
The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication

 , , and
, , and 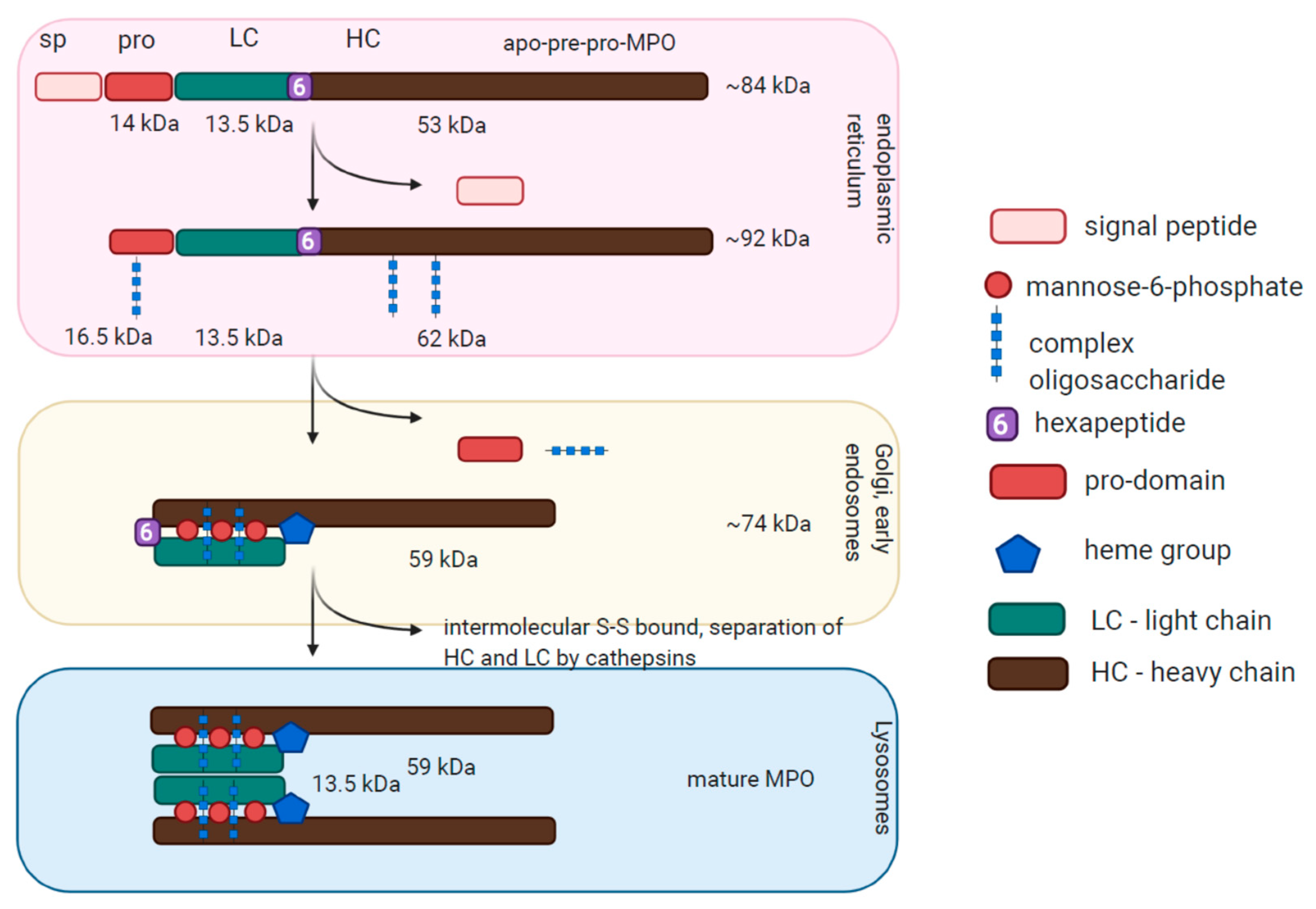{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction. MPO Conservation Across Species, Maturation in Myeloid Progenitors, and its Role in Immune Responses
2. Myeloperoxidase (MPO) Regulation of Gene Expression
3. Atypical Expression of MPO in Human Disease: Cancer and Neurodegenerative Disorders
4. Enzymatically Driven Role in Cardiovascular Disorders
5. Myeloperoxidase Is Important for NETs Formation
6. Non-Enzymatic Role of MPO
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Daiyasu, H.; Toh, H. Molecular evolution of the myeloperoxidase family. J. Mol. Evol. 2000, 51, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdbrügger, U.; Hellmark, T.; Bunch, D.O.; Alcorta, D.A.; Jennette, J.C.; Falk, R.J.; Nachman, P.H. Mapping of myeloperoxidase epitopes recognized by MPO-ANCA using human-mouse MPO chimers. Kidney Int. 2006, 69, 1799–1805. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W. Posttranslational processing of a human myeloid lysosomal protein, myeloperoxidase. Blood 1987, 70, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.; Nelson, A.; Nauseef, W.M. Proconvertase proteolytic processing of an enzymatically active myeloperoxidase precursor. Arch. Biochem. Biophys. 2012, 527, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M.; McCormick, S.; Yi, H. Roles of heme insertion and the mannose-6-phosphate receptor in processing of the human myeloid lysosomal enzyme, myeloperoxidase. Blood 1992, 80, 2622–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grishkovskaya, I.; Paumann-Page, M.; Tscheliessnig, R.; Stampler, J.; Hofbauer, S.; Soudi, M.; Sevcnikar, B.; Oostenbrink, C.; Furtmüller, P.G.; Djinović-Carugo, K.; et al. Structure of human promyeloperoxidase (proMPO) and the role of the propeptide in processing and maturation. J. Biol. Chem. 2017, 292, 8244–8261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretz, U.; Baggiolini, M. Biochemical and morphological characterization of azurophil and specific granules of human neutrophilic polymorphonuclear leukocytes. J. Cell Biol. 1974, 63, 251–269. [Google Scholar] [CrossRef] [Green Version]
- Adrover, J.M.; Aroca-Crevillén, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzón-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020. [Google Scholar] [CrossRef]
- Laura, R.P.; Dong, D.; Reynolds, W.F.; Maki, R.A. T47D cells expressing myeloperoxidase are able to process, traffic and store the mature protein in lysosomes: Studies in T47D cells reveal a role for Cys319 in MPO biosynthesis that precedes its known role in inter molecular disulfide bond formation. PLoS ONE 2016. [Google Scholar] [CrossRef] [Green Version]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebanoff, S.J. Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J. Bacteriol. 1968, 95, 2131–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase: A key regulator of neutrophil oxidant product. Redox Rep. 1997, 3, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J.; Hawkins, C.L.; Pattison, D.I.; Rees, M.D. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxidants Redox Signal. 2008, 10, 1199–1234. [Google Scholar] [CrossRef] [PubMed]
- Eiserich, J.P.; Baldus, S.; Brennan, M.L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef] [PubMed]
- Marquez, L.A.; Dunford, H.B. Kinetics of oxidation of tyrosine and dityrosine by myeloperoxidase compounds I and II: Implications for lipoprotein peroxidation studies. J. Biol. Chem. 1995, 270, 30434–30440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolphin, D.; Felton, R.H. ChemInform Abstract: The biochemical significance of porphyrin pi cation radicals. Chem. Informationsd. 1974, 5. [Google Scholar] [CrossRef]
