol induces abnormal differentiation and inhibits apoptosis in activated T cells. Cancer Res. 2008, 68, 4875–4881. [Google Scholar] [CrossRef]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Sripada, L.; Singh, R. Side effects of antibiotics during bacterial infection: Mitochondria, the main target in host cell. Mitochondrion 2014, 16, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.; Kannan, K.; Roncase, E.J.; Klepacki, D.; Kefi, A.; Orelle, C.; Vázquez-Laslop, N.; Mankin, A.S. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl. Acad. Sci. USA 2016, 113, 12150–12155. [Google Scholar] [CrossRef] [PubMed]
- Schlünzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef]
- Vázquez-Laslop, N.; Mankin, A.S. Context-Specific Action of Ribosomal Antibiotics. Annu. Rev. Microbiol. 2018, 72, 185–207. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Kouvela, E.C.; Magoulas, G.E.; Garnelis, T.; Panagoulias, I.; Rodi, M.; Papadopoulos, G.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; et al. Conjugation with polyamines enhances the antibacterial and anticancer activity of chloramphenicol. Nucleic Acids Res. 2014, 42, 8621–8634. [Google Scholar] [CrossRef] [Green Version]
- Magoulas, G.E.; Kostopoulou, O.N.; Garnelis, T.; Athanassopoulos, C.M.; Kournoutou, G.G.; Leotsinidis, M.; Dinos, G.P.; Papaioannou, D.; Kalpaxis, D.L. Synthesis and antimicrobial activity of chloramphenicol-polyamine conjugates. Bioorganic Med. Chem. 2015, 23, 3163–3174. [Google Scholar] [CrossRef]
- Dever, T.E.; Ivanov, I.P. Roles of polyamines in translation. J. Biol. Chem. 2018, 293, 18719–18729. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kashiwagi, K. Effects of polyamines on protein synthesis and growth of Escherichia coli. J. Biol. Chem. 2018, 293, 18702–18709. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Swiatlo, E. A multifaceted role for polyamines in bacterial pathogens. Mol. Microbiol. 2008, 68, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [Google Scholar] [CrossRef]
- Paz, E.A.; Lafleur, B.; Gerner, E.W. Polyamines are oncometabolites that regulate the LIN28/let-7 pathway in colorectal cancer cells. Mol. Carcinog. 2014, 53, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.J.; Wallace, H.M. The polyamine transport system as a target for anticancer drug development. Amino Acids 2010, 38, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Tsiakopoulos, N.; Damianakos, C.; Karigiannis, G.; Vahliotis, D.; Papaioannou, D.; Sindona, G. Syntheses of crowned polyamines using isolable succinimidyl esters of N-tritylated linear amino acids and peptides. Arkivoc 2002, 2002, 79–104. [Google Scholar]
