
Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer’s Disease Murine Models

 and
and
Abstract
:1. Introduction
1.1. Oxidative Stress Generated in the Central Nervous System
1.2. Alzheimer’s Disease
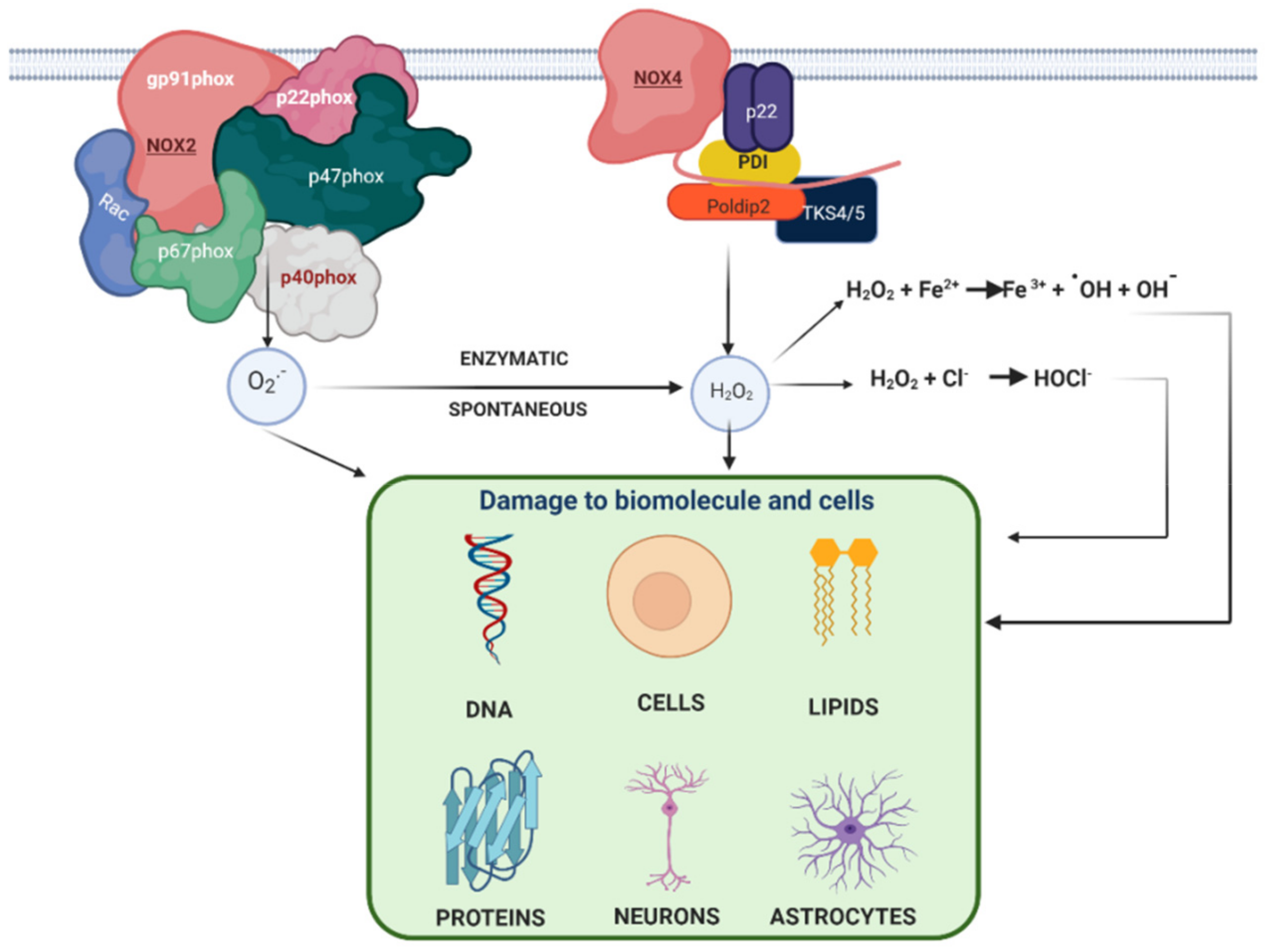
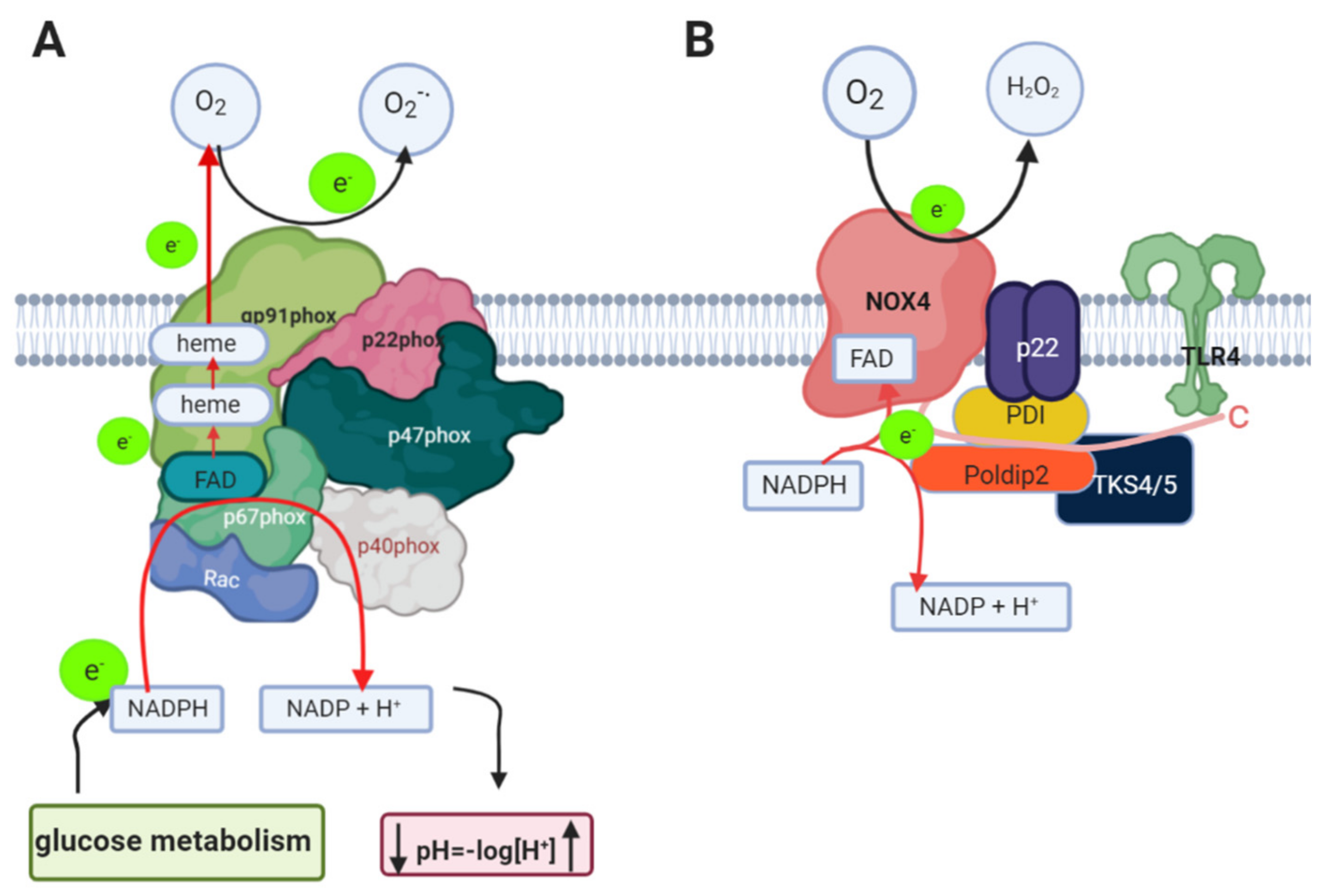
1.3. Nicotinamide Adenine Dinucleotide Phosphate Oxidase (NADPH Oxidase) in AD
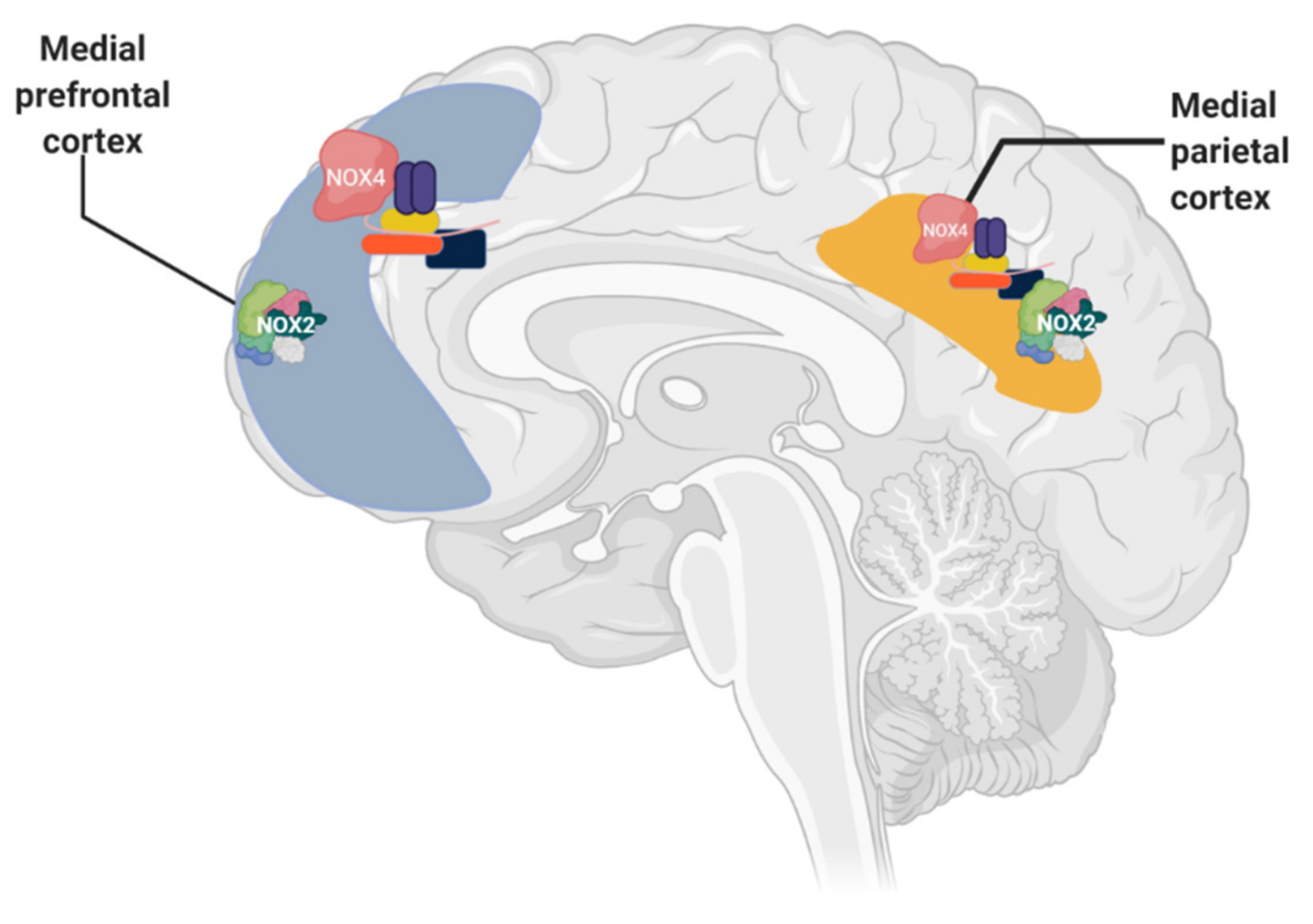
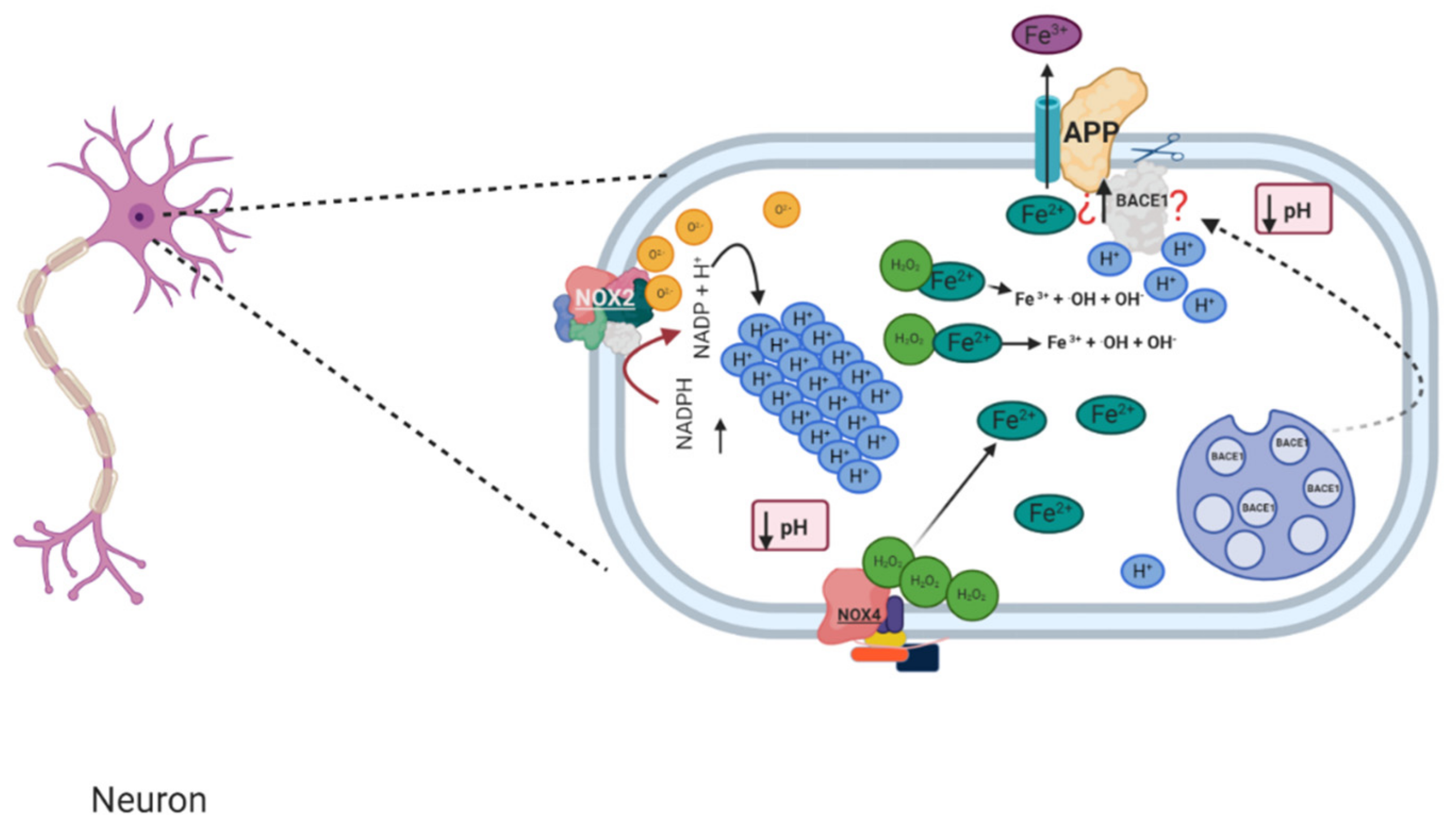
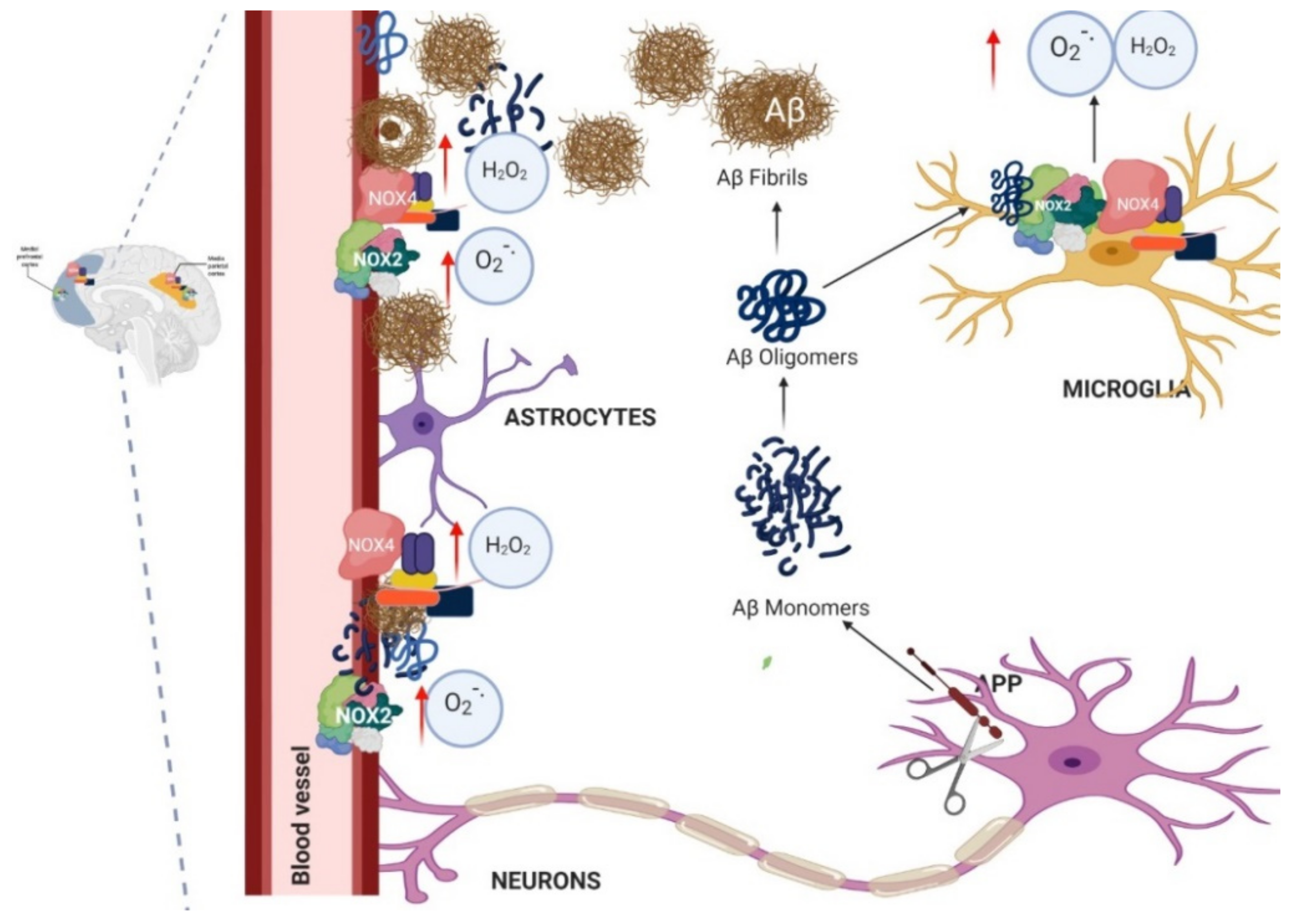
1.4. Relation between NADPH Oxidase and Alzheimer’s Disease
1.5. Murine Models of Alzheimer’s and Its Relation with NADPH Oxidase
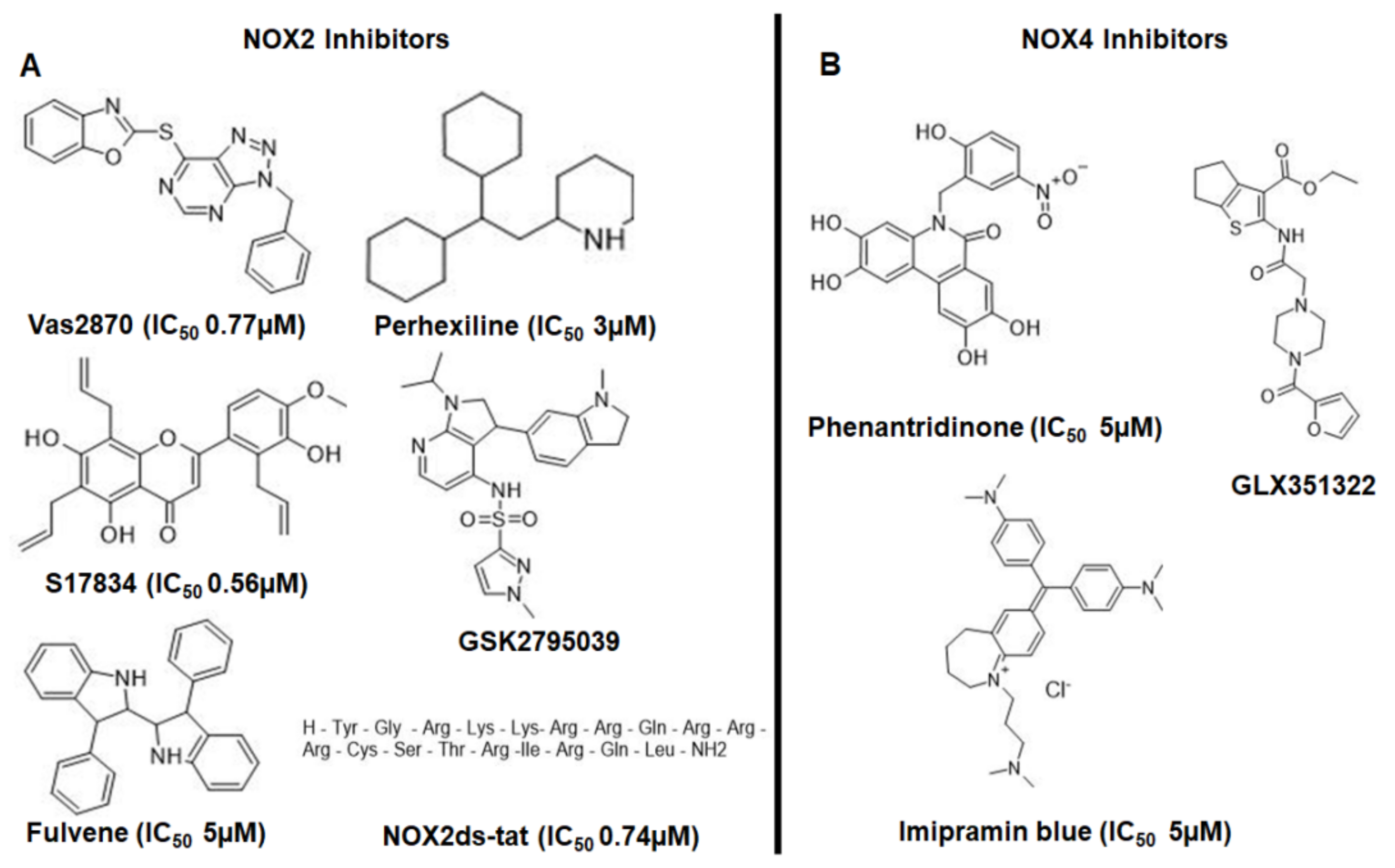
1.6. Drugs That Could Be Used as NADPH Oxidase Inhibitors in the Treatment of AD
1.6.1. VAS2870
1.6.2. Perhexiline
1.6.3. Nox2ds-tat (gp91ds-tat)
1.6.4. GSK2795039
1.6.5. GLX351322
1.6.6. M13
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative Stress in Alzheimer’s Disease: Why Did Antioxidant Therapy Fail? Oxidative Med. Cell. Longev. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Kabir, M.T. Oxidative Stress in Alzheimer’s Disease: Molecular Hallmarks of Underlying Vulnerability. In Biological, Diagnostic and Therapeutic Advances in Alzheimer’s Disease: Non-Pharmacological Therapies for Alzheimer’s Disease; Ashraf, G.M., Alexiou, A., Eds.; Springer: Singapore, 2019; pp. 91–115. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association between Apolipoprotein E Genotype and Alzheimer Disease. A Meta-Analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Rutten, B.P.F.; Steinbusch, H.W.M.; Korr, H.; Schmitz, C. Antioxidants and Alzheimer’s Disease: From Bench to Bedside (and Back Again). Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisen, P.S.; Davis, K.L. Inflammatory Mechanisms in Alzheimer’s Disease: Implications for Therapy. Am. J. Psychiatry 1994, 151, 1105–1113. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Santos, C.X.C.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R.M. Mechanisms and Implications of Reactive Oxygen Species Generation during the Unfolded Protein Response: Roles of Endoplasmic Reticulum Oxidoreductases, Mitochondrial Electron Transport, and NADPH Oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-Free Biology of Oxidative Stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.-K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.; Lipton, S.A. Aberrant Protein S-Nitrosylation Contributes to the Pathophysiology of Neurodegenerative Diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric Oxide, Superoxide, and Peroxynitrite: The Good, the Bad, and Ugly. Am. J. Physiol. 1996, 271 Pt 1, C1424–C1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.L.; Waite, T.D. Reduction of Organically Complexed Ferric Iron by Superoxide in a Simulated Natural Water. Environ. Sci. Technol. 2005, 39, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. Superoxide and Iron: Partners in Crime. IUBMB Life 1999, 48, 157–161. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide Anion Radical (O2●−), Superoxide Dismutases, and Related Matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aratani, Y. Myeloperoxidase: Its Role for Host Defense, Inflammation, and Neutrophil Function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Redox Chemistry: The Essentials. In Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [Green Version]
- Maraldi, T. Natural Compounds as Modulators of NADPH Oxidases. Oxidative Med. Cell. Longev. 2013, 2013, 271602. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Koju, N.; Taleb, A.; Zhou, J.; Lv, G.; Yang, J.; Cao, X.; Lei, H.; Ding, Q. Pharmacological Strategies to Lower Crosstalk between Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Mitochondria. Biomed. Pharmacother. 2019, 111, 1478–1498. [Google Scholar] [CrossRef]
