
Ubisol-Q10, a Nanomicellar and Water-Dispersible Formulation of Coenzyme-Q10 as a Potential Treatment for Alzheimer’s and Parkinson’s Disease

 , ,
, ,
Abstract
:1. Introduction
2. Standard Oil-Soluble Formulation of Coenzyme-Q10 in Neurodegenerative Diseases
2.1. Parkinson’s Disease
2.2. Alzheimer’s Disease
2.3. Huntington’s Disease
2.4. Amyotrophic Lateral Sclerosis
2.5. Other Neurodegenerative Diseases
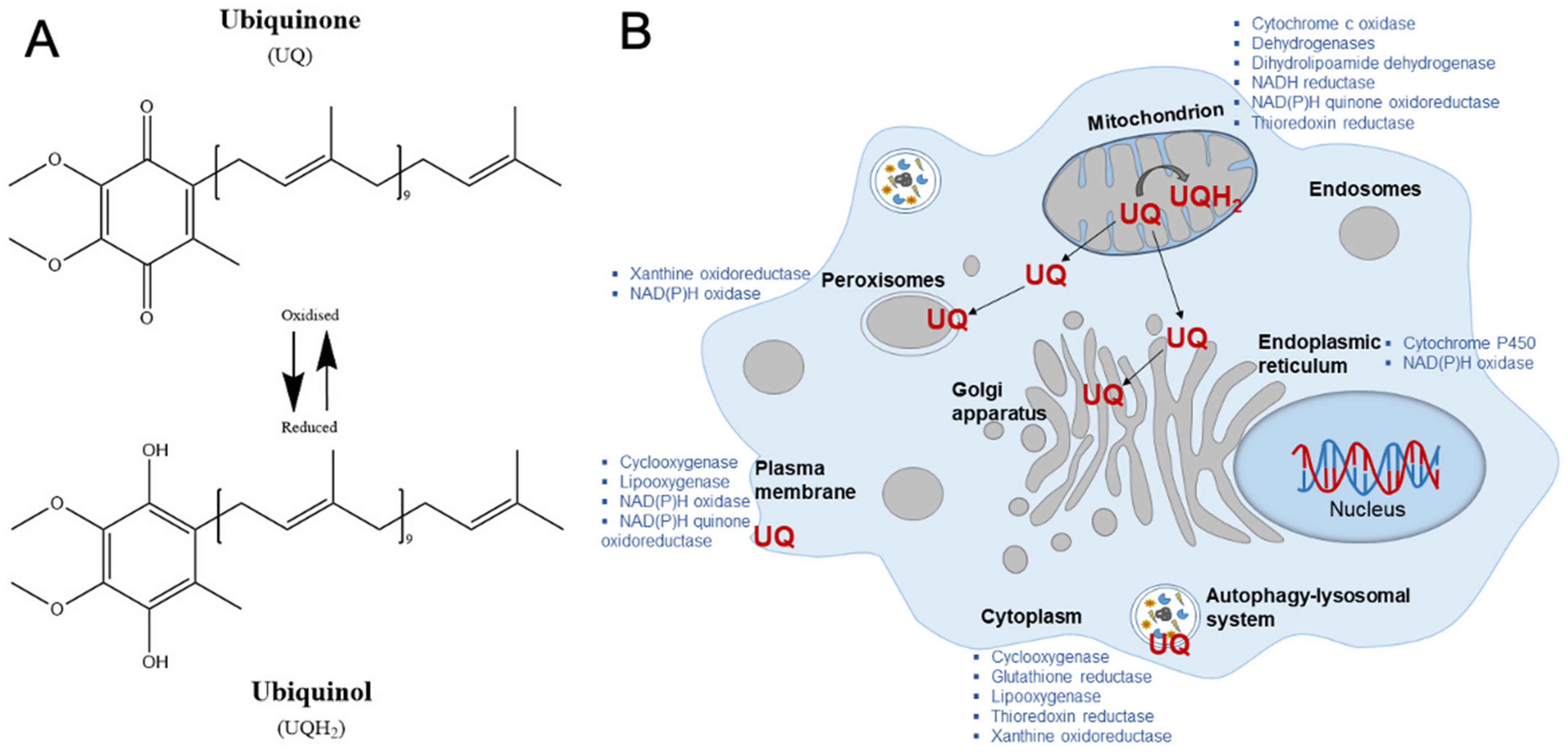
3. Water-Soluble Coenzyme-Q10
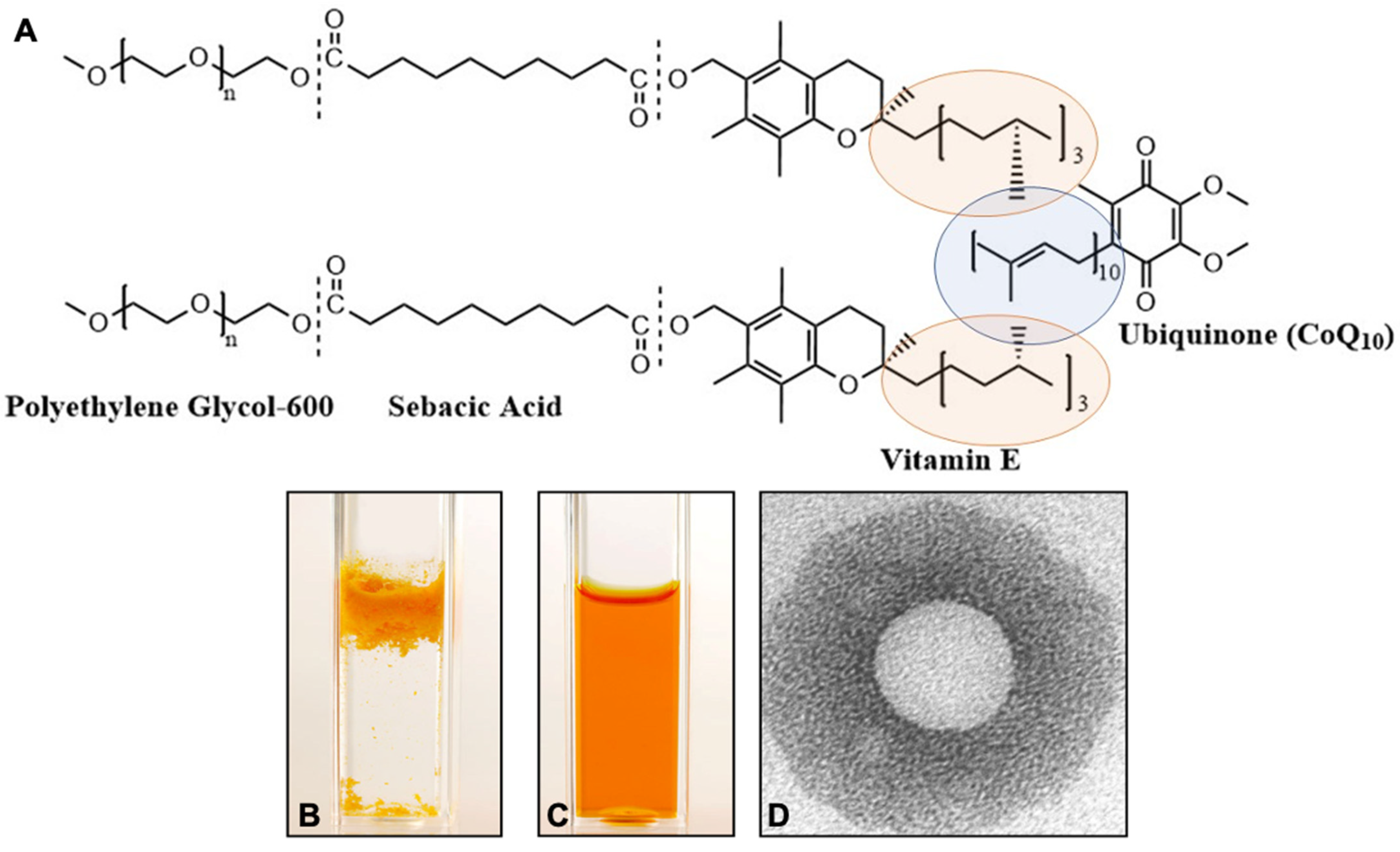
4. Ubisol-Q10 Formulation and Its Properties
5. Ubisol-Q10 Protects Differentiated Neuronal Cells from Oxidative Stress and Excitotoxicity
6. Ubisol-Q10 Inhibits Oxidative Stress and Premature Senescence while Inducing Autophagy in Human Fibroblasts Obtained from AD Patients
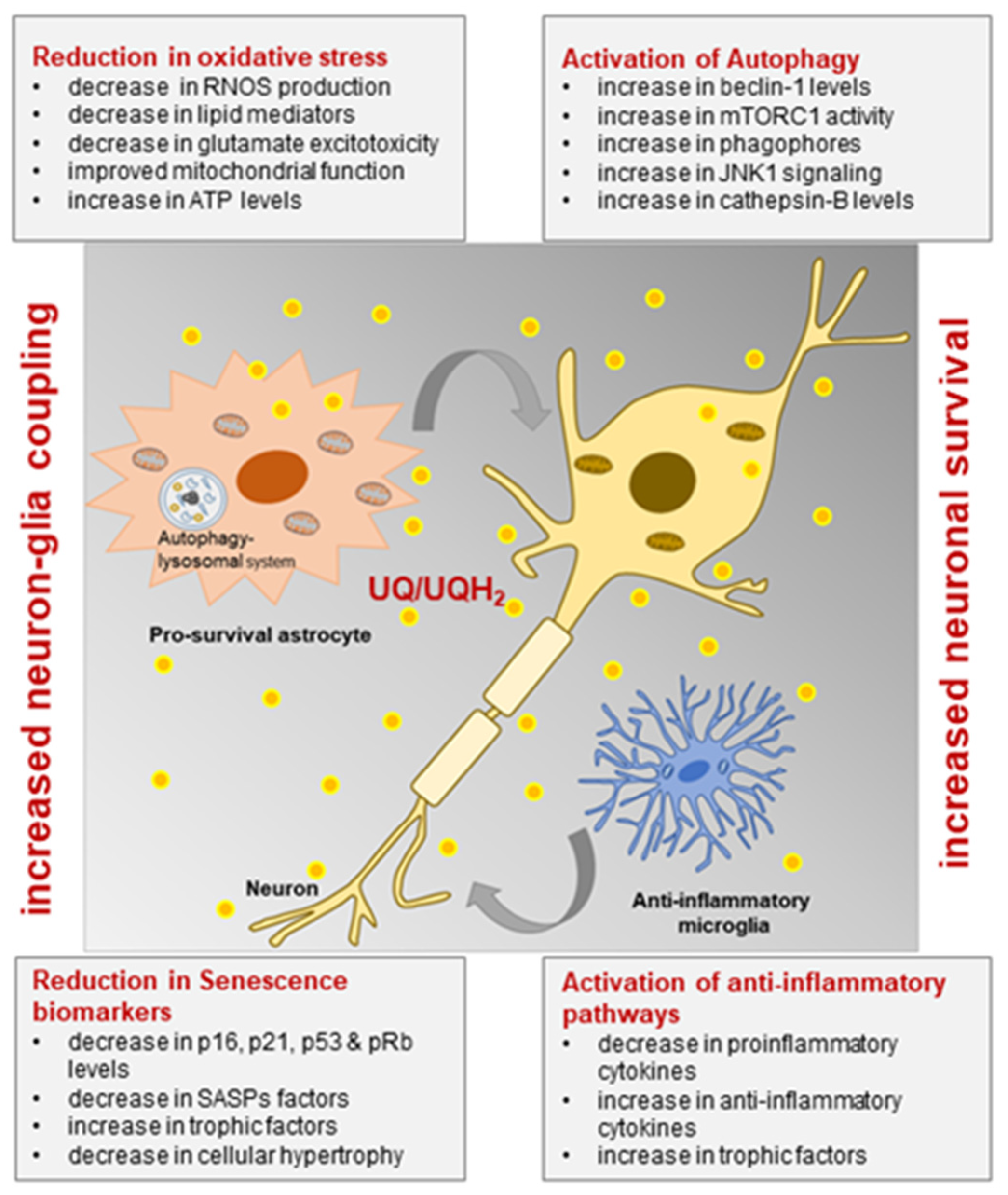
7. Ubisol-Q10 Prevents Neuronal Loss and Halts the Progression of Neurodegeneration in Rodent Models of PD
8. Ubisol-Q10 Halts Progression of AD Pathology and Symptoms by Inhibition of Oxidative Stress, Inflammation, and Activation of Autophagy
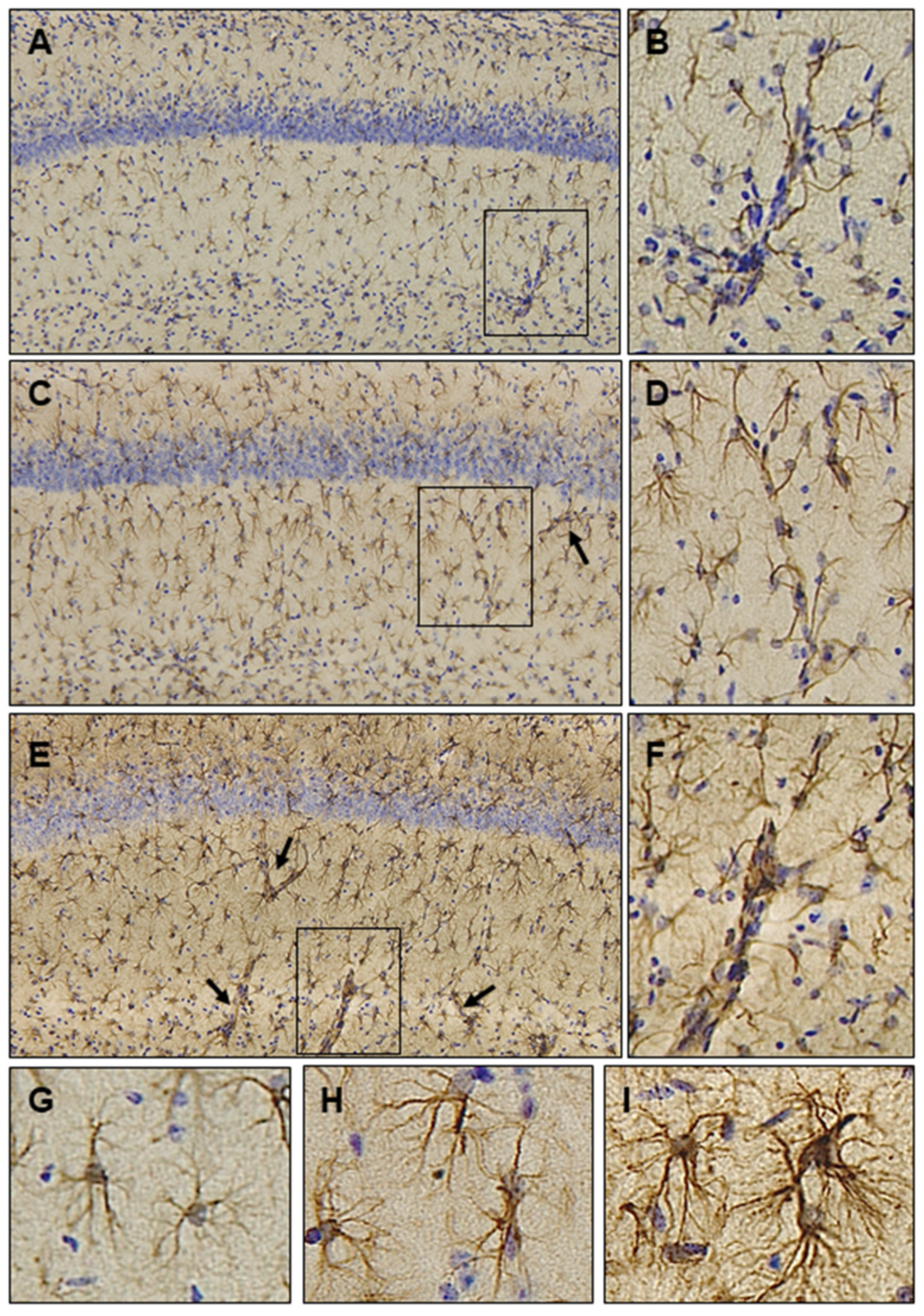
9. Astrocytic Responses in the Brains of Ubisol-Q10 Treated Animals
10. Ubisol-Q10 Effectiveness in Contrast to Other Coenzyme-Q10 Formulations
11. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jellinger, K.A. Dementia diagnosis. Am. J. Geriatr. Psychiatry 2010, 18, 92. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Lehmann, R. Modeling Human Disease. Science 2012, 337, 269. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Wu, C. Mitochondrial dynamics links PINCH-1 signaling to proline metabolic reprogramming and tumor growth. Cell Stress 2020, 5, 23–25. [Google Scholar] [CrossRef]
- Scarlett, D.-J.G.; Herst, P.M.; Berridge, M.V. Multiple proteins with single activities or a single protein with multiple activities: The conundrum of cell surface NADH oxidoreductases. Biochim. Biophys. Acta 2005, 1708, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Forsmark-Andrée, P.; Dallner, G.; Ernster, L. Endogenous ubiquinol prevents protein modification accompanying lipid peroxidation in beef heart submitochondrial particles. Free Radic. Biol. Med. 1995, 19, 749–757. [Google Scholar] [CrossRef]
- Frei, B.; Kim, M.C.; Ames, B.N. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc. Natl. Acad. Sci. USA 1990, 87, 4879–4883. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, P.; Lee, B.-J.; Oh, D.H.; Kim, J.O.; Lee, Y.-I.; Kim, D.-D.; Jee, J.-P.; Lee, Y.-B.; Woo, J.S.; Yong, C.S.; et al. Enhanced oral bioavailability of Coenzyme Q10 by self-emulsifying drug delivery systems. Int. J. Pharm. 2009, 374, 66–72. [Google Scholar] [CrossRef]
- Beg, S.; Javed, S.; Kohli, K. Bioavailability enhancement of coenzyme Q10: An extensive review of patents. Recent Patents Drug Deliv. Formul. 2010, 4, 245–255. [Google Scholar] [CrossRef]
- Masotta, N.E.; Martinefski, M.R.; Lucangioli, S.; Rojas, A.M.; Tripodi, V.P. High-dose coenzyme Q10-loaded oleogels for oral therapeutic supplementation. Int. J. Pharm. 2019, 556, 9–20. [Google Scholar] [CrossRef]
- Pastor-Maldonado, C.J.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10: Novel Formulations and Medical Trends. Int. J. Mol. Sci. 2020, 21, 8432. [Google Scholar] [CrossRef]