- Magoulas, G.E.; Garnelis, T.; Athanassopoulos, C.M.; Papaioannou, D.; Mattheolabakis, G.; Avgoustakis, K.; Hadjipavlou-Litina, D. Synthesis and antioxidative/anti-inflammatory activity of novel fullerene-polyamine conjugates. Tetrahedron 2012, 68, 7041–7049. [Google Scholar] [CrossRef]
- Ke, Y.; Reddel, R.R.; Gerwin, B.I.; Reddel, H.K.; Somers, A.N.; McMenamin, M.G.; LaVeck, M.A.; Stahel, R.A.; Lechner, J.F.; Harris, C.C. Establishment of a human in vitro mesothelial cell model system for investigating mechanisms of asbestos-induced mesothelioma. Am. J. Pathol. 1989, 134, 979–991. [Google Scholar] [PubMed]
- Kostopoulou, O.N.; Magoulas, G.E.; Papadopoulos, G.E.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; Kalpaxis, D.L. Synthesis and evaluation of chloramphenicol homodimers: Molecular target, antimicrobial activity, and toxicity against human cells. PLoS ONE 2015, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Xaplanteri, M.A.; Petropoulos, A.D.; Dinos, G.P.; Kalpaxis, D.L. Localization of spermine binding sites in 23S rRNA by photoaffinity labeling: Parsing the spermine contribution to ribosomal 50S subunit functions. Nucleic Acids Res. 2005, 33, 2792–2805. [Google Scholar] [CrossRef]
- Bougas, A.; Vlachogiannis, I.A.; Gatos, D.; Arenz, S.; Dinos, G.P. Dual effect of chloramphenicol peptides on ribosome inhibition. Amino Acids 2017, 49, 995–1004. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Wang, B.; Ao, J.; Yu, D.; Rao, T.; Ruan, Y.; Yao, X. Inhibition of mitochondrial translation effectively sensitizes renal cell carcinoma to chemotherapy. Biochem. Biophys. Res. Commun. 2017, 490, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and Action of Amino Acid Analogs of Chloramphenicol upon the Bacterial Ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Tian, F.; Wang, C.; Tang, M.; Li, J.; Cheng, X.; Zhang, S.; Ji, D.; Huang, Y.; Li, H.; Tian, F.; et al. The antibiotic chloramphenicol may be an effective new agent for inhibiting the growth of multiple myeloma. Oncotarget 2014, 7, 51934–51942. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.G.E.; Zomorodipour, A.; Andersson, J.O.; Sicheritz-Pontén, T.; Alsmark, U.C.M.; Podowski, R.M.; Näslund, A.K.; Eriksson, A.S.; Winkler, H.H.; Kurland, C.G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998, 396, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Amunts, A.; Bai, X.C.; Sugimoto, Y.; Edwards, P.C.; Murshudov, G.; Scheres, S.H.W.; Ramakrishnan, V. Structure of the large ribosomal subunit from human mitochondria. Science 2014, 346, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Douthwaite, S. Functional interactions within 23S rRNA involving the peptidyltransferase center. J. Bacteriol. 