- Burner, U.; Furtmüller, P.G.; Kettle, A.J.; Koppenol, W.H.; Obinger, C. Mechanism of reaction of myeloperoxidase with nitrite. J. Biol. Chem. 2000, 275, 20597–20601. [Google Scholar] [CrossRef] [Green Version]
- Burner, U.; Jantschko, W.; Obinger, C. Kinetics of oxidation of aliphatic and aromatic thiols by myeloperoxidase compounds I and II. FEBS Lett. 1999, 443, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Furtmüller, P.G.; Burner, U.; Jantschko, W.; Regelsberger, G.; Obinger, C. Two-electron reduction and one-electron oxidation of organic hydroperoxides by human myeloperoxidase. FEBS Lett. 2000, 484, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Van Der Veen, B.S.; De Winther, M.P.J.; Heeringa, P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxidants Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef]
- Kisic, B.; Miric, D.; Dragojevic, I.; Rasic, J.; Popovic, L. Role of Myeloperoxidase in Patients with Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [Green Version]
- Zgliczyński, J.M.; Stelmaszyńska, T.; Domański, J.; Ostrowski, W. Chloramines as intermediates of oxidation reaction of amino acids by myeloperoxidase. BBA Enzymol. 1971, 235, 419–424. [Google Scholar] [CrossRef]
- Zhang, R.; Brennan, M.L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 2002, 277, 46116–46122. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.C.; Frei, B. The nitric oxide congener nitrite inhibits myeloperoxidase/H2O2/Cl--mediated modification of low density lipoprotein. J. Biol. Chem. 2001, 276, 1822–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, N.; Nakano, K.; Aratani, Y.; Koyama, H.; Kodama, T.; Niki, E. Role of myeloperoxidase in the neutrophil-induced oxidation of low density lipoprotein as studied by myeloperoxidase-knockout mouse. J. Biochem. 2000, 127, 971–976. [Google Scholar] [CrossRef]
- Parry, M.F.; Root, R.K.; Metcalf, J.A.; Delaney, K.K.; Kaplow, L.S.; Richar, W.J. Myeloperoxidase deficiency. Prevalence and clinical significance. Ann. Intern. Med. 1981, 95, 293–301. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Cline, M.J. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: The role of myeloperoxidase in resistance to Candida infection. J. Clin. Investig. 1969, 48, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinauer, M.C. Primary immune deficiencies with defects in neutrophil function. Hematology 2016, 2016, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, R.I. Measurement of Candidacidal Activity of Specific Leukocyte Types in Mixed Cell Populations II. Normal and Chronic Granulomatous Disease Eosinophils. Infect. Immun. 1971, 3, 800–802. [Google Scholar] [CrossRef] [Green Version]
- Domingues-Ferreira, M.; Levy, A.; Barros, N.C.; Bertolini, D.L.; de Moraes Vasconcelos, D. Case report of myeloperoxidase deficiency associated with disseminated paracoccidioidomycosis and peritoneal tuberculosis. Rev. Soc. Bras. Med. Trop. 2017, 50, 568–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutter, D.; Devaquet, P.; Vanderstocken, G.; Paulus, J.M.; Marchal, V.; Gothot, A. Consequences of total and subtotal myeloperoxidase deficiency: Risk or benefit? Acta Haematol. 2000, 104, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.W.; Hughes, V.; Barlow, J.; Bucknall, R. Immunological detection of myeloperoxidase in synovial fluid from patients with rheumatoid arthritis. Biochem. J. 1988, 250, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Tseng, A.; Kim, K.; Li, J.; Cho, J. Myeloperoxidase negatively regulates neutrophil-endothelial cell interactions by impairing αMβ2 integrin function in sterile inflammation. Front. Med. 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stendahl, O.; Coble, B.I.; Dahlgren, C.; Hed, J.; Molin, L. Myeloperoxidase modulates the phagocytic activity of polymorphonuclear neutrophil leukocytes. Studies with cells from a myeloperoxidase-deficient patient. J. Clin. Investig. 1984, 73, 366–373. [Google Scholar] [CrossRef]
