Isolation and Characterization of Pectobacterium Phage vB_PatM_CB7: New Insights into the Genus Certrevirus

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Results
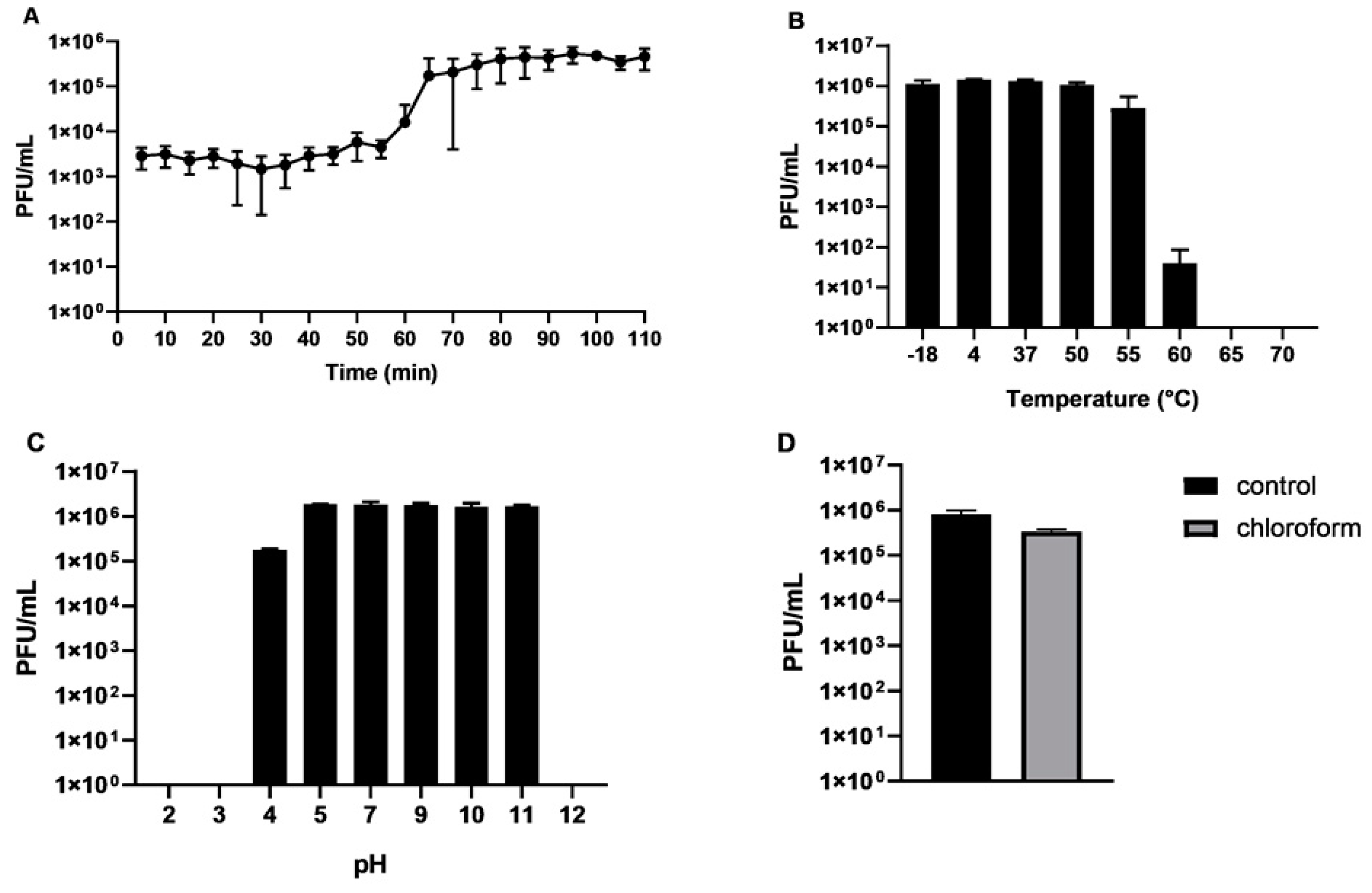
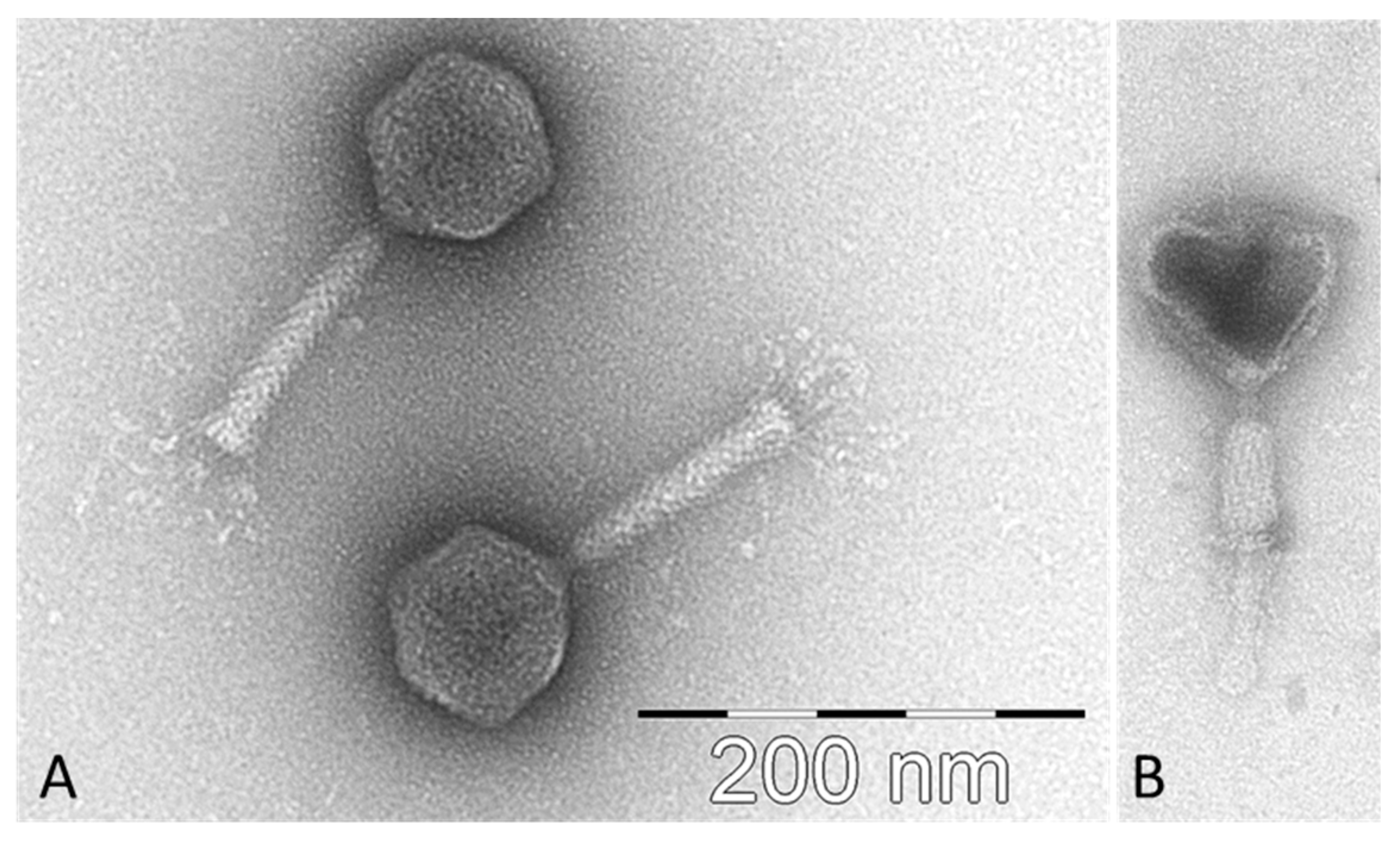
2.1. Isolation of CB7, Host Range and General Characteristics
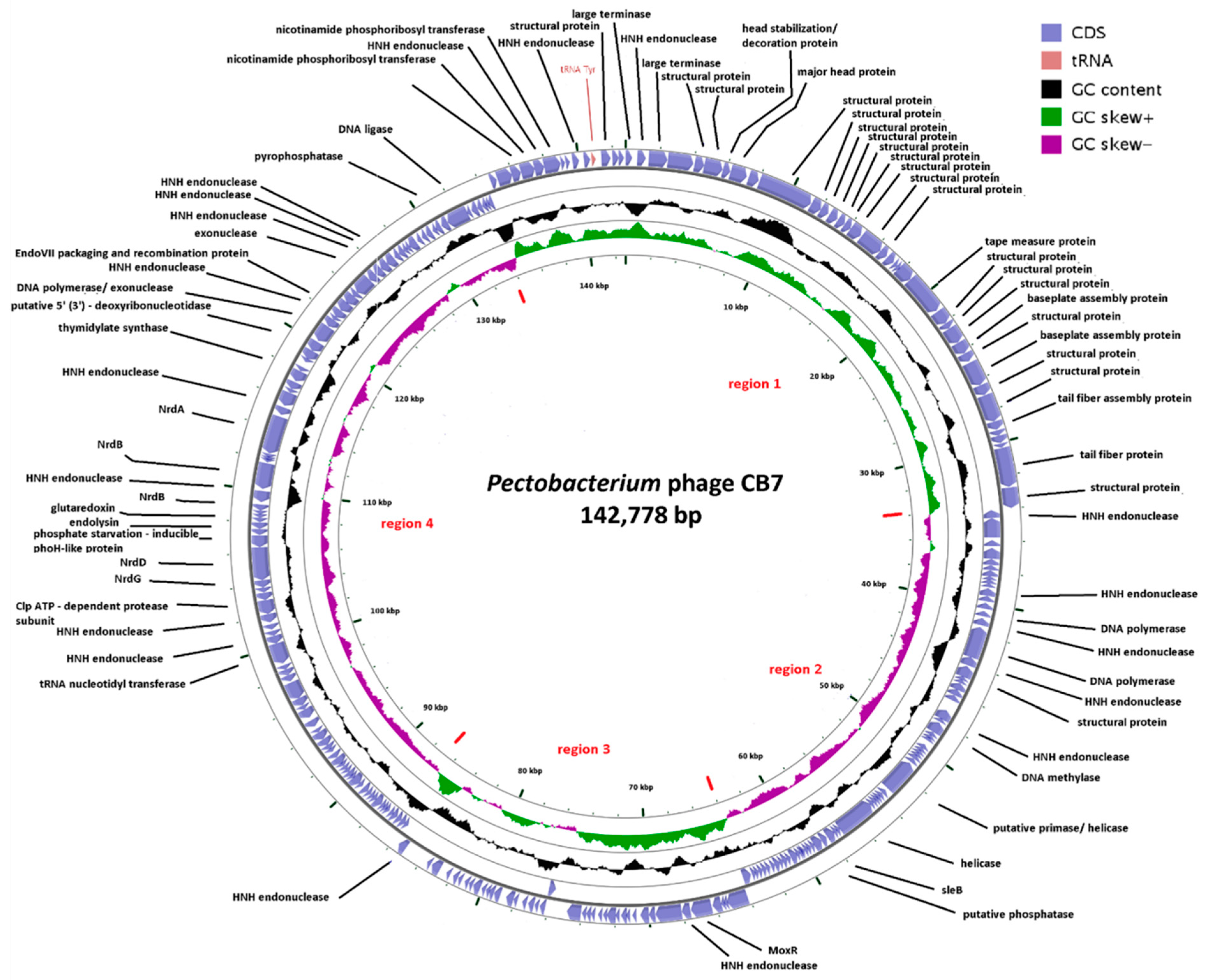
2.2. General Genome Characteristics of CB7
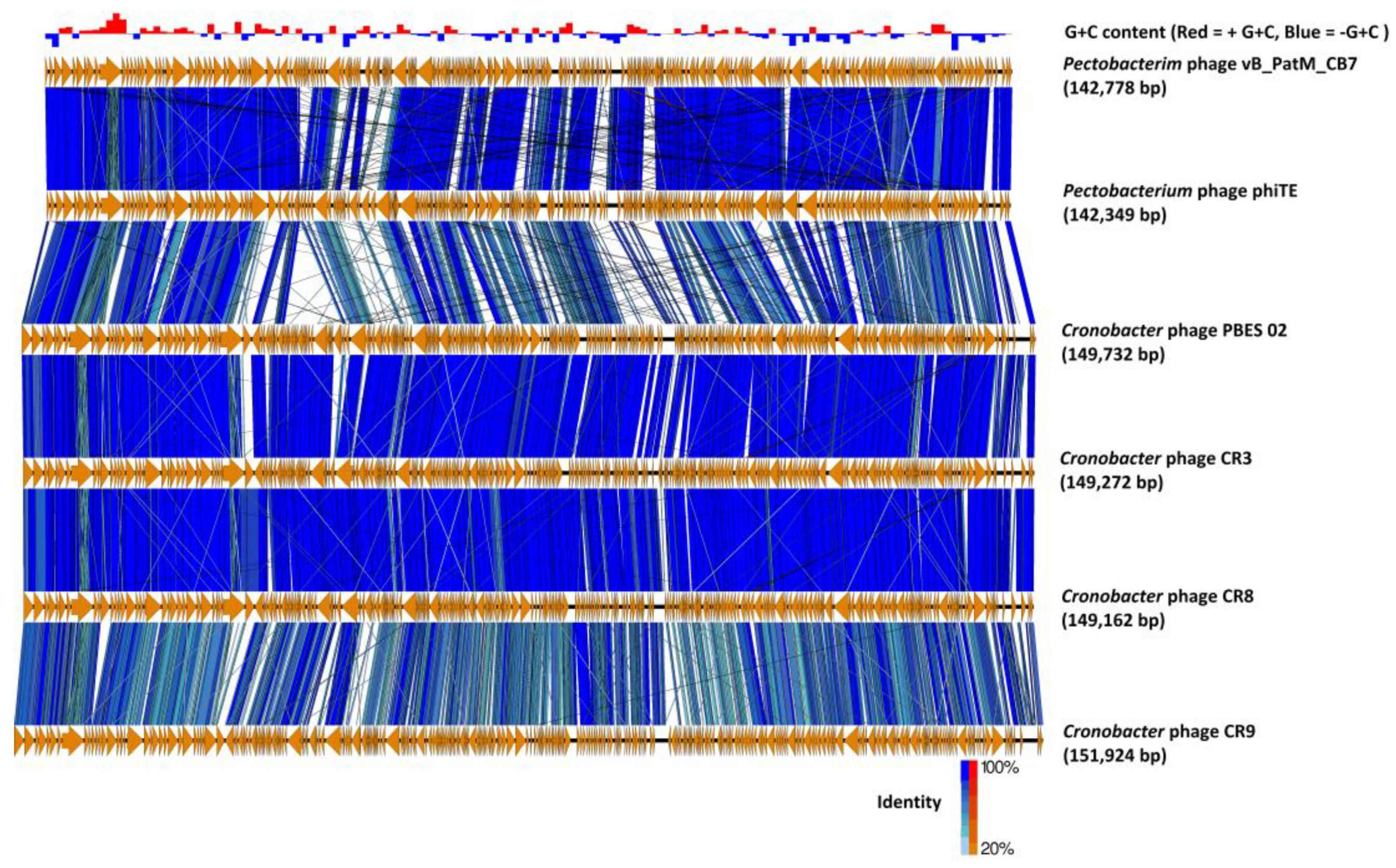
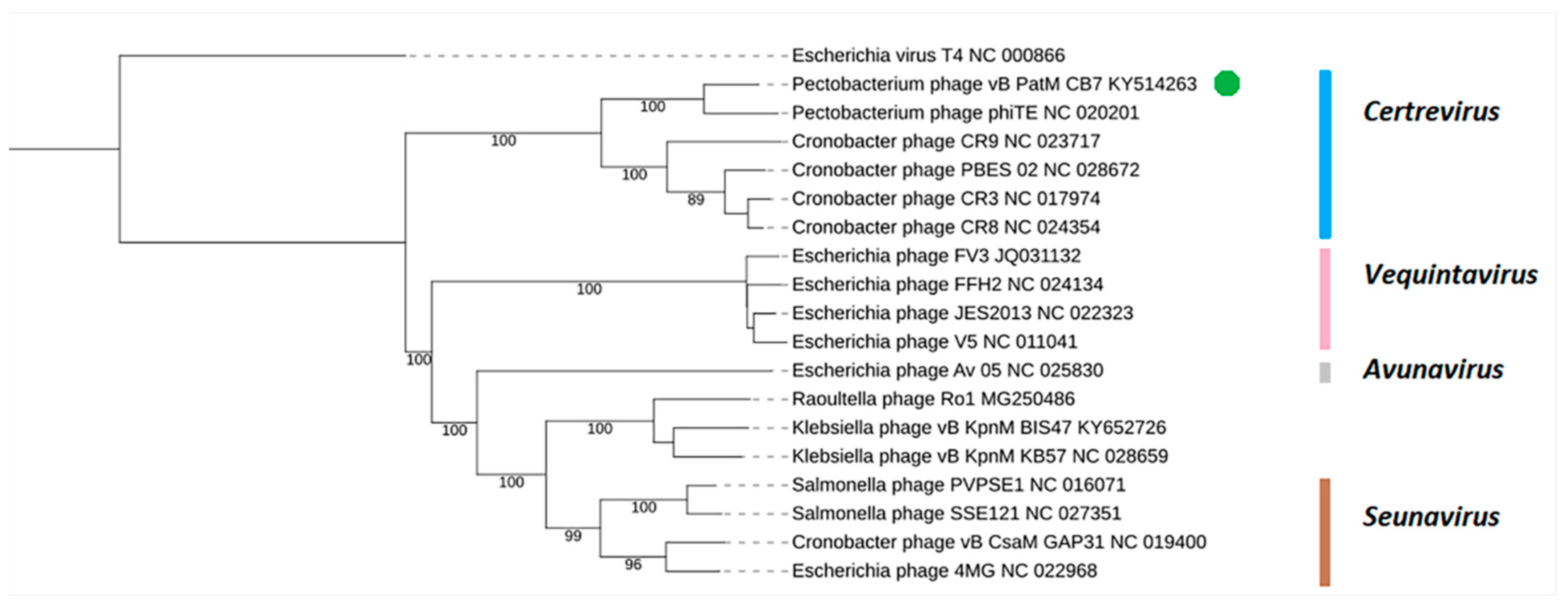
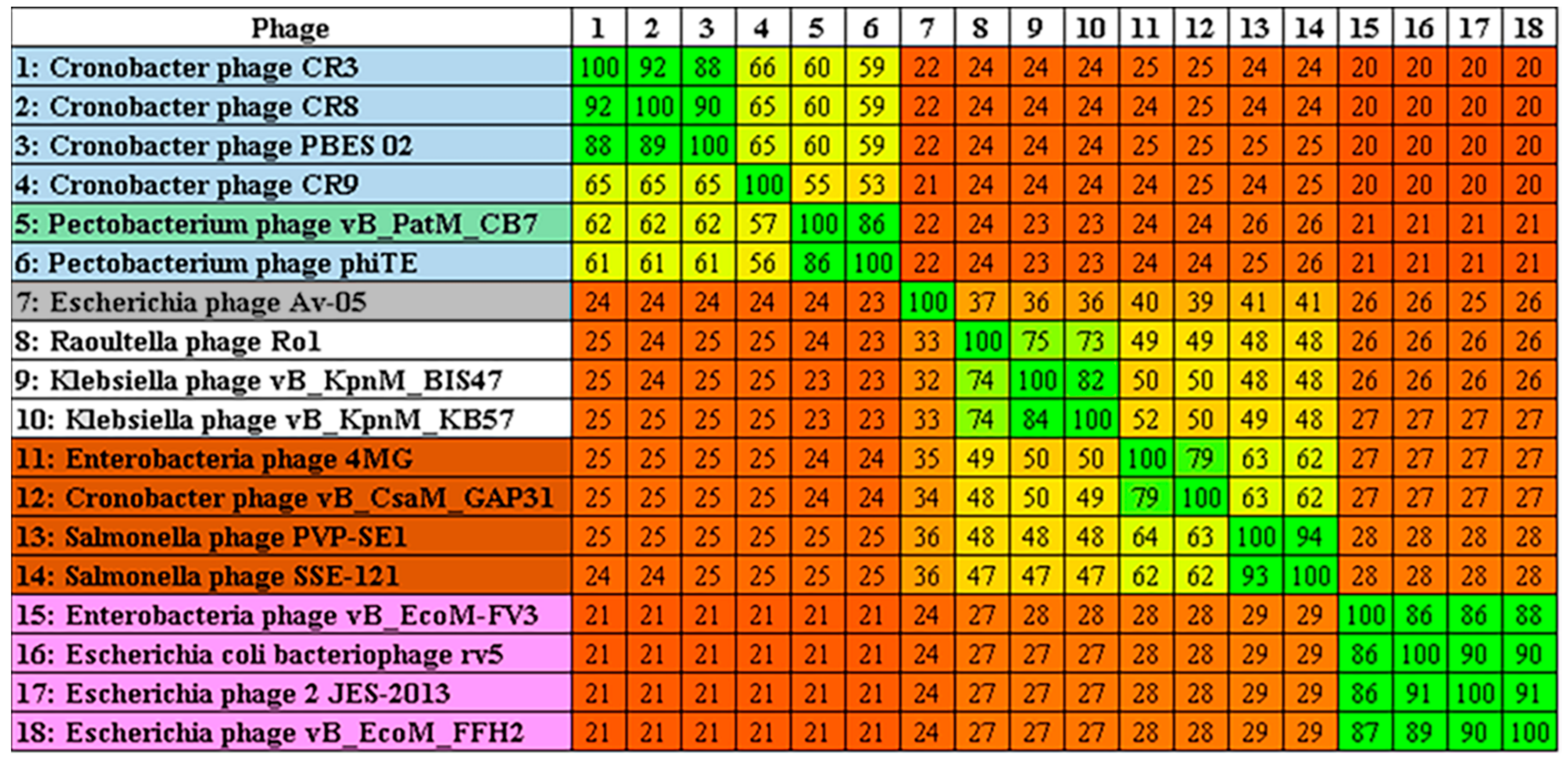
2.3. Phylogenetic Analysis—CB7 is A Member of Certrevirus and the Status of Vequintavirinae
2.4. Transcription, Promoters and Terminators
2.5. DNA Replication, Methylation and Nucleotide Metabolism
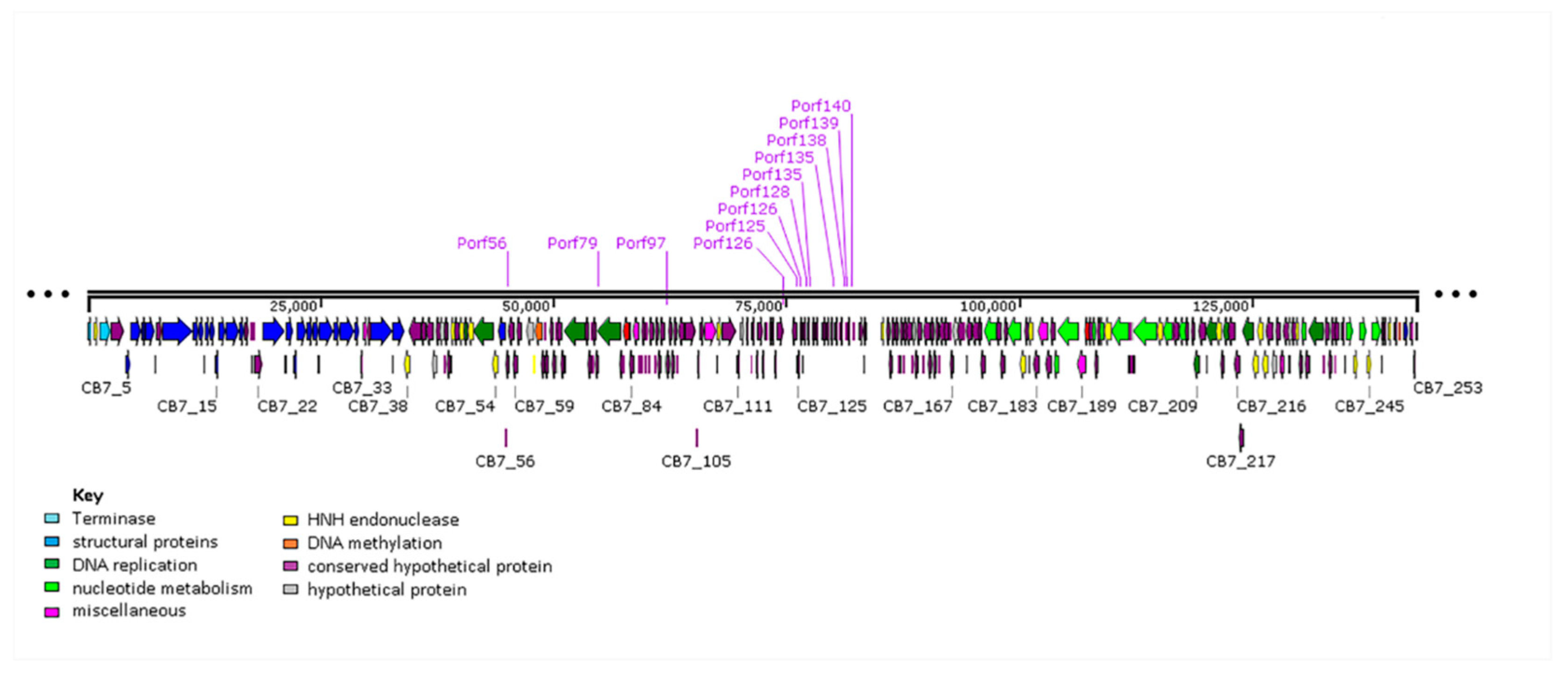
2.6. Selfish Genetic Elements within the Genome of CB7
2.7. Structural Proteome Analysis of Phage CB7 Particles
2.8. Cell Wall-Degrading Enzymes and Cell Lysis Proteins
3. Discussion
4. Materials and Methods
4.1. Phage Isolation
4.2. Host Range and General Characterization
4.3. Cesium Chloride Gradient Purification
4.4. Transmission Electron Microscopy
4.5. DNA Isolation, Restriction and Sequencing
4.6. BAL-31 Nuclease Treatment of Genomic DNA
4.7. Bioinformatic Analysis
4.8. Comparative Genomics
4.9. Comparative Genomics
4.10. Mass Spectrometric Analysis of the Phage CB7 Virion Proteome
4.11. Accession Number
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Toth, I.K.; Bell, K.S.; Holeva, M.C.; Birch, P.R.J. Soft rot Erwiniae: From genes to genomes. Mol. Plant Pathol. 2003, 4, 17–30. [Google Scholar] [CrossRef]
- Toth, I.K.; van der Wolf, J.M.; Saddler, G.; Lojkowska, E.; Hélias, V.; Pirhonen, M.; Tsror Lahkim, L.; Elphinstone, J.G. Dickeya species: An emerging problem for potato production in Europe. Plant Pathol. 2011, 60, 385–399. [Google Scholar] [CrossRef]
- Devaux, A.; Kromann, P.; Ortiz, O. Potatoes for sustainable global food security. Potato Res. 2014, 57, 185–199. [Google Scholar] [CrossRef]
- Charkowski, A.O. The changing face of bacterial soft-rot diseases. Annu. Rev. Phytopathol. 2018, 56, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yuan, L.; Shi, Y.; Xie, X.; Chai, A.; Wang, Q.; Li, B. Comparative genomic analysis of Pectobacterium carotovorum subsp. brasiliense SX309 provides novel insights into its genetic and phenotypic features. BMC Genomics 2019, 20, 486. [Google Scholar] [CrossRef] [Green Version]
- Pérombelon, M.C.M. Potato diseases caused by soft rot Erwinias: An overview of pathogenesis. Plant Pathol. 2002, 51, 1–12. [Google Scholar] [CrossRef]