1992, 174, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Mortimer, P.G.S.; Piddok, L.J.V. The accumulation of five antibacterial agents in porin-deficient mutants of escherichia coli. J. Antimicrob. Chemother. 1993, 32, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Shaw, W.V. O-Acetyltransferases for chloramphenicol and other natural products. Antimicrob. Agents Chemother. 1997, 41, 1–6. [Google Scholar] [CrossRef]
- Okusu, H.; Ma, D.; Nikaido, H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 1996, 178, 306–308. [Google Scholar] [CrossRef]
- Ghisalberti, D.; Masi, M.; Pagès, J.-M.; Chevalier, J. Chloramphenicol and expression of multidrug efflux pump in Enterobacter aerogenes. Biochem. Biophys. Res. Commun. 2005, 328, 1113–1118. [Google Scholar] [CrossRef]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [PubMed]
- Drainas, D.; Kalpaxis, D.L.; Coutsogeorgopoulos, C. Inhibition of ribosomal peptidyltransferase by chloramphenicol: Kinetic studies. Eur. J. Biochem. 1987, 164, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Drainas, D.; Mamos, P.; Coutsogeorgopoulos, C. Aminoacyl Analogs of Chloramphenicol: Examination of the Kinetics of Inhibition of Peptide Bond Formation. J. Med. Chem. 1993, 36, 3542–3545. [Google Scholar] [CrossRef]
- Michelinaki, M.; Mamos, P.; Coutsogeorgopoulos, C.; Kalpaxis, D.L. Aminoacyl and peptidyl analogs of chloramphenicol as slow-binding inhibitors of ribosomal peptidyltransferase: A new approach for evaluating their potency. Mol Pharmacol 1997, 51, 139–146. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, A.R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostinelli, E.; Marques, M.P.M.; Calheiros, R.; Gil, F.P.S.C.; Tempera, G.; Viceconte, N.; Battaglia, V.; Grancara, S.; Toninello, A. Polyamines: Fundamental characters in chemistry and biology. Amino Acids 2010, 38, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Maiti, P.; Barrientos, A. Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 2017, 47, 67–81. [Google Scholar] [CrossRef]
- Yusoff, A.A.M. Role of mitochondrial DNA mutations in brain tumors: A mini-review. J. Cancer Res. Ther. 2015, 11, 535–544. [Google Scholar] [CrossRef]
- Wallace, H.M.; Niiranen, K. Polyamine analogues—An update. Amino Acids 2007, 33, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E995–E1010. [Google Scholar] [CrossRef]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Mobile Phase A (%) | Mobile Phase B (%) |