- Schürmann, N.; Forrer, P.; Casse, O.; Li, J.; Felmy, B.; Burgener, A.V.; Ehrenfeuchter, N.; Hardt, W.D.; Recher, M.; Hess, C.; et al. Myeloperoxidase targets oxidative host attacks to Salmonella and prevents collateral tissue damage. Nat. Microbiol. 2017, 2. [Google Scholar] [CrossRef]
- Odobasic, D.; Kitching, A.R.; Yang, Y.; O’Sullivan, K.M.; Muljadi, R.C.M.; Edgtton, K.L.; Tan, D.S.Y.; Summers, S.A.; Morand, E.F.; Holdsworth, S.R. Neutrophil myeloperoxidase regulates T-cell2driven tissue inflammation in mice by inhibiting dendritic cell function. Blood 2013, 121, 4195–4204. [Google Scholar] [CrossRef]
- Bouali, H.; Nietert, P.; Nowling, T.M.; Pandey, J.; Dooley, M.A.; Cooper, G.; Harley, J.; Kamen, D.L.; Oates, J.; Gilkeson, G. Association of the G-463A myeloperoxidase gene polymorphism with renal disease in African Americans with systemic lupus erythematosus. J. Rheumatol. 2007, 34, 2028–2034. [Google Scholar]
- Weng, K.P.; Hsieh, K.S.; Huang, S.H.; Wu, H.W.; Chien, J.H.; Lin, C.C.; Tang, C.W.; Ou, S.F.; Huang, S.J.; Ger, L.P. Myeloperoxidase genetic polymorphisms and susceptibility to Kawasaki disease in Taiwanese children. J. Microbiol. Immunol. Infect. 2016, 49, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Chami, B.; Martin, N.J.J.; Dennis, J.M.; Witting, P.K. Myeloperoxidase in the inflamed colon: A novel target for treating inflammatory bowel disease. Arch. Biochem. Biophys. 2018, 645, 61–71. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Stegeman, C.A.; Cohen Tervaert, J.W. −463 G/A myeloperoxidase promoter polymorphism is associated with clinical manifestations and the course of disease in MPO-ANCA-associated vasculitis. Clin. Immunol. 2002, 103, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Gioffredi, A.; Maritati, F.; Oliva, E.; Buzio, C. Eosinophilic granulomatosis with polyangiitis: An overview. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennette, J.C.; Xiao, H.; Falk, R.; Gasim, A.M.H. Experimental models of vasculitis and glomerulonephritis induced by antineutrophil cytoplasmic autoantibodies. Contrib. Nephrol. 2011, 169, 211–220. [Google Scholar]
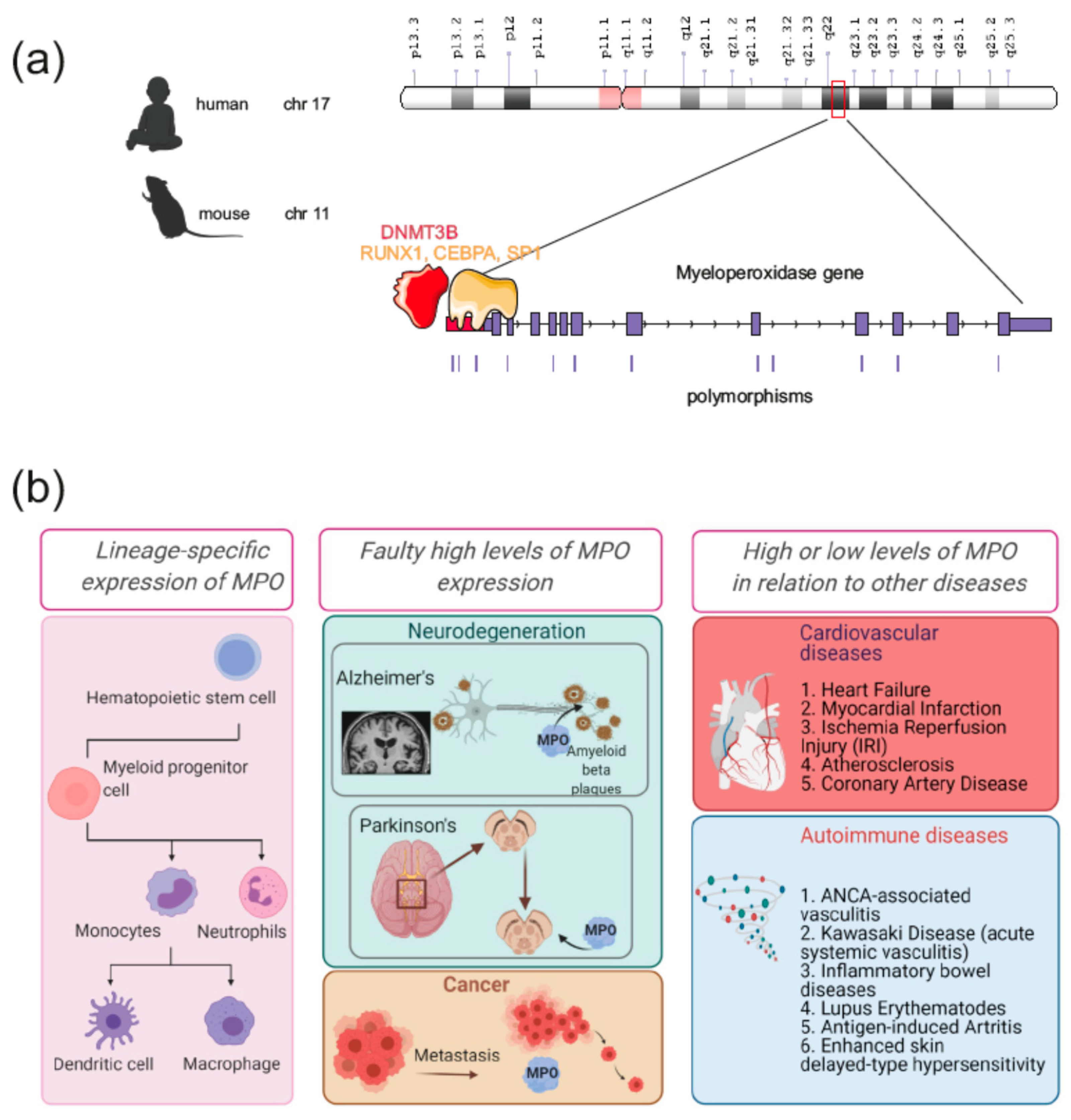
- Inazawa, J.; Inoue, K.; Nishigaki, H.; Tsuda, S.; Taniwaki, M.; Misawa, S.; Abe, T. Assignment of the human myeloperoxidase gene (MPO) to bands q213→q23 of chromosome 17. Cytogenet. Genome Res. 1989, 50, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.A.; Ford Bainton, D.; Farquhar, M.G. Differentiation of monocytes: Origin, nature, and fate of their azurophil granules. J. Cell Biol. 1971, 50, 498–515. [Google Scholar] [CrossRef]
- Bainton, D.F.; Ullyot, J.L.; Farquhar, M.G. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow: Origin and content of azurophil and specific granules. J. Exp. Med. 1971, 134, 907–934. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Cowland, J.B. Granules of the Human Neutrophilic Polymorphonuclear Leukocyte. Blood 1997, 89, 3503–3521. [Google Scholar] [CrossRef]
- Lübbert, M.; Ludwig, W.-D.; Ruszczysnski, R.; Mertelsmann, R.; Herrmann, F. Myeloperoxidase: Gene Demethylation and Expression in Acute Myeloid Leukemias. In Cytokines in Hemopoiesis, Oncology, and AIDS II; Springer: Berlin/Heidelberg, Germany, 1992; pp. 65–75. [Google Scholar]
- Tobler, A.; Miller, C.W.; Johnson, K.R.; Selsted, M.E.; Rovera, G.; Koeffler, H.P. Regulation of gene expression of myeloperoxidase during myeloid differentiation. J. Cell. Physiol. 1988, 136, 215–225. [Google Scholar] [CrossRef]
- Chumakov, A.M.; Chumakova, E.A.; Chih, D.; Koeffler, H.P. Molecular analysis of the human myeloperoxidase promoter region. Int. J. Oncol. 2000, 16, 401–411. [Google Scholar] [CrossRef]