- Czajkowski, R.; Pérombelon, M.C.M.; van Veen, J.A.; van der Wolf, J.M. Control of blackleg and tuber soft rot of potato caused by Pectobacterium and Dickeya species: A review. Plant Pathol. 2011, 60, 999–1013. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Van Vaerenbergh, J.; Vandenheuvel, D.; Dunon, V.; Ceyssens, P.-J.; De Proft, M.; Kropinski, A.M.; Noben, J.-P.; Maes, M.; Lavigne, R. T4-Related bacteriophage LIMEstone isolates for the control of soft rot on potato caused by ‘Dickeya solani’. PLoS ONE 2012, 7, e33227. [Google Scholar] [CrossRef] [Green Version]
- Czajkowski, R.; Ozymko, Z.; de Jager, V.; Siwinska, J.; Smolarska, A.; Ossowicki, A.; Narajczyk, M.; Lojkowska, E. Genomic, proteomic and morphological characterization of two novel broad host lytic bacteriophages ΦPD10.3 and ΦPD23.1 infecting pectinolytic Pectobacterium spp. and Dickeya spp. PLoS ONE 2015, 10, e0119812. [Google Scholar] [CrossRef] [Green Version]
- Smolarska, A.; Rabalski, L.; Narajczyk, M.; Czajkowski, R. Isolation and phenotypic and morphological characterization of the first Podoviridae lytic bacteriophages ϕA38 and ϕA41 infecting Pectobacterium parmentieri (former Pectobacterium wasabiae). Eur. J. Plant Pathol. 2018, 150, 413–425. [Google Scholar] [CrossRef]
- Buttimer, C.; Hendrix, H.; Lucid, A.; Neve, H.; Noben, J.-P.; Franz, C.; O’Mahony, J.; Lavigne, R.; Coffey, A.; O’Mahony, J.; et al. Novel N4-like bacteriophages of Pectobacterium atrosepticum. Pharmaceuticals 2018, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaczek-Moczydłowska, M.A.; Young, G.K.; Trudgett, J.; Plahe, C.; Fleming, C.C.; Campbell, K.; O’ Hanlon, R. Phage cocktail containing Podoviridae and Myoviridae bacteriophages inhibits the growth of Pectobacterium spp. under in vitro and in vivo conditions. PLoS ONE 2020, 15, e0230842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttimer, C.; Lucid, A.; Neve, H.; Franz, C.; O’Mahony, J.; Turner, D.; Lavigne, R.; Coffey, A. Pectobacterium atrosepticum phage vB_PatP_CB5: A member of the proposed genus ‘Phimunavirus’. Viruses 2018, 10, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blower, T.R.; Evans, T.J.; Przybilski, R.; Fineran, P.C.; Salmond, G.P.C. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 2012, 8, e1003023. [Google Scholar] [CrossRef]
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: Update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef]
- Shin, H.; Lee, J.-H.; Kim, Y.; Ryu, S. Complete genome sequence of Cronobacter sakazakii bacteriophage CR3. J. Virol. 2012, 86, 6367–6368. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Kim, W.I.; Kwon, Y.C.; Cha, K.E.; Kim, M.; Myung, H. A newly isolated bacteriophage, PBES 02, infecting Cronobacter sakazakii. J. Microbiol. Biotechnol. 2016, 26, 1629–1635. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, H.W. Frequency of morphological phage descriptions in the year 2000. Brief review. Arch. Virol. 2001, 146, 843–857. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. In Bacteriophages: Methods and Protocols, Volume 2 Molecular and Applied Aspects; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 502, pp. 91–111. ISBN 9781603275651. [Google Scholar]