- Cummings, J.L. Cognitive and Behavioral Heterogeneity in Alzheimer’s Disease: Seeking the Neurobiological Basis. Neurobiol. Aging 2000, 21, 845–861. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D. Alzheimer Disease. Clin. Geriatr. Med. 2018, 34, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease: Genes, Proteins, and Therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M. Alzheimer’s Disease and Other Dementias: Advances in 2013. Lancet Neurol. 2014, 13, 3–5. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic Risk Factors of Sporadic Alzheimer’s Disease: Implications in the Pathology, Pathogenesis and Treatment. Aging Dis. 2015, 6, 282–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-C.; Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An Emerging Therapeutic Target for Alzheimer’s Disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.; Borchelt, D.R.; Chakrabarty, P. Therapeutic Approaches Targeting Apolipoprotein E Function in Alzheimer’s Disease. Mol. Neurodegener. 2020, 15, 8. [Google Scholar] [CrossRef] [Green Version]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 2017, 11, 680. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Sonnen, J.A.; Santa Cruz, K.; Hemmy, L.S.; Woltjer, R.; Leverenz, J.B.; Montine, K.S.; Jack, C.R.; Kaye, J.; Lim, K.; Larson, E.B.; et al. Ecology of the Aging Human Brain. Arch Neurol. 2011, 68, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R.; Wiste, H.J.; Lesnick, T.G.; Weigand, S.D.; Knopman, D.S.; Vemuri, P.; Pankratz, V.S.; Senjem, M.L.; Gunter, J.L.; Mielke, M.M.; et al. Brain β-Amyloid Load Approaches a Plateau. Neurology 2013, 80, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Jack, C.R.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Mattsson, N.; Schöll, M.; Hansson, O.; Zetterberg, H. Amyloid Biomarkers in Alzheimer’s Disease. Trends Pharmacol. Sci. 2015, 36, 297–309. [Google Scholar] [CrossRef]
- Rüb, U.; Stratmann, K.; Heinsen, H.; Seidel, K.; Bouzrou, M.; Korf, H.-W. Alzheimer’s Disease: Characterization of the Brain Sites of the Initial Tau Cytoskeletal Pathology Will Improve the Success of Novel Immunological Anti-Tau Treatment Approaches. J. Alzheimers Dis. 2017, 57, 683–696. [Google Scholar] [CrossRef]
- Segal, A.W. The Function of the NADPH Oxidase of Phagocytes and Its Relationship to Other NOXs in Plants, Invertebrates, and Mammals. Int. J. Biochem. Cell Biol. 2008, 40, 604–618. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.A.; Drummond, G.R.; Sobey, C.G. Novel Isoforms of NADPH-Oxidase in Cerebral Vascular Control. Pharmacol. Ther. 2006, 111, 928–948. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive Oxygen Species and the Central Nervous System. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Lim, H.-W.; More, S.V.; Kim, B.-W.; Koppula, S.; Kim, I.S.; Choi, D.-K. The Role of Free Radicals in the Aging Brain and Parkinson’s Disease: Convergence and Parallelism. Int. J. Mol. Sci. 2012, 13, 10478–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of Increased Oxidative Damage in Subjects with Mild Cognitive Impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, S.; Neri, M.; Mhillaj, E.; Morgese, M.G.; Cantatore, S.; Bove, M.; Riezzo, I.; Tucci, P.; Pomara, C.; Turillazzi, E.; et al. The NADPH Oxidase NOX2 as a Novel Biomarker for Suicidality: Evidence from Human Post Mortem Brain Samples. Transl. Psychiatry 2016, 6, e813. [Google Scholar] [CrossRef] [Green Version]
- Shimohama, S.; Tanino, H.; Kawakami, N.; Okamura, N.; Kodama, H.; Yamaguchi, T.; Hayakawa, T.; Nunomura, A.; Chiba, S.; Perry, G.; et al. Activation of NADPH Oxidase in Alzheimer’s Disease Brains. Biochem. Biophys. Res. Commun. 2000, 273, 5–9. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH Oxidase in Brain Injury and Neurodegenerative Disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Tian, F.; Shao, Z.; Shen, X.; Qi, X.; Li, H.; Wang, Z.; Chen, G. Expression and Clinical Significance of Non-Phagocytic Cell Oxidase 2 and 4 after Human Traumatic Brain Injury. Neurol. Sci. 2015, 36, 61–71. [Google Scholar] [CrossRef]