- Saha, S.P.; Whayne, T.F., Jr. Coenzyme Q-10 in Human Health: Supporting Evidence? South. Med. J. 2016, 109, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Muztaba, M.; Khan, M.I.; Khalid, M.; Tarique, M.; Akhtar, J.; Ahmad, M. CoQ10: A miraculous and clinically vital coenzyme for normal body functions, prevention and treatment of diseases. J. Pharm. Sci. Res. 2018, 10, 976–982. [Google Scholar]
- Manzar, H.; Abdulhussein, D.; Yap, T.E.; Cordeiro, M.F. Francesca Cellular consequences of coenzyme Q10 deficiency in neurodegeneration of the retina and brain. Int. J. Mol. Sci. 2020, 21, 9299. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Xu, H.; Luo, X.; Yu, J.; Liu, J.; Chuen-Chung, R. Neuroprotection of Coenzyme Q10 in Neurodegenerative Diseases. Curr. Top. Med. Chem. 2015, 16, 858–866. [Google Scholar] [CrossRef]
- Chang, K.-H.; Cheng, M.-L.; Chiang, M.-C.; Chen, C.-M. Lipophilic antioxidants in neurodegenerative diseases. Clin. Chim. Acta 2018, 485, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Pelegay, E.C.; Puzzo, F.; Yilmazer, A.; Cagin, U. Targeting Mitochondrial Defects to Increase Longevity in Animal Models of Neurodegenerative Diseases. Single Mol. Single Cell Seq. 2019, 1134, 89–110. [Google Scholar] [CrossRef]
- Oertel, W.; Schulz, J.B. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J. Neurochem. 2016, 139, 325–337. [Google Scholar] [CrossRef]
- Negida, A.; Menshawy, A.; El Ashal, G.; Elfouly, Y.; Hani, Y.; Hegazy, Y.; El Ghonimy, S.; Fouda, S.; Rashad, Y. Coenzyme Q10 for Patients with Parkinson’;s Disease: A Systematic Review and Meta-Analysis. CNS Neurol. Disord. Drug Targets 2016, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-G.; Sun, M.-X.; Zhang, W.-L.; Wang, W.-W.; Jin, Y.-M.; Xie, C.-L. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: A meta-analysis of randomized controlled trials. Neurol. Sci. 2016, 38, 215–224. [Google Scholar] [CrossRef]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 1995, 1271, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F.; Matthews, R.T.; Tieleman, A.; Shults, C.W. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998, 783, 109–114. [Google Scholar] [CrossRef]
- Cleren, C.; Yang, L.; Lorenzo, B.; Calingasan, N.Y.; Schomer, A.; Sireci, A.; Wille, E.J.; Beal, M.F. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem. 2008, 104, 1613–1621. [Google Scholar] [CrossRef]
- Beal, M.F. Neuroprotective effects of creatine. Amino Acids 2011, 40, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Flint Beal, M. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Gehrke, S.; Yang, Y.; Yang, L.; Beal, M.F.; Lu, B. Neuroprotective effects of compounds with antioxidant and anti-inflammatory properties in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2009, 10, 109. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A Randomized Clinical Trial of High-Dosage Coenzyme Q10 in Early Parkinson Disease. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Bin Choy, Y. Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 9572. [Google Scholar] [CrossRef]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Matsudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994, 269, 13623–13628. [Google Scholar] [CrossRef]