- Lin, K.M.; Austin, G.E. Functional activity of three distinct myeloperoxidase (MPO) promoters in human myeloid cells. Leukemia 2002, 16, 1143–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Ma, O.; Speck, N.A.; Friedman, A.D. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood 2012, 119, 4408–4418. [Google Scholar] [CrossRef] [Green Version]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.M.; Bennett, C.A.; Healy, L.E.; Towatari, M.; Greaves, M.F.; Enver, T. Regulation of the myeloperoxidase enhancer binding proteins Pu1, C-EBPα, -β, and -δ during granulocyte-lineage specification. Proc. Natl. Acad. Sci. USA 1996. [Google Scholar] [CrossRef] [Green Version]
- Gombart, A.F.; Koeffler, H.P. Neutrophil specific granule deficiency and mutations in the gene encoding transcription factor C/EBPε. Curr. Opin. Hematol. 2002. [Google Scholar] [CrossRef]
- Itonaga, H.; Imanishi, D.; Wong, Y.F.; Sato, S.; Ando, K.; Sawayama, Y.; Sasaki, D.; Tsuruda, K.; Hasegawa, H.; Imaizumi, Y.; et al. Expression of myeloperoxidase in acute myeloid leukemia blasts mirrors the distinct DNA methylation pattern involving the downregulation of DNA methyltransferase DNMT3B. Leukemia 2014, 28, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Javier Piedrafita, F.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 1996, 271, 14412–14420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coit, P.; Direskeneli, H.; Sawalha, A.H. An update on the role of epigenetics in systemic vasculitis. Curr. Opin. Rheumatol. 2018, 30, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ge, H.; Poulton, C.J.; Hogan, S.L.; Hu, Y.; Jones, B.E.; Henderson, C.D.; McInnis, E.A.; Pendergraft, W.F.; Jennette, J.C.; et al. Histone modification signature at myeloperoxidase and proteinase 3 in patients with anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Clin. Epigenetics 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.G.; Lu, J.P.; Regmi, A.; Austin, G.E. Identification and functional analysis of multiple murine myeloperoxidase (MPO) promoters and comparison with the human MPO promoter region. Leukemia 1997, 11, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzow, J.; Friedman, A.D. The murine myeloperoxidase promoter contains several functional elements, one of which binds a cell type-restricted transcription factor, myeloid nuclear factor 1 (MyNF1). Mol. Cell. Biol. 1993, 13, 2141–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzhalin, A.E.; Kutikhin, A.G. Common genetic variants in the myeloperoxidase and paraoxonase genes and the related cancer risk: A review. J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev. 2012, 30, 287–322. [Google Scholar] [CrossRef]
- Cascorbi, I.; Brockmoller, J.; Roots, I. A C4887A polymorphism in exon 7 of human CYP1A1: Population frequency, mutation linkages, and impact on lung cancer susceptibility. Cancer Res. 1996, 56, 4965–4969. [Google Scholar] [PubMed]
- Castillo-Tong, D.C.; Pils, D.; Heinze, G.; Braicu, I.; Sehouli, J.; Reinthaller, A.; Schuster, E.; Wolf, A.; Watrowski, R.; Maki, R.A.; et al. Association of myeloperoxidase with ovarian cancer. Tumor Biol. 2014, 35, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008, 18, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Guruvayoorappan, C. Myeloperoxidase: The yin and yang in tumour progression. J. Exp. Ther. Oncol. 2011, 9, 93–100. [Google Scholar]
- London, S.J.; Lehman, T.A.; Taylor, J.A. Myeloperoxidase genetic polymorphism and lung cancer risk. Cancer Res. 1997, 57, 5001–5003. [Google Scholar]
- Zou, S.; Pan, X.; Hua, C.; Wu, M.; He, B.; Chen, Z. Myeloperoxidase −463 G/A polymorphism is associated with lung cancer risk: A meta-analysis with 7420 cases and 9132 controls. J. Cancer Res. Ther. 2018, 14, S282–S287. [Google Scholar]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of immune cells in the central nervous system. J. Clin. Investig. 2010, 120, 1368–1379. [Google Scholar] [CrossRef] [Green Version]