- Vilhardt, F.; Plastre, O.; Sawada, M.; Suzuki, K.; Wiznerowicz, M.; Kiyokawa, E.; Trono, D.; Krause, K.-H. The HIV-1 Nef Protein and Phagocyte NADPH Oxidase Activation. J. Biol. Chem. 2002, 277, 42136–42143. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, M.C.; Malech, H.L.; Holland, S.M.; Leto, T.L. Genetic Requirement of P47phox for Superoxide Production by Murine Microglia. FASEB J. 2001, 15, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.M.; Koh, J.Y. Induction and Activation by Zinc of NADPH Oxidase in Cultured Cortical Neurons and Astrocytes. J. Neurosci. 2000, 20, RC111. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Lambeth, J.D.; Nauseef, W. The Neutrophil NADPH Oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox Proteins in Signal Transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Breitenbach, M.; Rinnerthaler, M.; Weber, M.; Breitenbach-Koller, H.; Karl, T.; Cullen, P.; Basu, S.; Haskova, D.; Hasek, J. The Defense and Signaling Role of NADPH Oxidases in Eukaryotic Cells: Review. Wien. Med. Wochenschr. 2018, 168, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Taylor, W.R.; Jones, D.T.; Segal, A.W. A Structural Model for the Nucleotide Binding Domains of the Flavocytochrome B-245 Beta-Chain. Protein Sci. 1993, 2, 1675–1685. [Google Scholar] [CrossRef]
- Lee, C.F.; Qiao, M.; Schröder, K.; Zhao, Q.; Asmis, R. Nox4 Is a Novel Inducible Source of Reactive Oxygen Species in Monocytes and Macrophages and Mediates Oxidized Low Density Lipoprotein-Induced Macrophage Death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Kang, J.-H.; Zheng, R.-L. NADPH Oxidase Produces Reactive Oxygen Species and Maintains Survival of Rat Astrocytes. Cell Biochem. Funct. 2005, 23, 93–100. [Google Scholar] [CrossRef]
- Coucha, M.; Abdelsaid, M.; Li, W.; Johnson, M.H.; Orfi, L.; El-Remessy, A.B.; Fagan, S.C.; Ergul, A. Nox4 Contributes to the Hypoxia-Mediated Regulation of Actin Cytoskeleton in Cerebrovascular Smooth Muscle. Life Sci. 2016, 163, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH Oxidases: An Overview from Structure to Innate Immunity-Associated Pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.C.; Laurindo, F.R.M. Regulation of NAD(P)H Oxidase by Associated Protein Disulfide Isomerase in Vascular Smooth Muscle Cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, B.; Shani, G.; Pass, I.; Anderson, D.; Quintavalle, M.; Courtneidge, S.A. Tks5-Dependent, Nox-Mediated Generation of Reactive Oxygen Species Is Necessary for Invadopodia Formation. Sci. Signal. 2009, 2, ra53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Löhneysen, K.; Noack, D.; Jesaitis, A.J.; Dinauer, M.C.; Knaus, U.G. Mutational Analysis Reveals Distinct Features of the Nox4-P22 Phox Complex. J. Biol. Chem. 2008, 283, 35273–35282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-Loop Is Involved in Hydrogen Peroxide Formation by the NADPH Oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [Green Version]