- Frederikse, P.H.; Garland, D.; Zigler, J.S.; Piatigorsky, J. Oxidative Stress Increases Production of β-Amyloid Precursor Protein and β-Amyloid (Aβ) in Mammalian Lenses, and Aβ Has Toxic Effects on Lens Epithelial Cells (∗). J. Biol. Chem. 1996, 271, 10169–10174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadsworth, T.L.; Bishop, J.A.; Pappu, A.S.; Woltjer, R.L.; Quinn, J.F. Evaluation of Coenzyme Q as an Antioxidant Strategy for Alzheimer’s Disease. J. Alzheimers Dis. 2008, 14, 225–234. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.-G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by α-, β- and γ-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Sadli, N.; Barrow, C.J.; McGee, S.; Suphioglu, C. Effect of DHA and CoenzymeQ10 Against Aβ- and Zinc-Induced Mitochondrial Dysfunction in Human Neuronal Cells. CPB 2013, 32, 243–252. [Google Scholar]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Preformed β-amyloid fibrils are destabilized by coenzyme Q10 in vitro. Biochem. Biophys. Res. Commun. 2005, 330, 111–116. [Google Scholar] [CrossRef]
- Dumont, M.; Kipiani, K.; Yu, F.; Wille, E.; Katz, M.; Calingasan, N.Y.; Gouras, G.K.; Lin, M.T.; Beal, M.F. Coenzyme Q10 Decreases Amyloid Pathology and Improves Behavior in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 211–223. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta (BBA)—Bioenerg. 1998, 1366, 211–223. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, ra97–ra142. [Google Scholar] [CrossRef] [Green Version]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic Effects of Coenzyme Q10 and Remacemide in Transgenic Mouse Models of Huntington’s Disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stack, E.C.; Smith, K.M.; Ryu, H.; Cormier, K.; Chen, M.; Hagerty, S.W.; Del Signore, S.J.; Cudkowicz, M.E.; Friedlander, R.M.; Ferrante, R.J. Combination therapy using minocycline and coenzyme Q10 in R6/2 transgenic Huntington’s disease mice. Biochim. Biophys. Acta 2006, 1762, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Stack, E.C.; Kubilus, J.K.; Smith, K.; Cormier, K.; Del Signore, S.J.; Guelin, E.; Ryu, H.; Hersch, S.M.; Ferrante, R.J. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington’s disease transgenic mice. J. Comp. Neurol. 2005, 490, 354–370. [Google Scholar] [CrossRef]
- Hickey, M.A.; Zhu, C.; Medvedeva, V.; Franich, N.R.; Levine, M.S.; Chesselet, M.-F. Evidence for behavioral benefits of early dietary supplementation with CoEnzymeQ10 in a slowly progressing mouse model of Huntington’s disease. Mol. Cell Neurosci. 2012, 49, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Lucchetti, J.; Marino, M.; Papa, S.; Tortarolo, M.; Guiso, G.; Pozzi, S.; Bonetto, V.; Caccia, S.; Beghi, E.; Bendotti, C.; et al. A Mouse Model of Familial ALS Has Increased CNS Levels of Endogenous Ubiquinol9/10 and Does Not Benefit from Exogenous Administration of Ubiquinol10. PLoS ONE 2013, 8, e69540. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.D.; Rohrer, J.D.; Rossor, M.N. Frontotemporal dementia. BMJ 2013, 347, f4827. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Elipenahli, C.; Stack, C.; Jainuddin, S.; Gerges, M.; Yang, L.; Starkov, A.; Beal, M.F.; Dumont, M. Behavioral Improvement after Chronic Administration of Coenzyme Q10 in P301S Transgenic Mice. J. Alzheimer’s Dis. 2012, 28, 173–182. [Google Scholar] [CrossRef]