- Bell, K.S.; Sebaihia, M.; Pritchard, L.; Holden, M.T.G.; Hyman, L.J.; Holeva, M.C.; Thomson, N.R.; Bentley, S.D.; Churcher, L.J.C.; Mungall, K.; et al. Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp. atroseptica and characterization of virulence factors. Proc. Natl. Acad. Sci. USA 2004, 101, 11105–11110. [Google Scholar] [CrossRef] [Green Version]
- Nikolaichik, Y.; Gorshkov, V.; Gogolev, Y.; Valentovich, L.; Evtushenkov, A. Genome sequence of Pectobacterium atrosepticum strain 21A. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, A.; Xia, X. GC skew in protein-coding genes between the leading and lagging strands in bacterial genomes: New substitution models incorporating strand bias. J. Theor. Biol. 2008, 253, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Hinton, D.M. Transcriptional control in the prereplicative phase of T4 development. Virol. J. 2010, 7, 289. [Google Scholar] [CrossRef] [Green Version]
- Shcherbakov, V.P.; Plugina, L.; Shcherbakova, T. Endonuclease VII is a key component of the mismatch repair mechanism in bacteriophage T4. DNA Repair (Amst). 2011, 10, 356–362. [Google Scholar] [CrossRef]
- Lundin, D.; Torrents, E.; Poole, A.M.; Sjöberg, B.-M. RNRdb, a curated database of the universal enzyme family ribonucleotide reductase, reveals a high level of misannotation in sequences deposited to Genbank. BMC Genomics 2009, 10, 589. [Google Scholar] [CrossRef] [Green Version]
- Feeney, M.A.; Ke, N.; Beckwith, J. Mutations at several loci cause increased expression of ribonucleotide reductase in Escherichia coli. J. Bacteriol. 2012, 194, 1515–1522. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, B.S.; Stoddard, B.L. Homing endonucleases: Structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001, 29, 3757–3774. [Google Scholar] [CrossRef]
- Edgell, D.R.; Belfort, M.; Shub, D.A. Barriers to intron promiscuity in bacteria. J. Bacteriol. 2000, 182. [Google Scholar] [CrossRef] [Green Version]
- Pope, W.H.; Jacobs-Sera, D.; Best, A.A.; Broussard, G.W.; Connerly, P.L.; Dedrick, R.M.; Kremer, T.A.; Offner, S.; Ogiefo, A.H.; Pizzorno, M.C.; et al. Cluster J Mycobacteriophages: Intron splicing in capsid and tail genes. PLoS ONE 2013, 8, e69273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kala, S.; Cumby, N.; Sadowski, P.D.; Hyder, B.Z.; Kanelis, V.; Davidson, A.R.; Maxwell, K.L. HNH proteins are a widespread component of phage DNA packaging machines. Proc. Natl. Acad. Sci. USA 2014, 111, 6022–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgell, D.R.; Gibb, E.A.; Belfort, M. Mobile DNA elements in T4 and related phages. Virol. J. 2010, 7, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, N.; McKillop, T.J.; Jowitt, T.A.; Howard, M.; Davies, H.; Holmes, D.F.; Roberts, I.S.; Bella, J. Collagen-like proteins in pathogenic E. coli strains. PLoS ONE 2012, 7, e37872. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-like domains on bacteriophages: A tale of promiscuity and deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef]