- De Simoni, A.; Allen, N.J.; Attwell, D. Charge Compensation for NADPH Oxidase Activity in Microglia in Rat Brain Slices Does Not Involve a Proton Current. Eur. J. Neurosci. 2008, 28, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Mermelstein, D.J.; McCammon, J.A.; Walker, R.C. PH-Dependent Conformational Dynamics of Beta-Secretase 1: A Molecular Dynamics Study. J. Mol. Recognit. 2019, 32, e2765. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashier, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-Anchored Aspartyl Protease with Alzheimer’s Disease Beta-Secretase Activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Barrera-Ocampo, A.; Lopera, F.; Barrera-Ocampo, A.; Lopera, F. Amyloid-Beta Immunotherapy: The Hope for Alzheimer Disease? Colomb. Médica 2016, 47, 203–212. [Google Scholar] [CrossRef]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-Export Ferroxidase Activity of β-Amyloid Precursor Protein Is Inhibited by Zinc in Alzheimer’s Disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, S.; Rogers, J.T. Alzheimer’s Disease Therapeutics Targeted to the Control of Amyloid Precursor Protein Translation: Maintenance of Brain Iron Homeostasis. Biochem. Pharmacol. 2014, 88, 486–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, H.; Rao, R. Amyloid Clearance Defect in ApoE4 Astrocytes Is Reversed by Epigenetic Correction of Endosomal PH. Proc. Natl. Acad. Sci. USA 2018, 115, E6640–E6649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, R.-S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH Homeostasis in Cancer: Functions, Mechanisms and Therapeutic Implications. Signal. Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Gao, H.-M.; Zhou, H.; Hong, J.-S. NADPH Oxidases: Novel Therapeutic Targets for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2012, 33, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Bruce-Keller, A.J.; Gupta, S.; Parrino, T.E.; Knight, A.G.; Ebenezer, P.J.; Weidner, A.M.; LeVine, H.; Keller, J.N.; Markesbery, W.R. NOX Activity Is Increased in Mild Cognitive Impairment. Antioxid. Redox Signal. 2010, 12, 1371–1382. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S.M.; Wands, J.R. Molecular Indices of Oxidative Stress and Mitochondrial Dysfunction Occur Early and Often Progress with Severity of Alzheimer’s Disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Krause, K.-H. NOX Enzymes in the Central Nervous System: From Signaling to Disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. NADPH-Oxidase Activation and Cognition in Alzheimer Disease Progression. Free Radic. Biol. Med. 2011, 51, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.-P.; Szanto, I. Neuronal Expression of the NADPH Oxidase NOX4, and Its Regulation in Mouse Experimental Brain Ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Infanger, D.W.; Sharma, R.V.; Davisson, R.L. NADPH Oxidases of the Brain: Distribution, Regulation, and Function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar] [CrossRef] [PubMed]
- Serrano, F.; Kolluri, N.S.; Wientjes, F.B.; Card, J.P.; Klann, E. NADPH Oxidase Immunoreactivity in the Mouse Brain. Brain Res. 2003, 988, 193–198. [Google Scholar] [CrossRef]
- Tejada-Simon, M.V.; Serrano, F.; Villasana, L.E.; Kanterewicz, B.I.; Wu, G.-Y.; Quinn, M.T.; Klann, E. Synaptic Localization of a Functional NADPH Oxidase in the Mouse Hippocampus. Mol. Cell Neurosci. 2005, 29, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, J.; Ago, T.; Nishimura, A.; Nakamura, K.; Matsuo, R.; Wakisaka, Y.; Kamouchi, M.; Kitazono, T. Nox4 Is a Major Source of Superoxide Production in Human Brain Pericytes. J. Vasc. Res. 2014, 51, 429–438. [Google Scholar] [CrossRef]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Dal Prà, I.; Rossi, F. Beta-Amyloid Activates the O-2 Forming NADPH Oxidase in Microglia, Monocytes, and Neutrophils. A Possible Inflammatory Mechanism of Neuronal Damage in Alzheimer’s Disease. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.; Choi, M.L.; Yi, J.-H.; Kim, S.-C.; Drews, A.; George-Hyslop, P.S.; Bryant, C.; Gandhi, S.; Cho, K.; Klenerman, D. Beta Amyloid Aggregates Induce Sensitised TLR4 Signalling Causing Long-Term Potentiation Deficit and Rat Neuronal Cell Death. Commun. Biol. 2020, 3, 79. [Google Scholar] [CrossRef]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting Edge: Direct Interaction of TLR4 with NAD(P)H Oxidase 4 Isozyme Is Essential for Lipopolysaccharide-Induced Production of Reactive Oxygen Species and Activation of NF-kB. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the Toll-like Receptor 4 in Neuroinflammation in Alzheimer’s Disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; García-Rodríguez, C.; Villalobos, C.; Núñez, L. Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front. Immunol. 2020, 11, 1588. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH Oxidase 4 in Lipopolysaccharide-Induced Proinflammatory Responses by Human Aortic Endothelial Cells. Cardiovasc. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef]
- Knowles, R.B.; Gomez-Isla, T.; Hyman, B.T. Abeta Associated Neuropil Changes: Correlation with Neuronal Loss and Dementia. J. Neuropathol. Exp. Neurol. 1998, 57, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.; Lippa, C.F. Beta-Amyloid, Neuronal Death and Alzheimer’s Disease. Curr. Mol. Med. 2001, 1, 733–737. [Google Scholar] [CrossRef]
- Ikegaya, Y.; Matsuura, S.; Ueno, S.; Baba, A.; Yamada, M.K.; Nishiyama, N.; Matsuki, N. Beta-Amyloid Enhances Glial Glutamate Uptake Activity and Attenuates Synaptic Efficacy. J. Biol. Chem. 2002, 277, 32180–32186. [Google Scholar] [CrossRef] [Green Version]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.-H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid Beta Peptide and NMDA Induce ROS from NADPH Oxidase and AA Release from Cytosolic Phospholipase A2 in Cortical Neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated Alpha-Synuclein Activates Microglia: A Process Leading to Disease Progression in Parkinson’s Disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Park, L.; Zhou, P.; Pitstick, R.; Capone, C.; Anrather, J.; Norris, E.H.; Younkin, L.; Younkin, S.; Carlson, G.; McEwen, B.S.; et al. Nox2-Derived Radicals Contribute to Neurovascular and Behavioral Dysfunction in Mice Overexpressing the Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1347–1352. [Google Scholar] [CrossRef] [Green Version]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C. NADPH-Oxidase-Derived Reactive Oxygen Species Mediate the Cerebrovascular Dysfunction Induced by the Amyloid Beta Peptide. J. Neurosci. 2005, 25, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Rostagno, A.; Ghiso, J.; Frangione, B. Sporadic and Familial Cerebral Amyloid Angiopathies. Brain Pathol. 2002, 12, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.-L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of Reactive Oxygen Species to Cerebral Amyloid Angiopathy, Vasomotor Dysfunction, and Microhemorrhage in Aged Tg2576 Mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagust, W. Is Amyloid-β Harmful to the Brain? Insights from Human Imaging Studies. Brain 2016, 139 Pt 1, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Hölscher, C. Development of Beta-Amyloid-Induced Neurodegeneration in Alzheimer’s Disease and Novel Neuroprotective Strategies. Rev. Neurosci. 2005, 16, 181–212. [Google Scholar] [CrossRef]
- Geerts, H.; Spiros, A.; Roberts, P. Impact of Amyloid-Beta Changes on Cognitive Outcomes in Alzheimer’s Disease: Analysis of Clinical Trials Using a Quantitative Systems Pharmacology Model. Alzheimers Res. Ther. 2018, 10, 14. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Mohammad, D.; Trépanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of Microglia in Post-Mortem Brain Samples from Patients with Alzheimer’s Disease: A Systematic Review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef]