- Lopes-Ramos, C.; Pereira, T.; Dogini, D.; Gilioli, R.; Lopes-Cendes, I. Lithium carbonate and coenzyme Q10 reduce cell death in a cell model of Machado-Joseph disease. Braz. J. Med. Biol. Res. 2016, 49, e5805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I.; et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef]
- Nakamoto, F.K.; Okamoto, S.; Mitsui, J.; Sone, T.; Ishikawa, M.; Yamamoto, Y.; Kanegae, Y.; Nakatake, Y.; Imaizumi, K.; Ishiura, H.; et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci. Rep. 2018, 8, 14215. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Burn, D.J.; Jaros, E. Multiple system atrophy: Cellular and molecular pathology. Mol. Pathol. 2001, 54, 419–426. [Google Scholar]
- Schottlaender, L.V.; Bettencourt, C.; Kiely, A.P.; Chalasani, A.; Neergheen, V.; Holton, J.L.; Hargreaves, I.; Houlden, H. Coenzyme Q10 Levels Are Decreased in the Cerebellum of Multiple-System Atrophy Patients. PLoS ONE 2016, 11, e0149557. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Stokes, K.; Mahngar, K.; Domazet-Damjanov, D.; Sikorska, M.; Pandey, S. Inhibition of stress induced premature senescence in presenilin-1 mutated cells with water soluble Coenzyme Q10. Mitochondrion 2014, 17, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Muthukumaran, K.; Kanwar, A.; Vegh, C.; Marginean, A.; Elliott, A.; Guilbeault, N.; Badour, A.; Sikorska, M.; Cohen, J.; Pandey, S. Ubisol-Q10 (a Nanomicellar Water-Soluble Formulation of CoQ10) Treatment Inhibits Alzheimer-Type Behavioral and Pathological Symptoms in a Double Transgenic Mouse (TgAPEswe, PSEN1dE9) Model of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 221–236. [Google Scholar] [CrossRef]
- Vegh, C.; Pupulin, S.; Wear, D.; Culmone, L.; Huggard, R.; Ma, D.; Pandey, S. Resumption of Autophagy by Ubisol-Q10 in Presenilin-1 Mutated Fibroblasts and Transgenic AD Mice: Implications for Inhibition of Senescence and Neuroprotection. Oxid. Med. Cell. Longev. 2019, 2019, 7404815. [Google Scholar] [CrossRef] [Green Version]
- Somayajulu-Niţu, M.; Sandhu, J.K.; Cohen, J.; Sikorska, M.; Sridhar, T.S.; Matei, A.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress, neuronal loss in substantia nigra region and parkinsonism in adult rats: Neuroprotection and amelioration of symptoms by water-soluble formulation of coenzyme Q10. BMC Neurosci. 2009, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Muthukumaran, K.; Smith, J.; Jasra, H.; Sikorska, M.; Sandhu, J.K.; Cohen, J.; Lopatin, D.; Pandey, S. Genetic susceptibility model of Parkinson’s disease resulting from exposure of DJ-1 deficient mice to MPTP: Evaluation of neuroprotection by Ubisol-Q10. J. Park. Dis. 2014, 4, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Muthukumaran, K.; Leahy, S.; Harrison, K.; Sikorska, M.; Sandhu, J.K.; Cohen, J.; Keshan, C.; Lopatin, D.; Miller, H.; Borowy-Borowski, H.; et al. Orally delivered water soluble Coenzyme Q10 (Ubisol-Q10) blocks on-going neurodegeneration in rats exposed to paraquat: Potential for therapeutic application in Parkinson’s disease. BMC Neurosci. 2014, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- Sikorska, M.; Lanthier, P.; Miller, H.; Beyers, M.; Sodja, C.; Zurakowski, B.; Gangaraju, S.; Pandey, S.; Sandhu, J.K. Nanomicellar formulation of coenzyme Q10 (Ubisol-Q10) effectively blocks ongoing neurodegeneration in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model: Potential use as an adjuvant treatment in Parkinson’s disease. Neurobiol. Aging 2014, 35, 2329–2346. [Google Scholar] [CrossRef] [Green Version]