- Santos, S.B.; Kropinski, A.M.; Ceyssens, P.-J.; Ackermann, H.-W.; Villegas, A.; Lavigne, R.; Krylov, V.N.; Carvalho, C.M.; Ferreira, E.C.; Azeredo, J. Genomic and proteomic characterization of the broad-host-range Salmonella phage PVP-SE1: Creation of a new phage genus. J. Virol. 2011, 85, 11265–11273. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.M.; Waddell, T.; Meng, J.; Franklin, K.; Ackermann, H.W.; Ahmed, R.; Mazzocco, A.; Yates, J.; Lingohr, E.J.; Johnson, R.P. The host-range, genomics and proteomics of Escherichia coli O157:H7 bacteriophage rV5. Virol. J. 2013, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Piya, D.; Lessor, L.; Koehler, B.; Stonecipher, A.; Cahill, J.; Gill, J.J. Genome-wide screens reveal Escherichia coli genes required for growth of T1-like phage LL5 and rV5-like phage LL12. Sci Rep 2018, 10. [Google Scholar]
- Oliveira, H.; São-José, C.; Azeredo, J. Phage-derived peptidoglycan degrading enzymes: Challenges and future prospects for in vivo therapy. Viruses 2018, 10, 292. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk, B.; Bienkowska-Szewczyk, K.; Kozloff, L.M. Identification of T4 gene 25 product, a component of the tail baseplate, as a 15K lysozyme. Mol. Gen. Genet. 1986, 202, 363–367. [Google Scholar] [CrossRef]
- Summer, E.J.; Berry, J.; Tran, T.A.T.; Niu, L.; Struck, D.K.; Young, R. Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. J. Mol. Biol. 2007, 373, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Barenboim, M.; Chang, C.Y.; dib Hajj, F.; Young, R. Characterization of the dual start motif of a class II holin gene. Mol. Microbiol. 1999, 32, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Paddison, P.; Abedon, S.T.; Dressman, H.K.; Gailbreath, K.; Tracy, J.; Mosser, E.; Neitzel, J.; Guttman, B.; Kutter, E. The roles of the bacteriophage T4 r genes in lysis inhibition and fine-structure genetics: A new perspective. Genetics 1998, 148, 1539–1550. [Google Scholar] [PubMed]
- Evans, T.J.; Trauner, A.; Komitopoulou, E.; Salmond, G.P.C. Exploitation of a new flagellatropic phage of Erwinia for positive selection of bacterial mutants attenuated in plant virulence: Towards phage therapy. J. Appl. Microbiol. 2010, 108, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.J.; Ind, A.; Komitopoulou, E.; Salmond, G.P.C. Phage-selected lipopolysaccharide mutants of Pectobacterium atrosepticum exhibit different impacts on virulence. J. Appl. Microbiol. 2010, 109, 505–514. [Google Scholar] [PubMed]