- Moxon-Emre, I.; Schlichter, L.C. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J. Neuropathol. Exp. Neurol. 2011, 70, 218–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Üllen, A.; Singewald, E.; Konya, V.; Fauler, G.; Reicher, H.; Nusshold, C.; Hammer, A.; Kratky, D.; Heinemann, A.; Holzer, P.; et al. Myeloperoxidase-Derived Oxidants Induce Blood-Brain Barrier Dysfunction In Vitro and In Vivo. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Maki, R.A.; Holzer, M.; Motamedchaboki, K.; Malle, E.; Masliah, E.; Marsche, G.; Reynolds, W.F. Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-α-synuclein-A53T mouse model, correlating with increased nitration and aggregation of α-synuclein and exacerbation of motor impairment. Free Radic. Biol. Med. 2019, 141, 115–140. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Kalogiannis, M.; Krasnikov, B.F.; Gomlin, I.; Peltier, M.R.; Moran, G.R. Linking inflammation and parkinson disease: Hypochlorous acid generates parkinsonian poisons. Toxicol. Sci. 2016, 151, 388–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.K.; Pennathur, S.; Perier, C.; Tieu, K.; Teismann, P.; Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Vonsattel, J.P.; Heinecke, J.W.; et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J. Neurosci. 2005, 25, 6594–6600. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef]
- Maki, R.A.; Tyurin, V.A.; Lyon, R.C.; Hamilton, R.L.; Dekosky, S.T.; Kagan, V.E.; Reynolds, W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 3158–3169. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Brennan, M.L.; Fu, X.; Aviles, R.J.; Pearce, G.L.; Penn, M.S.; Topol, E.J.; Sprecher, D.L.; Hazen, S.L. Association between myeloperoxidase levels and risk of coronary artery disease. J. Am. Med. Assoc. 2001, 286, 2136–2142. [Google Scholar] [CrossRef]
- Teng, N.; Maghzal, G.J.; Talib, J.; Rashid, I.; Lau, A.K.; Stocker, R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. 2017, 22, 51–73. [Google Scholar] [CrossRef] [Green Version]
- Scharnagl, H.; Kleber, M.E.; Genser, B.; Kickmaier, S.; Renner, W.; Weihrauch, G.; Grammer, T.; Rossmann, C.; Winkelmann, B.R.; Boehm, B.O.; et al. Association of myeloperoxidase with total and cardiovascular mortality in individuals undergoing coronary angiography—The LURIC study. Int. J. Cardiol. 2014, 174, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Sun, M.; Sader, S. Matrix metalloproteinases in cardiovascular disease. Can. J. Cardiol. 2006, 22, 25B–30B. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Parks, W.C.; Heinecke, J.W. Activation and silencing of matrix metalloproteinases. Semin. Cell Dev. Biol. 2008, 19, 2–13. [Google Scholar] [CrossRef]
- Rudolph, V.; Andrié, R.P.; Rudolph, T.K.; Friedrichs, K.; Klinke, A.; Hirsch-Hoffmann, B.; Schwoerer, A.P.; Lau, D.; Fu, X.; Klingel, K.; et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat. Med. 2010, 16, 470–474. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.L.; Anderson, M.M.; Shih, D.M.; Qu, X.D.; Wang, X.; Mehta, A.C.; Lim, L.L.; Shi, W.; Hazen, S.L.; Jacob, J.S.; et al. Increased atherosclerosis in myeloperoxidase-deficient mice. J. Clin. Investig. 2001, 107, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Rashid, I.; Maghzal, G.J.; Chen, Y.C.; Cheng, D.; Talib, J.; Newington, D.; Ren, M.; Vajandar, S.K.; Searle, A.; Maluenda, A.; et al. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur. Heart J. 2018. [Google Scholar] [CrossRef]
- Van Der Wal, A.C.; Becker, A.E. Atherosclerotic plaque rupture—Pathologic basis of plaque stability and instability. Cardiovasc. Res. 1999, 41, 334–344. [Google Scholar] [CrossRef]