- Prosek, M.; Butinar, J.; Lukanc, B.; Fir, M.M.; Milivojevic, L.; Krizman, M.; Smidovnik, A. Bioavailability of water-soluble CoQ10 in beagle dogs. J. Pharm. Biomed. Anal. 2008, 47, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Banshoya, K.; Nakamura, T.; Tanaka, T.; Kaneo, Y. Coenzyme Q10-Polyethylene Glycol Monostearate Nanoparticles: An Injectable Water-Soluble Formulation. Antioxidants 2020, 9, 86. [Google Scholar] [CrossRef] [Green Version]
- Bergamini, C.; Moruzzi, N.; Sblendido, A.; Lenaz, G.; Fato, R. A Water Soluble CoQ10 Formulation Improves Intracellular Distribution and Promotes Mitochondrial Respiration in Cultured Cells. PLoS ONE 2012, 7, e33712. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chen, G.; Ma, W.; Li, P.-A.A. Water-Soluble Coenzyme Q10 Inhibits Nuclear Translocation of Apoptosis Inducing Factor and Cell Death Caused by Mitochondrial Complex I Inhibition. Int. J. Mol. Sci. 2014, 15, 13388–13400. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Luo, K.; Quan, Y.; Lim, S.W.; Shin, Y.J.; Lee, K.E.; Kim, H.L.; Ko, E.J.; Kim, J.H.; Chung, S.J.; et al. Water-soluble coenzyme Q10 provides better protection than lipid-soluble coenzyme Q10 in a rat model of chronic tacrolimus nephropathy. Korean J. Intern. Med. 2021, 12. [Google Scholar] [CrossRef]
- Di Pierro, F.; Rossi, A.; Consensi, A.; Giacomelli, C.; Bazzichi, L. Role for a water-soluble form of CoQ10 in female subjects affected by fibromyalgia. A preliminary study. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 105), S20–S27. [Google Scholar]
- Guastini, L.; Mora, R.; Dellepiane, M.; Santomauro, V.; Giorgio, M.; Salami, A. Water-soluble coenzyme Q10 formulation in presbycusis: Long-term effects. Acta Oto-Laryngol. 2011, 131, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.; Tiwari, R.; Bannerjee, S.K.; Bhati, L.; Pandey, S.; Pandey, P.; Sriwastawa, B. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Lan, J.; Li, X.; Xin, M.; Wang, H.; Zhang, F.; Lu, X.; Zhuang, Z.; Wu, X. Novel ultra-small micelles based on ginsenoside Rb1: A potential nanoplatform for ocular drug delivery. Drug Deliv. 2019, 26, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q10: An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Micellization of coenzyme Q by the fungicide caspofungin allows for safe intravenous administration to reach extreme supraphysiological concentrations. Redox Biol. 2020, 36, 101680. [Google Scholar] [CrossRef]
- Borowy-Borowski, H.; Sikorska, M.; Walker, P.R. Water-Soluble Composition of Bioactive Lipophilic Compounds. U.S. Patent 6045826A, 4 April 2000. [Google Scholar]
- Borowy-Borowski, H.; Sikorska, M.; Walker, P.R. Water-Soluble Compositions of Bioactive Lipophilic Compounds. U.S. Patent 6191172B1, 20 February 2001. [Google Scholar]
- Borowy-Borowski, H.; Sikorska, M.; Walker, P.R. Delivery Compositions of Bioactive Compounds. U.S. Patent 6632443, 14 October 2003. [Google Scholar]