- Thierauf, A.; Perez, G.; Maloy, S. Generalized transduction. In Bacteriophages. Methods in Molecular Biology; Clokie, M., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 267–286. ISBN 9781603275644. [Google Scholar]
- Buttimer, C.; McAuliffe, O.; Ross, R.P.P.P.; Hill, C.; O’Mahony, J.; Coffey, A.; O’Mahony, J.; Coffey, A.; O’Mahony, J.; Coffey, A. Bacteriophages and bacterial plant diseases. Front. Microbiol. 2017, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruttin, A.; Brüssow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [Green Version]
- Denou, E.; Bruttin, A.; Barretto, C.; Ngom-Bru, C.; Brüssow, H.; Zuber, S. T4 phages against Escherichia coli diarrhea: Potential and problems. Virology 2009, 388, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Vandersteegen, K.; Kropinski, A.M.; Nash, J.H.E.; Noben, J.-P.J.-P.; Hermans, K.; Lavigne, R. Romulus and Remus, two phage isolates representing a distinct clade within the Twortlikevirus genus, display suitable properties for phage therapy applications. J. Virol. 2013, 87, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Picking bacteriophage λ plaques. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 1, p. 2.32. ISBN 0879695773. [Google Scholar]
- Sambrook, J.; Russell, D.W. Plating bacteriophage λ. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 1, p. 2.25. [Google Scholar]
- Park, M.; Lee, J.-H.; Shin, H.; Kim, M.; Choi, J.; Kang, D.-H.; Heu, S.; Ryu, S. Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl. Environ. Microbiol. 2012, 78, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liang, L.; Lin, S.; Jia, S. Isolation and characterization of a virulent bacteriophage AB1 of Acinetobacter baumannii. BMC Microbiol. 2010, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, D.W. Purification of bacteriophage lamda particles by isopycnic centrifugation through CsCl gradients. In Molecular Cloning: A Laboratory Manual, Volume 1; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 1, p. 2.47. ISBN 0879695773. [Google Scholar]
- Pickard, D.J.J. Preparation of bacteriophage lysates and pure DNA. In Bacteriophages. Methods in Molecular Biology: Isolation, Chatacterization, and Interactions; Clokie, M., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; ISBN 9781603271646. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.A.; Dvorkin, M.; Kulikov, A.S.S.; Lesin, V.M.M.; Nikolenko, S.I.I.; Pham, S.; Prjibelski, A.D.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klumpp, J.; Dorscht, J.; Lurz, R.; Bielmann, R.; Wieland, M.; Zimmer, M.; Calendar, R.; Loessner, M.J. The terminally redundant, nonpermuted genome of Listeria bacteriophage A511: A model for the SPO1-like myoviruses of Gram-positive bacteria. J. Bacteriol. 2008, 190, 5753–5765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2014, 43, D213–D221. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- Juncker, A.S.; Willenbrock, H.; von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Naville, M.; Ghuillot-Gaudeffroy, A.; Marchais, A.; Gautheret, D. ARNold: A web tool for the prediction of Rho-independent transcription terminators. RNA Biol. 2011, 8, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Reynolds, D.; Seto, D.; Mahadevan, P. CoreGenes3.5: A webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res. Notes 2013, 6, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.-A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis comparison tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-Based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47. [Google Scholar] [CrossRef] [Green Version]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef]
- Landthaler, M.; Begley, U.; Lau, N.C.; Shub, D.A. Two self-splicing group I introns in the ribonucleotide reductase large subunit gene of Staphylococcus aureus phage Twort. Nucleic Acids Res. 2002, 30, 1935–1943. [Google Scholar] [CrossRef]
- Ceyssens, P.-J.; Hertveldt, K.; Ackermann, H.-W.; Noben, J.-P.; Demeke, M.; Volckaert, G.; Lavigne, R. The intron-containing genome of the lytic Pseudomonas phage LUZ24 resembles the temperate phage PaP3. Virology 2008, 377, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, A.; Ceyssens, P.-J.; De Smet, J.; Hendrix, H.; Bellon, H.; Leimer, N.; Wagemans, J.; Delattre, A.-S.; Cenens, W.; Aertsen, A.; et al. Systematic identification of hypothetical bacteriophage proteins targeting key protein complexes of Pseudomonas aeruginosa. J. Proteome Res. 2014, 13, 4446–4456. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Sensitivity |
|---|---|---|
| Pectobacterium atrosepticum | DSMZ 18077 (type strain) | - |
| DSMZ 30184 | - | |
| DSMZ 30185 | + | |
| DSMZ 30186 | + * | |
| CB BL1-1 | - | |
| CB BL2-1 | + | |
| CB BL3-1 | + | |
| CB BL4-1 | + | |
| CB BL5-1 | - | |
| CB BL7-1 | - | |
| CB BL9-1 | - | |
| CB BL11-1 | - | |
| CB BL12-2 | - | |
| CB BL13-1 | - | |
| CB BL14-1 | - | |
| CB BL15-1 | - | |
| CB BL16-1 | - | |
| CB BL18-1 | - | |
| CB BL19-1 | - | |
| Pectobacterium carotovorum subsp. carotovorum | DSMZ 30168 (type strain) | - |
| DSMZ 30169 | - | |
| DSMZ 30170 | - | |
| CB BL19-1-37 | - | |
| Dickeya chrysanthemi bv. chrysanthemi | LMG 2804 | - |
| Dickeya dianthicola | PD 482 | - |
| PD 2174 | - | |
| GBBC 1538 | - | |
| Dickeya solani | sp. PRI 2222 (D36) | - |
| LMG 25865 (D10) | - | |
| GBBC 1502 | - | |
| GBBC 1586 | - | |
| Erwinia amylovora | LMG 2024 | - |
| GBBC 403 | - | |
| Erwinia mallotivora | LMG 1271 | - |
| Phage | Accession No. | Genome Size (bp) | G+C Content (%) | ORFs | tRNA | Identity (%) * | Shared Proteins (%) ** |
|---|---|---|---|---|---|---|---|
| Cronobacter phage CR3 | JQ691612 | 149,273 | 50.9 | 265 | 18 | 100 | 100 |