- Von Leitner, E.C.; Klinke, A.; Atzler, D.; Slocum, J.L.; Lund, N.; Kielstein, J.T.; Maas, R.; Schmidt-Haupt, R.; Pekarova, M.; Hellwinkel, O.; et al. Pathogenic cycle between the endogenous nitric oxide synthase inhibitor asymmetrical dimethylarginine and the leukocyte-derived hemoprotein myeloperoxidase. Circulation 2011. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Talib, J.; Stanley, C.P.; Rashid, I.; Michaëlsson, E.; Lindstedt, E.L.; Croft, K.D.; Kettle, A.J.; Maghzal, G.J.; Stocker, R. Inhibition of MPO (myeloperoxidase) attenuates endothelial dysfunction in mouse models of vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1448–1457. [Google Scholar] [CrossRef]
- Rudolph, T.K.; Wipper, S.; Reiter, B.; Rudolph, V.; Coym, A.; Detter, C.; Lau, D.; Klinke, A.; Friedrichs, K.; Rau, T.; et al. Myeloperoxidase deficiency preserves vasomotor function in humans. Eur. Heart J. 2012, 33, 1625–1634. [Google Scholar] [CrossRef]
- Baldus, S.; Rudolph, V.; Roiss, M.; Ito, W.D.; Rudolph, T.K.; Eiserich, J.P.; Sydow, K.; Lau, D.; Szöcs, K.; Klinke, A.; et al. Heparins increase endothelial nitric oxide bioavailability by liberating vessel-immobilized myeloperoxidase. Circulation 2006, 113, 1871–1878. [Google Scholar] [CrossRef] [Green Version]
- Epstein, F.H.; Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond Cholesterol. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar] [CrossRef]
- Hazen, S.L.; Crowley, J.R.; Mueller, D.M.; Heinecke, J.W. Mass spectrometric quantification of 3-chlorotyrosine in human tissues with attomole sensitivity: A sensitive and specific marker for myeloperoxidase-catalyzed chlorination at sites of inflammation. Free Radic. Biol. Med. 1997, 23, 909–916. [Google Scholar] [CrossRef]
- Esterbauer, H.; Gebicki, J.; Puhl, H.; Jürgens, G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic. Biol. Med. 1992, 13, 341–390. [Google Scholar] [CrossRef]
- Gießauf, A.; Steiner, E.; Esterbauer, H. Early destruction of tryptophan residues of apolipoprotein B is a vitamin E-independent process during copper-mediated oxidation of LDL. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1995, 1256, 221–232. [Google Scholar]
- Carr, A.C.; Myzak, M.C.; Stocker, R.; McCall, M.R.; Frei, B. Myeloperoxidase binds to low-density lipoprotein: Potential implications for atherosclerosis. FEBS Lett. 2000, 487, 176–180. [Google Scholar] [CrossRef] [Green Version]
- Hazell, L.J.; Arnold, L.; Flowers, D.; Waeg, G.; Malle, E.; Stocker, R. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J. Clin. Investig. 1996. [Google Scholar] [CrossRef]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Myeloperoxidase: An oxidative pathway for generating dysfunctional high-density lipoprotein. Chem. Res. Toxicol. 2010, 23, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and function impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004. [Google Scholar] [CrossRef] [Green Version]
- Pennathur, S.; Bergt, C.; Shao, B.; Byun, J.; Kassim, S.Y.; Singh, P.; Green, P.B.; McDonald, T.O.; Brunzell, J.; Chait, A.; et al. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J. Biol. Chem. 2004, 279, 42977–42983. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Mathew, A.V.; Yu, H.; Li, L.; He, L.; Gao, W.; Liu, X.; Guo, Y.; Byun, J.; Zhang, J.; et al. Myeloperoxidase mediated HDL oxidation and HDL proteome changes do not contribute to dysfunctional HDL in Chinese subjects with coronary artery disease. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.A.; Davies, M.J. Fragmentation of extracellular matrix by hypochlorous acid. Biochem. J. 2003, 376, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Nybo, T.; Cai, H.; Chuang, C.Y.; Gamon, L.F.; Rogowska-Wrzesinska, A.; Davies, M.J. Chlorination and oxidation of human plasma fibronectin by myeloperoxidase-derived oxidants, and its consequences for smooth muscle cell function. Redox Biol. 2018, 19, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chuang, C.Y.; Vanichkitrungruang, S.; Hawkins, C.L.; Davies, M.J. Hypochlorous acid-modified extracellular matrix contributes to the behavioral switching of human coronary artery smooth muscle cells. Free Radic. Biol. Med. 2019, 134, 516–526. [Google Scholar] [CrossRef]