- Shen, Z.; Bao, X.; Wang, R. Clinical PET Imaging of Microglial Activation: Implications for Microglial Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 314. [Google Scholar] [CrossRef]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 Drives M1-like Microglial/Macrophage Activation and Neurodegeneration Following Experimental Traumatic Brain Injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeloni, C.; Prata, C.; Dalla Sega, F.V.; Piperno, R.; Hrelia, S. Traumatic Brain Injury and NADPH Oxidase: A Deep Relationship. Oxidative Med. Cell. Longev. 2015, 2015, 370312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Pagano, P.J. Microvascular NADPH Oxidase in Health and Disease. Free Radic. Biol. Med. 2017, 109, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s Disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s Disease: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F. Alzheimer-Type Neuropathology in Transgenic Mice Overexpressing V717F Beta-Amyloid Precursor Protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Abeta Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Irizarry, M.C.; McNamara, M.; Fedorchak, K.; Hsiao, K.; Hyman, B.T. APPSw Transgenic Mice Develop Age-Related A Beta Deposits and Neuropil Abnormalities, but No Neuronal Loss in CA1. J. Neuropathol. Exp. Neurol. 1997, 56, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Lanz, T.A.; Carter, D.B.; Merchant, K.M. Dendritic Spine Loss in the Hippocampus of Young PDAPP and Tg2576 Mice and Its Prevention by the ApoE2 Genotype. Neurobiol. Dis. 2003, 13, 246–253. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two Amyloid Precursor Protein Transgenic Mouse Models with Alzheimer Disease-like Pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, M.E.; Wiederhold, K.H.; Abramowski, D.; Phinney, A.L.; Probst, A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Jucker, M. Neuron Loss in APP Transgenic Mice. Nature 1998, 395, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.T.; Bondolfi, L.; Herzig, M.C.; Jann, L.; Calhoun, M.E.; Wiederhold, K.H.; Tolnay, M.; Staufenbiel, M.; Jucker, M. Spontaneous Hemorrhagic Stroke in a Mouse Model of Cerebral Amyloid Angiopathy. J. Neurosci. 2001, 21, 1619–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andorfer, C.; Kress, Y.; Espinoza, M.; de Silva, R.; Tucker, K.L.; Barde, Y.-A.; Duff, K.; Davies, P. Hyperphosphorylation and Aggregation of Tau in Mice Expressing Normal Human Tau Isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Djukic, B.; Taneja, P.; Yu, G.-Q.; Lo, I.; Davis, A.; Craft, R.; Guo, W.; Wang, X.; Kim, D.; et al. Expression of A152T Human Tau Causes Age-Dependent Neuronal Dysfunction and Loss in Transgenic Mice. EMBO Rep. 2016, 17, 530–551. [Google Scholar] [CrossRef] [PubMed]
- Hernandes, M.S.; Britto, L.R.G. NADPH Oxidase and Neurodegeneration. Curr. Neuropharmacol. 2012, 10, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Martín-de-Saavedra, M.D.; Parada, E.; Romero, A.; Del Barrio, L.; Rosa, A.O.; García, A.G.; López, M.G. Galantamine Elicits Neuroprotection by Inhibiting INOS, NADPH Oxidase and ROS in Hippocampal Slices Stressed with Anoxia/Reoxygenation. Neuropharmacology 2012, 62, 1082–1090. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef]
- Dao, V.T.-V.; Elbatreek, M.H.; Altenhöfer, S.; Casas, A.I.; Pachado, M.P.; Neullens, C.T.; Knaus, U.G.; Schmidt, H.H.H.W. Isoform-Selective NADPH Oxidase Inhibitor Panel for Pharmacological Target Validation. Free Radic. Biol. Med. 2020, 148, 60–69. [Google Scholar] [CrossRef]
- Reis, J.; Massari, M.; Marchese, S.; Ceccon, M.; Aalbers, F.S.; Corana, F.; Valente, S.; Mai, A.; Magnani, F.; Mattevi, A. A Closer Look into NADPH Oxidase Inhibitors: Validation and Insight into Their Mechanism of Action. Redox Biol. 2020, 32, 101466. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [Green Version]
- Wingler, K.; Altenhoefer, S.A.; Kleikers, P.W.M.; Radermacher, K.A.; Kleinschnitz, C.; Schmidt, H.H.H.W. VAS2870 Is a Pan-NADPH Oxidase Inhibitor. Cell Mol. Life Sci. 2012, 69, 3159–3160. [Google Scholar] [CrossRef] [PubMed]