| Cronobacter phage CR8 | KC954774 | 149,162 | 50.8 | 269 | 17 | 94 | 80 |
| Cronobacter phage CR9 | JQ691611 | 151,924 | 50.6 | 281 | 17 | 70 | 89 |
| Cronobacter phage PBES 02 | KT353109 | 149,732 | 50.7 | 270 | 14 | 64 | 90 |
| Pectobacterium phage DU_PP_I | MF979560 | 144,959 | 50.1 | 267 | 8 | 73 | 80 |
| Pectobacterium phage DU_PP_IV | MF979563 | 145,233 | 50.3 | 268 | 8 | 73 | 80 |
| Pectobacterium phage vB_PatM_CB7 | KY514263 | 142,778 | 50.1 | 253 | 1 | 64 | 70 |
| Pectobacterium phage ΦTE | JQ015307 | 142,349 | 50.1 | 242 | 2 | 62 | 63 |
| ORF/Associated ORF | Selfish Genetic Element | Homing Endonuclease Family | Gene Product Function of Targeted Gene | Does Splicing Occur (mRNA Level)? | Shared with ΦTE | Shared with Cr3 |
|---|---|---|---|---|---|---|
| CB7_2 | intron associated with homing endonuclease | HNH | Large terminase (CB7_1,3) | Yes | Shared | No |
| CB7_38 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_48 | free-standing homing endonuclease | HNH | _ | _ | No | No |
| CB7_50 | intron associated with homing endonuclease | HNH | DNA polymerase (CB7_49, 51) | Yes | Shared | No |
| CB7_52 | intron associated with homing endonuclease | HNH | DNA polymerase (CB7_51, 53) | Yes | Shared | No |
| CB7_54 | free-standing homing endonuclease | HNH | _ | No | shared | No |
| CB7_62 | free-standing homing endonuclease | HNH | _ | _ | No | No |
| CB7_80 | intein associated with homing endonuclease | LAGLIDADG | Putative helicase (CB7_80) | _ | Shared | _ |
| CB7_109 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_143 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_180 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_182 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_196 | intron associated with mobile genetic element | HNH | Ribonucleotide reductase NrdB, part 1 and 2 (CB7_195, 197) | Yes | No | No |
| CB7_197 | intein with no homing endonuclease | _ | Ribonucleotide reductase NrdB, part 2 (CB_197) | _ | Shared | _ |
| CB7_201 | intein associated with homing endonuclease | LAGLIDADG | Ribonucleotide reductase NrdA, part 1 (CB7_201) | _ | Shared | _ |
| CB7_202 | intron associated with homing endonuclease | HNH | Ribonucleotide reductase NrdA (CB7_201, 203) | Yes | Shared | No |
| CB7_212 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_219 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_220 | free-standing homing endonuclease | HNH | _ | _ | Shared | Shared |
| CB7_221 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_230 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_243 | free-standing homing endonuclease | HNH | _ | _ | Shared | No |
| CB7_245 | intron associated with homing endonuclease | HNH | Nicotinamide phosphoribosyl Transferase (CB7_245) | No | Shared | No |
| CB7_249 | free-standing homing endonuclease | HNH | _ | _ | No | No |