- Cai, H.; Chuang, C.Y.; Hawkins, C.L.; Davies, M.J. Binding of myeloperoxidase to the extracellular matrix of smooth muscle cells and subsequent matrix modification. Sci. Rep. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolarova, H.; Klinke, A.; Kremserova, S.; Adam, M.; Pekarova, M.; Baldus, S.; Eiserich, J.P.; Kubala, L. Myeloperoxidase induces the priming of platelets. Free Radic. Biol. Med. 2013, 61, 357–369. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Askari, A.T.; Brennan, M.L.; Zhou, X.; Drinko, J.; Morehead, A.; Thomas, J.D.; Topol, E.J.; Hazen, S.L.; Penn, M.S. Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J. Exp. Med. 2003, 197, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Vasilyev, N.; Williams, T.; Brennan, M.L.; Unzek, S.; Zhou, X.; Heinecke, J.W.; Spitz, D.R.; Topol, E.J.; Hazen, S.L.; Pen, M.S. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 2005, 112, 2812–2820. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, M.; Friedrichs, K.; Lange, M.; Gesenberg, J.; Remane, L.; Kerkenpaß, C.; Krause, J.; Schneider, J.; Ravekes, T.; Maass, M.; et al. Myeloperoxidase Mediates Postischemic Arrhythmogenic Ventricular Remodeling. Circ. Res. 2017, 121. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Pulli, B.; Courties, G.; Tricot, B.; Sebas, M.; Iwamoto, Y.; Hilgendorf, I.; Schob, S.; Dong, A.; Zheng, W.; et al. Myeloperoxidase Inhibition Improves Ventricular Function and Remodeling After Experimental Myocardial Infarction. JACC Basic to Transl. Sci. 2016, 1, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Reichlin, T.; Socrates, T.; Egli, P.; Potocki, M.; Breidthardt, T.; Arenja, N.; Meissner, J.; Noveanu, M.; Reiter, M.; Twerenbold, R.; et al. Use of myeloperoxidase for risk stratification in acute heart failure. Clin. Chem. 2010, 56, 944–951. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, V.; Rudolph, T.K.; Hennings, J.C.; Blankenberg, S.; Schnabel, R.; Steven, D.; Haddad, M.; Knittel, K.; Wende, S.; Wenzel, J.; et al. Activation of polymorphonuclear neutrophils in patients with impaired left ventricular function. Free Radic. Biol. Med. 2007, 43, 1189–1196. [Google Scholar] [CrossRef]
- Deuschl, F.G.; Klinke, A.; Friedrichs, K.; Knappe, D.; Weinberger, F.; Müllerleile, K.; Westermann, D.; Reichenspurner, H.; Blankenberg, S.; Baldus, S. Myeloperoxidase is Critically Linked to the Development of Diastolic Heart Failure Following Pressure Overload. J. Hear. Lung Transplant. 2014, 33, S164. [Google Scholar] [CrossRef]
- Nussbaum, C.; Klinke, A.; Adam, M.; Baldus, S.; Sperandio, M. Myeloperoxidase: A leukocyte-derived protagonist of inflammation and cardiovascular disease. Antioxidants Redox Signal. 2013, 18, 692–713. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef] [Green Version]
- Forghani, R.; Kim, H.J.; Wojtkiewicz, G.R.; Bure, L.; Wu, Y.; Hayase, M.; Wei, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J. Perinatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Klinke, A.; Berghausen, E.; Friedrichs, K.; Molz, S.; Lau, D.; Remane, L.; Berlin, M.; Kaltwasser, C.; Adam, M.; Mehrkens, D.; et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight 2018, 3, e97530. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H.U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Remijsen, Q.; Berghe, T.V.