- Borowy-Borowski, H.; Sikorska, M.; Walker, P.R. Water-Soluble Compositions of Bioactive Lipophilic Compounds. U.S. Patent 7645816, 12 January 2010. [Google Scholar]
- Sikorska, M.; Borowy-Borowski, H.; Zurakowski, B.; Walker, P.R. Derivatised alpha-tocopherol as a CoQ10 carrier in a novel water-soluble formulation. Biofactors 2003, 18, 173–183. [Google Scholar] [CrossRef]
- Graves, S.; Sikorska, M.; Borowy-Borowski, H.; Ho, R.J.; Bui, T.; Woodhouse, C. Analysis of coenzyme Q10 content in human plasma and other biological samples. Methods Mol. Biol. 1998, 108, 353–365. [Google Scholar]
- Borowy-Borowski, H.; Sodja, C.; Docherty, J.; Walker, P.R.; Sikorska, M. Unique technology for solubilization and delivery of highly lipophilic bioactive molecules. J. Drug Target. 2004, 12, 415–424. [Google Scholar] [CrossRef]
- Somayajulu, M.; McCarthy, S.; Hung, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by coenzyme Q10. Neurobiol. Dis. 2005, 18, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Pandey, S.; Ribecco-Lutkiewicz, M.; Monette, R.; Borowy-Borowski, H.; Walker, P.R.; Sikorska, M. Molecular mechanisms of glutamate neurotoxicity in mixed cultures of NT2-derived neurons and astrocytes: Protective effects of coenzyme Q10. J. Neurosci. Res. 2003, 72, 691–703. [Google Scholar] [CrossRef]
- McCarthy, S.; Somayajulu, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10. Toxicol. Appl. Pharmacol. 2004, 201, 21–31. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer’s-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Naderi, J.; Somayajulu-Niţu, M.; Mukerji, A.; Sharda, P.; Sikorska, M.; Borowy-Borowski, H.; Antonsson, B.; Pandey, S. Water-soluble formulation of Coenzyme Q10 inhibits Bax-induced destabilization of mitochondria in mammalian cells. Apoptosis 2006, 11, 1359–1369. [Google Scholar] [CrossRef]
- Vegh, C.; Wear, D.; Okaj, I.; Huggard, R.; Culmone, L.; Eren, S.; Cohen, J.; Rishi, A.; Pandey, S. Combined Ubisol-Q10 and Ashwagandha Root Extract Target Multiple Biochemical Mechanisms and Halt Neurodegeneration in a Paraquat Induced Rat Model of Parkinson’s Disease. Antioxidants 2021, 10, 563. [Google Scholar] [CrossRef]
- Berry, C.; La Vecchia, C.; Nicotera, P. Paraquat and Parkinson’s disease. Cell Death Differ. 2010, 17, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease: A case–control study in Taiwan. Neurology 1997, 48, 1583–1588. [Google Scholar] [CrossRef]
- Gatto, N.M.; Cockburn, M.; Bronstein, J.; Manthripragada, A.D.; Ritz, M. Well-water consumption and Parkinson’s Disease in rural California. Environ. Health Perspect. 2009, 117, 1912–1918. [Google Scholar] [CrossRef] [Green Version]
- Spindler, M.; Beal, M.F.; Henchcliffe, C. Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatr. Dis. Treat. 2009, 5, 597–610. [Google Scholar]
- Olanow, C.W.; Lees, A.; Obeso, J. Levodopa therapy for Parkinson’s disease: Challenges and future prospects.le first published online. Mov. Disord. 2008, 23, S495–S496. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Galani, R.; Weiss, I.; Cassel, J.-C.; Kelche, C. Spatial memory, habituation, and reactions to spatial and nonspatial changes in rats with selective lesions of the hippocampus, the entorhinal cortex or the subiculum. Behav. Brain Res. 1998, 96, 1–12. [Google Scholar] [CrossRef]