- Gatto, G.J.; Ao, Z.; Kearse, M.G.; Zhou, M.; Morales, C.R.; Daniels, E.; Bradley, B.T.; Goserud, M.T.; Goodman, K.B.; Douglas, S.A.; et al. NADPH Oxidase-Dependent and -Independent Mechanisms of Reported Inhibitors of Reactive Oxygen Generation. J. Enzym. Inhib. Med. Chem. 2013, 28, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological Characterization of the Seven Human NOX Isoforms and Their Inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Zhao, W.; Yu, X.-J.; Li, F.-X.; Liu, Z.-T.; Li, L.; Xu, S.-Y. Activation of P47phox as a Mechanism of Bupivacaine-Induced Burst Production of Reactive Oxygen Species and Neural Toxicity. Oxidative Med. Cell. Longev. 2017, 2017, 8539026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, J.A.; Beck-Oldach, K.; McFadden-Lewis, K.; Murphy, G.A.; Wong, Y.W.; Zhang, Y.; Horowitz, J.D. Effect of the Anti-Anginal Agent, Perhexiline, on Neutrophil, Valvular and Vascular Superoxide Formation. Eur. J. Pharmacol. 2006, 531, 13–19. [Google Scholar] [CrossRef]
- Roberts, R.K.; Cohn, D.; Petroff, V.; Seneviratne, B. Liver disease induced by perhexiline maleate. Med. J. Aust. 1981, 2, 553–554. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Guastella, A.R.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Perhexiline Demonstrates FYN-Mediated Antitumor Activity in Glioblastoma. Mol. Cancer Ther. 2020, 19, 1415–1422. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Zhai, P.; Liu, T.; Ikeda, S.; Nagarajan, N.; Oka, S.-I.; Yokota, T.; Kinugawa, S.; Hsu, C.-P.; et al. Tyrosine Kinase FYN Negatively Regulates NOX4 in Cardiac Remodeling. J. Clin. Investig. 2016, 126, 3403–3416. [Google Scholar] [CrossRef]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel Competitive Inhibitor of NAD(P)H Oxidase Assembly Attenuates Vascular O(2)(-) and Systolic Blood Pressure in Mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Csányi, G.; Cifuentes-Pagano, E.; Al Ghouleh, I.; Ranayhossaini, D.J.; Egaña, L.; Lopes, L.R.; Jackson, H.M.; Kelley, E.E.; Pagano, P.J. Nox2 B-Loop Peptide, Nox2ds, Specifically Inhibits the NADPH Oxidase Nox2. Free Radic. Biol. Med. 2011, 51, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Askarova, S.; Yang, X.; Sheng, W.; Sun, G.Y.; Lee, J.C.-M. Role of Aβ-Receptor for Advanced Glycation Endproducts Interaction in Oxidative Stress and Cytosolic Phospholipase A2 Activation in Astrocytes and Cerebral Endothelial Cells. Neuroscience 2011, 199, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Devine, I.; Walker, S.; Pham, H.; Ondrasik, R.; Patel, H.; Chau, W.; Parker, C.W.; Bartol, K.D.; Riahi, S.; et al. Nox2ds-Tat, A Peptide Inhibitor of NADPH Oxidase, Exerts Cardioprotective Effects by Attenuating Reactive Oxygen Species During Ischemia/Reperfusion Injury. Am. J. Biomed. Sci. 2016, 208–227. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Destée, A.; Gressier, B.; Chartier-Harlin, M.-C. NADPH Oxidases in Parkinson’s Disease: A Systematic Review. Mol. Neurodegener. 2017, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, G.M.; Dourron, H.M.; Liu, J.; Carretero, O.A.; Reddy, D.J.; Andrzejewski, T.; Pagano, P.J. Novel NAD(P)H Oxidase Inhibitor Suppresses Angioplasty-Induced Superoxide and Neointimal Hyperplasia of Rat Carotid Artery. Circ. Res. 2003, 92, 637–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, K.; Chen, W.S.; Chueng, A.L.W.; Dunne, A.A.; Seredenina, T.; Filippova, A.; Ramachandran, S.; Bridges, A.; Chaudry, L.; Pettman, G.; et al. Discovery of GSK2795039, a Novel Small Molecule NADPH Oxidase 2 Inhibitor. Antioxid. Redox Signal. 2015, 23, 358–374. [Google Scholar] [CrossRef] [Green Version]