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc. Biol. 2012, 91, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Laube, B.; Abed, U.A.; Goosmann, C.; Zychlinsky, A. Neutrophil extracellular traps: How to generate and visualize them. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães-Costa, A.B.; Nascimento, M.T.C.; Wardini, A.B.; Pinto-Da-Silva, L.H.; Saraiva, E.M. ETosis: A microbicidal mechanism beyond cell death. J. Parasitol. Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Winterbourn, C.C. Reactive oxidants and myeloperoxidase and their involvement in neutrophil extracellular traps. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone acetylation promotes neutrophil extracellular trap formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchanda, K.; Kolarova, H.; Kerkenpaß, C.; Mollenhauer, M.; Vitecek, J.; Rudolph, V.; Kubala, L.; Baldus, S.; Adam, M.; Klinke, A. MPO (Myeloperoxidase) Reduces Endothelial Glycocalyx Thickness Dependent on Its Cationic Charge. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1859–1867. [Google Scholar] [CrossRef]
- Adam, M.; Gajdova, S.; Kolarova, H.; Kubala, L.; Lau, D.; Geisler, A.; Ravekes, T.; Rudolph, V.; Tsao, P.S.; Blankenberg, S.; et al. Red blood cells serve as intravascular carriers of myeloperoxidase. J. Mol. Cell. Cardiol. 2014, 74, 353–363. [Google Scholar] [CrossRef]
- Baldus, S.; Eiserich, J.P.; Mani, A.; Castro, L.; Figueroa, M.; Chumley, P.; Ma, W.; Tousson, A.; White, C.R.; Bullard, D.C.; et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J. Clin. Investig. 2001, 108, 1759–1770. [Google Scholar] [CrossRef]
- Astern, J.M.; Pendergraft, W.F.; Falk, R.J.; Jennette, J.C.; Schmaier, A.H.; Mahdi, F.; Preston, G.A. Myeloperoxidase Interacts with Endothelial Cell-Surface Cytokeratin 1 and Modulates Bradykinin Production by the Plasma Kallikrein-Kinin System. Am. J. Pathol. 2007, 171, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Yogalingam, G.; Lee, A.R.; Mackenzie, D.S.; Maures, T.J.; Rafalko, A.; Prill, H.; Berguig, G.Y.; Hague, C.; Christianson, T.; Bell, S.M.; et al. Cellular uptake and delivery of myeloperoxidase to lysosomes promote lipofuscin degradation and lysosomal stress in retinal cells. J. Biol. Chem. 2017, 292, 4255–4265. [Google Scholar] [CrossRef] [Green Version]
- Bradley, P.P.; Christensen, R.D.; Rothstein, G. Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood 1982, 60, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.J.; Preston, G.A.; Pendergraft, W.F.; Segelmark, M.; Heeringa, P.; Hogan, S.L.; Jennette, C.J.; Falk, R.J. Internalization of proteinase 3 is concomitant with endothelial cell apoptosis and internalization of myeloperoxidase with generation of intracellular oxidants. Am. J. Pathol. 2001, 158, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Lefkowitz, D.L.; Roberts, E.; Grattendick, K.; Schwab, C.; Stuart, R.; Lincoln, J.; Allen, R.C.; Moguilevsky, N.; Bollen, A.; Lefkowitz, S.S. The endothelium and cytokine secretion: The role of peroxidases as immunoregulators. Cell. Immunol. 2000, 202, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; Louwe, P.A.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kargapolova, Y.; Geißen, S.; Zheng, R.; Baldus, S.; Winkels, H.; Adam, M. The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication. Antioxidants 2021, 10, 562. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040562
Kargapolova Y, Geißen S, Zheng R, Baldus S, Winkels H, Adam M. The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication. Antioxidants. 2021; 10(4):562. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040562
Chicago/Turabian StyleKargapolova, Yulia, Simon Geißen, Ruiyuan Zheng, Stephan Baldus, Holger Winkels, and Matti Adam. 2021. "The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication" Antioxidants 10, no. 4: 562. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040562