- Platel, A.; Porsolt, R.D. Habituation of exploratory activity in mice: A screening test for memory enhancing drugs. Psychopharmacology 1982, 78, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Murphy, E.S.; Elijah, I.E.; Holtfreter, K.L.; Davis, C.J.; Olson, M.L.; Muhunthan, K.; Harding, J.W. Influence of hippocampectomy on habituation, exploratory behavior, and spatial memory in rats. Brain Res. 2004, 1023, 1–14. [Google Scholar] [CrossRef]
- Aggleton, J.; Hunt, P.; Rawlins, J. The effects of hippocampal lesions upon spatial and non-spatial tests of working memory. Behav. Brain Res. 1986, 19, 133–146. [Google Scholar] [CrossRef]
- Broadbent, N.J.; Squire, L.R.; Clark, R.E. Spatial memory, recognition memory, and the hippocampus. Proc. Natl. Acad. Sci. USA 2004, 101, 14515–14520. [Google Scholar] [CrossRef] [Green Version]
- Forwood, S.; Winters, B.; Bussey, T. Hippocampal lesions that abolish spatial maze performance spare object recognition memory at delays of up to 48 hours. Hippocampus 2004, 15, 347–355. [Google Scholar] [CrossRef]
- Save, E.; Poucet, B.; Foreman, N.; Buhot, M.C. Object exploration and reactions to spatial and nonspatial changes in hooded rats following damage to parietal cortex or hippocampal formation. Behav. Neurosci. 1992, 106, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Stupien, G.; Florian, C.; Roullet, P. Involvement of the hippocampal CA3-region in acquisition and in memory consolidation of spatial but not in object information in mice. Neurobiol. Learn. Mem. 2003, 80, 32–41. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology 2001, 57, 397–404. [Google Scholar]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Cordero, M.D.; Alcocer-Gómez, E.; Culic, O.; Carrión, A.M.; De Miguel, M.; Díaz-Parrado, E.; Pérez-Villegas, E.M.; Bullón, P.; Battino, M.; Sánchez-Alcazar, J.A. NLRP3 inflammasome is activated in fibromyalgia: The effect of coenzyme Q10. Antioxid. Redox Sign. 2014, 20, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Wang, H.; Gong, Z.; Li, X.; He, L.; Shen, Q.; Pan, J.; Peng, Y. Idebenone attenuates cerebral inflammatory injury in ischemia and reperfusion via dampening NLRP3 inflammasome activity. Mol. Immunol. 2020, 123, 74–87. [Google Scholar] [CrossRef]
- Banker, G.A. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 1980, 209, 809–810. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of Glutamate Transporters Reveals a Major Role for Astroglial Transport in Excitotoxicity and Clearance of Glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Koehler, R.C.; Roman, R.J.; Harder, D.R. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009, 32, 160–169. [Google Scholar] [CrossRef]
- Yeager, M.; Harris, A.L. Gap junction channel structure in the early 21st century: Facts and fantasies. Curr. Opin. Cell Biol. 2007, 19, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Guevara, G.; Sanchez, K.R.; Sun, J.; Harrington, M.A.; Temburni, M.; Biol., Delaware State Univ., Dover, DE. Molecular Mechanisms of Astrocyte-Neuron Interactions in the Development of Synchronized Activity in Neuronal Networks. Program No. 652.29. 2018 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience. 2018. Available online: https://www.abstractsonline.com/pp8/#!/4649/presentation/23270 (accessed on 18 April 2021).