|---|---|---|
| 0 | 45 | 55 |
| 14 | 80 | 20 |
| 15 | 90 | 10 |
| Compound | EC50 (μΜ) | ||
|---|---|---|---|
| Escherichia coli K12 | Escherichia coli ΔTolC | Staphylococcus aureus | |
| CAM | 6.2 ± 0.5 | 2.3 ± 0.6 | 3.4 ± 0.3 |
| 3 | 11.0 ± 0.9 | 8.9 ± 0.7 | 6.8 ± 0.5 |
| 4 | >200 | >200 | >200 |
| 5 | >200 | >200 | >200 |
| 6 | >200 | >200 | >200 |
| 7 | >200 | >200 | >200 |
| Compound | Ki (μΜ) |
|---|---|
| CAM | 1.5 ± 0.1 |
| 3 | 0.8 ± 0.1 |
| 4 | 1.1 ± 0.1 |
| 5 | 2.6 ± 0.3 |
| 6 | 1.3 ± 0.2 |
| 7 | 1.0 ± 0.1 |
| (nmol/mg Protein) | |||||
|---|---|---|---|---|---|
| Cell line | Compound | Concentration (μΜ) | PUT | SPD | SPM |
| ZL34 | - | - | 3.18 ± 1.82 | 15.16 ± 3.63 | 35.40 ± 5.16 |
| 3 | 5.96 ± 0.92 | 1.58 ± 0.13 | 4.18 ± 1.87 1 | 2.71 ± 1.17 2 | |
| 4 | 29.00 ± 2.53 | 2.07 ± 0.91 | 5.36 ± 2.15 1 | 3.17 ± 1.70 2 | |
| Met5A | - | - | 7.29 ± 3.17 | 12.87 ± 3.34 | 35.96 ± 11.72 |
| 3 | 3.75 ± 0.60 | 7.38 ± 1.93 | 7.38 ± 1.85 | 22.50 ± 3.11 | |
| 4 | 7.40 ± 0.80 | 5.60 ± 1.10 | 8.14 ± 2.16 | 25.80 ± 6.57 | |
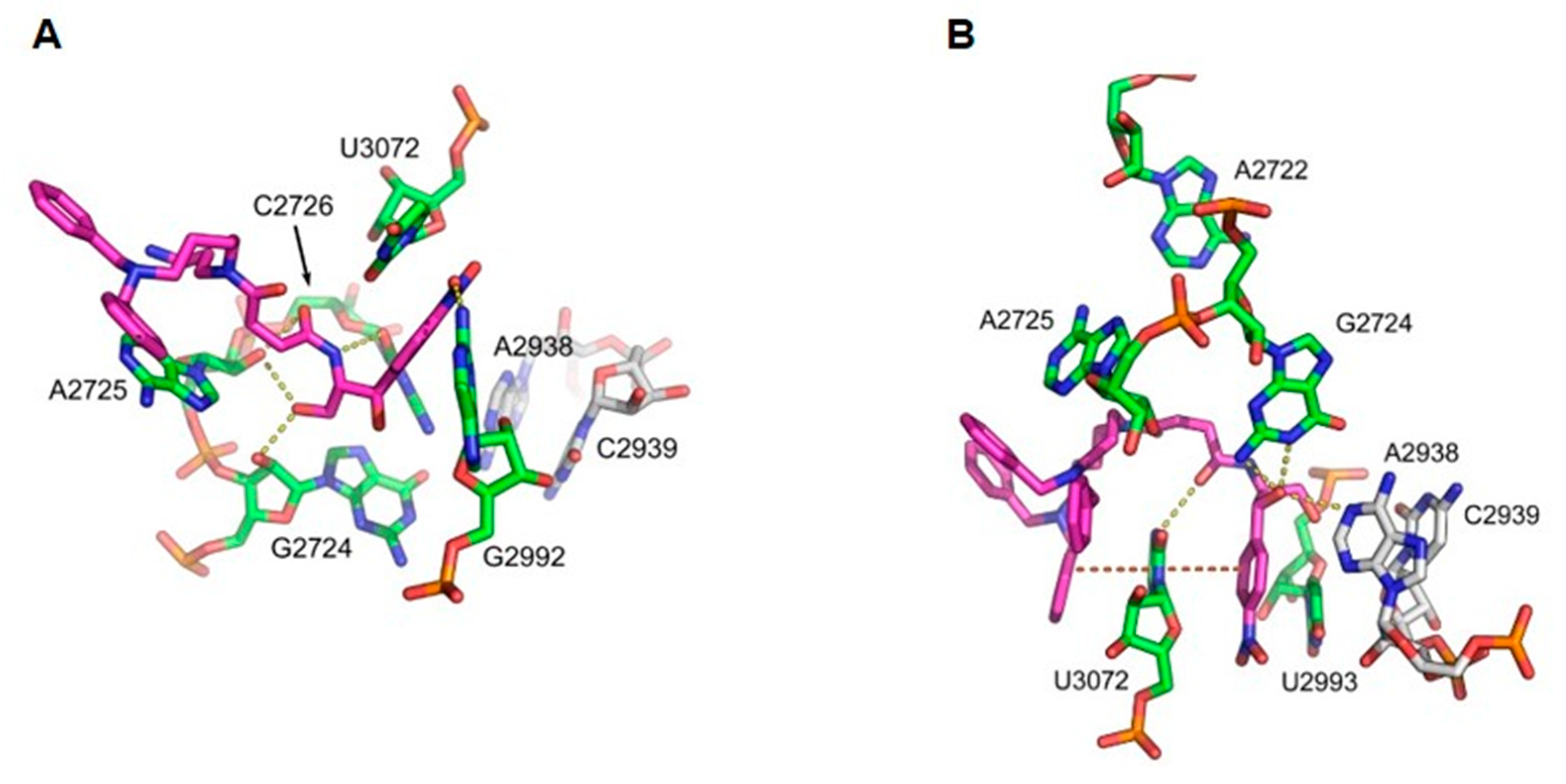
| rRNA | Comp. 3 | rRNA | Comp. 4 |
|---|---|---|---|
| N2: G2992 (G2505) | O1 | N3: U3072 (U2585) | O5 |
| O2: C2726 (C2063) | N2 | N2: G2724 (G2061) | O3 |
| O2′: G2724 (G2061) | O4 | N1: G2724 (G2061) | O3 |
| O2′: A2725 (A2062) | O4 | N1: A2938 (A2451) | O3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannopoulou, P.C.; Missiri, D.A.; Kournoutou, G.G.; Sazakli, E.; Papadopoulos, G.E.; Papaioannou, D.; Dinos, G.P.; Athanassopoulos, C.M.; Kalpaxis, D.L. New Chloramphenicol Derivatives from the Viewpoint of Anticancer and Antimicrobial Activity. Antibiotics 2019, 8, 9. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8010009
Giannopoulou PC, Missiri DA, Kournoutou GG, Sazakli E, Papadopoulos GE, Papaioannou D, Dinos GP, Athanassopoulos CM, Kalpaxis DL. New Chloramphenicol Derivatives from the Viewpoint of Anticancer and Antimicrobial Activity. Antibiotics. 2019; 8(1):9. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8010009
Chicago/Turabian StyleGiannopoulou, Panagiota C., Dionissia A. Missiri, Georgia G. Kournoutou, Eleni Sazakli, Georgios E. Papadopoulos, Dionissios Papaioannou, George P. Dinos, Constantinos M. Athanassopoulos, and Dimitrios L. Kalpaxis. 2019. "New Chloramphenicol Derivatives from the Viewpoint of Anticancer and Antimicrobial Activity" Antibiotics 8, no. 1: 9. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8010009