| ORF | Predicted Function | pVOG | Protein Molecular Weight (kDa) | No. of Unique Peptides | Sequence Coverage % |
|---|---|---|---|---|---|
| CB7_4 | putative portal protein | VOG1356 | 55.37 | 35 | 75 |
| CB7_5 | putative prohead core protein protease | VOG4722 | 21.35 | 2 | 16 |
| CB7_6 | unknown structural protein | VOG1355 | 40.57 | 3 | 14 |
| CB7_7 | putative head stabilization/decoration protein | VOG5255 | 15.6 | 16 | 96 |
| CB7_8 | putative major head protein | VOG0976 | 37.6 | 31 | 93 |
| CB7_10 | putative tail fibre protein | VOG9955 | 111.95 | 39 | 46 |
| CB7_11 | unknown structural protein | VOG5488 | 24.54 | 7 | 30 |
| CB7_12 | unknown structural protein | _ | 17.19 | 8 | 47 |
| CB7_13 | unknown structural protein | VOG5152 | 20.14 | 4 | 31 |
| CB7_15 | unknown structural protein | VOG1428 | 16.58 | 5 | 44 |
| CB7_16 | unknown structural protein | VOG5266 | 26.47 | 3 | 16 |
| CB7_17 | putative tail sheath protein | VOG1352 | 50.69 | 27 | 90 |
| CB7_18 | putative tail tube protein | VOG4699 | 17.2 | 6 | 45 |
| CB7_23 | putative tape measure protein | VOG10480 | 87.72 | 30 | 41 |
| CB7_24 | unknown structural protein | VOG1956 | 31.67 | 5 | 24 |
| CB7_25 | unknown structural protein | VOG1433 | 14.38 | 1 | 15 |
| CB7_26 | putative tail protein | VOG1348 | 36.5 | 1 | 5 |
| CB7_27 | putative baseplate protein | VOG0573 | 26.42 | 9 | 63 |
| CB7_28 | putative tail lysozyme | VOG4550 | 20.45 | 1 | 8 |
| CB7_29 | putative baseplate wedge protein | VOG4691 | 54.37 | 10 | 22 |
| CB7_30 | putative baseplate protein | VOG4620 | 24.2 | 10 | 56 |
| CB7_31 | putative tail collar protein | VOG8134 | 59.21 | 19 | 46 |
| CB7_36 | putative tail fibre protein | VOG4546 | 81.89 | 19 | 34 |
| CB7_37 | unknown structural protein | _ | 43.48 | 4 | 19 |
| CB7_55 | unknown structural protein | VOG7652 | 26.53 | 4 | 24 |
| CB7_251 | unknown structural protein | _ | 19.68 | 1 | 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buttimer, C.; Lynch, C.; Hendrix, H.; Neve, H.; Noben, J.-P.; Lavigne, R.; Coffey, A. Isolation and Characterization of Pectobacterium Phage vB_PatM_CB7: New Insights into the Genus Certrevirus. Antibiotics 2020, 9, 352. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060352
Buttimer C, Lynch C, Hendrix H, Neve H, Noben J-P, Lavigne R, Coffey A. Isolation and Characterization of Pectobacterium Phage vB_PatM_CB7: New Insights into the Genus Certrevirus. Antibiotics. 2020; 9(6):352. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060352
Chicago/Turabian StyleButtimer, Colin, Caoimhe Lynch, Hanne Hendrix, Horst Neve, Jean-Paul Noben, Rob Lavigne, and Aidan Coffey. 2020. "Isolation and Characterization of Pectobacterium Phage vB_PatM_CB7: New Insights into the Genus Certrevirus" Antibiotics 9, no. 6: 352. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060352