- Malkov, A.; Popova, I.; Ivanov, A.; Jang, S.-S.; Yoon, S.Y.; Osypov, A.; Huang, Y.; Zilberter, Y.; Zilberter, M. Aβ-Induced NOX2 Activation Underlies Oxidative Stress Leading to Brain Hypometabolism and Hyperactivity in Alzheimer’s Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Anvari, E.; Wikström, P.; Walum, E.; Welsh, N. The Novel NADPH Oxidase 4 Inhibitor GLX351322 Counteracts Glucose Intolerance in High-Fat Diet-Treated C57BL/6 Mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Yu, L.; Shu, S.; Liu, Y.; Zhuang, Z.; Xu, S.; Bao, X.; Gu, Y.; Cai, F.; Song, W.; et al. MiR-204-3p/Nox4 Mediates Memory Deficits in a Mouse Model of Alzheimer’s Disease. Mol. Ther. 2020. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NADPH Oxidase Isoform | Species | ||
|---|---|---|---|
| Rat | Mouse | Human | |
| NOX2 | Medulla, superior colliculus, thalamus, hippocampus, hypothalamus, substantia nigra, striatum and cortex [86,87] | Thalamus, hippocampus, cerebellum, forebrain, midbrain, hindbrain, hypothalamus, substantia nigra, amygdala, striatum, cortex [86,88,89] (Cerebral cortex, olfactory region, basal ganglia, hypothalamus, thalamus, pons and medulla, hippocampus, amygdala, midbrain, retina, corpus callosum and cerebellum) * | (Cerebral cortex, olfactory region, basal ganglia, thalamus, hypothalamus, pons and medulla, hippocampus, amygdala, midbrain and cerebellum) * |
| NOX4 | Hippocampus, cortex cerebellum, forebrain, midbrain, hindbrain, hypothalamus [86] (Cerebral cortex, olfactory region, basal ganglia, thalamus, hypothalamus, pons and medulla and pituitary gland) * | Brain cells [61,90] (Cerebral cortex, olfactory region, basal ganglia, hypothalamus, pons and medulla, hippocampus, amygdala, midbrain and cerebellum) * | |
| Species Model | Gene Expression | Disease | Phenotype Characterization | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse | Rat | APP | PSEN 1 | MAPT | Plaques Aβ42 | Tangles | Neuronal Loss | Gliosis | Synaptic Loss | Changes In LTP/LTD | Cognitive Impairment | ||
| PDAPP [118] | ✓ | ✓ | X | X | AD | ✓ | X | X | ✓ | ✓ | ✓ | ✓ | |
| Tg2576 [119,120,121] | ✓ | ✓ | X | X | AD | ✓ | X | X | ✓ | ✓ | ✓ | ✓ | |
| APP23 [122,123,124] | ✓ | ✓ | X | X | AD/CAA * | ✓ | X | ✓ | ✓ | X | X | ✓ | |
| J20 (PDGF-APPSw, Ind) | ✓ | ✓ | X | X | AD | ✓ | X | ✓ | ✓ | ✓ | ✓ | ✓ | |
| APP/PS1 | ✓ | ✓ | ✓ | X | AD | ✓ | X | ✓ | ✓ | ✓ | ✓ | ✓ | |
| APPswe/PS1dE9 | ✓ | ✓ | ✓ | X | AD | ✓ | X | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Tg-SwDI | ✓ | ✓ | X | X | AD/CAA/HCH ** | ✓ | X | Unknown | ✓ | Unknown | X | ✓ | |
| APPE693-Δ-Tg | ✓ | ✓ | X | X | AD | X | X | ✓ | ✓ | ✓ | ✓ | ✓ | |
| APP NL-G-F knock-in | ✓ | ✓ | X | X | AD | ✓ | X | X | ✓ | ✓ | Unknown | ✓ | |
| 3xTg | ✓ | ✓ | ✓ | ✓ | AD | ✓ | ✓ | Unknown | ✓ | X | ✓ | ✓ | |
| 5xFAD | ✓ | ✓ | ✓ | X | AD | ✓ | X | ✓ | ✓ | ✓ | ✓ | ✓ | |
| PS/APP | ✓ | ✓ | ✓ | X | AD | ✓ | X | ✓ | ✓ | Unknown | Unknown | ✓ | |
| TgF344-AD | ✓ | ✓ | ✓ | X | AD | ✓ | ✓ | ✓ | ✓ | X | X | ✓ | |
| McGill-R-ThyI-APP | ✓ | ✓ | X | X | AD | ✓ | X | ✓ | ✓ | ✓ | ✓ | ✓ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fragoso-Morales, L.G.; Correa-Basurto, J.; Rosales-Hernández, M.C. Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer’s Disease Murine Models. Antioxidants 2021, 10, 218. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020218
Fragoso-Morales LG, Correa-Basurto J, Rosales-Hernández MC. Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer’s Disease Murine Models. Antioxidants. 2021; 10(2):218. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020218
Chicago/Turabian StyleFragoso-Morales, Leticia Guadalupe, José Correa-Basurto, and Martha Cecilia Rosales-Hernández. 2021. "Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer’s Disease Murine Models" Antioxidants 10, no. 2: 218. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020218