- Tukker, A.M.; Wijnolts, F.M.J.; de Groot, A.; Westerink, R.H.S. Human iPSC-derived neuronal models for in vitro neurotoxicity assessment. Neurotoxicology 2018, 67, 215–225. [Google Scholar] [CrossRef]
- Kuijlaars, J.; Oyelami, T.; Diels, A.; Rohrbacher, J.; Versweyveld, S.; Meneghello, G.; Tuefferd, M.; Verstraelen, P.; Detrez, J.R.; Verschuuren, M.; et al. Sustained synchronized neuronal network activity in a human astrocyte co-culture system. Sci. Rep. 2016, 6, 36529. [Google Scholar] [CrossRef] [Green Version]
- Paavilainen, T.; Pelkonen, A.; Mäkinen, M.E.-L.; Peltola, M.; Huhtala, H.; Fayuk, D.; Narkilahti, S. Effect of prolonged differentiation on functional maturation of human pluripotent stem cell-derived neuronal cultures. Stem Cell Res. 2018, 27, 151–161. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Petzold, G.C.; Murthy, V.N. Role of astrocytes in neurovascular coupling. Neuron 2011, 71, 782–797. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, P.A.; Aizenman, E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett. 1989, 103, 162–168. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Jing, L.; He, M.-T.; Chang, Y.; Mehta, S.L.; He, Q.-P.; Zhang, J.-Z.; Li, P.A. Coenzyme Q10 protects astrocytes from ROS-induced damage through inhibition of mitochondria-mediated cell death pathway. Int. J. Biol. Sci. 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valverde, Y.; Benson, B.; Gupta, M.; Gupta, K. Spinal glial activation and oxidative stress are alleviated by treatment with curcumin or coenzyme Q in sickle mice. Haematologica 2016, 101, e44–e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelzer, C.; Lorenz, G.; Lindner, I.; Rimbach, G.; Niklowitz, P.; Menke, T.; Döring, F. Effects of Coenzyme Q10 on TNF-alpha secretion in human and murine monocytic cell lines. Biofactors 2007, 31, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, C.; Lorenz, G.; Rimbach, G.; Döring, F. In Vitro Effects of the Reduced Form of Coenzyme Q(10) on Secretion Levels of TNF-alpha and Chemokines in Response to LPS in the Human Monocytic Cell Line THP-1. J. Clin. Biochem. Nutr. 2009, 44, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xu, D.; Lin, J.; Zhang, D.; Wang, G.; Sui, L.; Ding, H.; Du, J. Coenzyme Q10 attenuated β-amyloid25-35-induced inflammatory responses in PC12 cells through regulation of the NF-κB signaling pathway. Brain Res. Bull. 2017, 131, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neurodegenerative Disease | Model | Effective Dose | Mode of Administration | Major Outcomes | Reference | |
|---|---|---|---|---|---|---|
| Oil-Soluble CoQ10 | Alzheimer’s Disease | In-Vivo (Mice) | 10 g/kg diet | Oral | - Protection against neurotoxicity & oxidative stress - Mitochondrial stabilization - Reduced Aβ plaques - Improved cognitive performance | Wadsworth 2008 [30] |
| In-Vitro | 6.25 µM | Media Supplementation | Wadsworth 2008 [30] | |||
| In-Vivo (Mice) | 0.4% or 2.4% | Oral | Dumont 2011 [35] | |||
| In-Vitro | 10 µM | Media Supplementation | Sadli 2013 [33] | |||
| Amyotrophic Lateral Sclerosis (ALS) | In-Vivo (Rats & Mice) | 200 mg/kg/ day | Oral | - Anti-oxidative effects - Preserved mitochondrial function - Increased lifespan | Matthews 1998 [39] | |
| In-Vivo (Mice) | 200 mg/kg/day (no effect) | Oral (Gavage) | Lucchetti 2013 [44] | |||
| Frontotemporal Dementia | In-Vivo (Mice) | 0.5% of Diet | Oral | - Improved behaviour & survival | Elipenahli 2012 [50] | |
| Huntington’s Disease | In-Vivo (Rats) | 200 mg/kg/day | Oral | - Improved motor performance & survival - Delayed weight loss - Prevented striatal neuron intranuclear inclusion formation - Slowed striatal neuron atrophy - Reduced HTT aggregate formation - Reduced oxidative damage | Matthews 1998 [39] | |
| In-Vivo (Mice) | 400 mg/kg/day | Oral | Ferrante 2002 [40] | |||
| In-Vivo (Mice) | 0.2% of Diet | Oral | Stack 2006 [41] | |||
| In-Vivo (Mice, Rats) | 1600–2000 mg/kg/day | Oral | Yang 2009 [24] | |||
| In-Vivo (Mice) | 0.2% of Diet | Oral | Hickey 2012 [43] | |||
| Machado-Joseph Disease | In-Vitro | 10 µM, 30 µM, 90 µM | Media Supplementation | - Improved cell viability & reduced apoptosis - Prevented ATX3 protein aggregation | Lopes-Ramos 2016 [51] | |
| Multiple-System Atrophy | In-Vitro | 25 µM | Media Supplementation | - Improved oxidative metabolism - Reduced apoptosis | Nakamoto 2018 [53] | |
| Parkinson’s Disease | In-Vivo (Mice) | 200 mg/kg/day | Oral | - Dopaminergic neurons saved in striatum and SNpc - Clearance of α-synuclein aggregates - Limited oxidative damage - Reduced pro-inflammatory cytokines | Beal 1998 [21] | |
| In-Vivo (Mice) | 200–1600 mg/kg/day | Oral | Cleren 2008 [22] | |||
| In-Vivo (Mice) | 1% of Diet | Oral | Yang 2009 [24] | |||
| In-Vivo (Drosophila) | 100 mg/mL (no effect) | Oral | Faust 2009 [25] | |||
| In-Vivo (Rats) | 25 µg/mL | Intrastriatal Injection | Park 2020 [27] | |||
| Ubisol-Q10 | Alzheimer’s Disease | In-Vitro | 50 µg/mL | Media Supplementation | - Inhibited oxidative stress - Upregulated autophagy - Maintained MMP - Reduced cell cycle arrest protein expression - Prevented SIPS onset - Inhibited apoptosis - Improved memory - Reduced hippocampal neurodegeneration - Cleared Aβ plaques - Increased astrocyte activity - Reduced microglial activity | Ma 2014 [57] |
| In-Vivo (Mice) | 6 mg/kg/day | Oral | Muthukumaran 2018 [58] | |||
| In-Vivo (Mice) | 50 µg/mL | Oral | Vegh 2019 [59] | |||
| In-Vitro | 50 µg/mL | Media Supplementation | Vegh 2019 [59] | |||
| Parkinson’s Disease | In-Vivo (Rats) | 50 µg/mL | Oral | - Reduced oxidative stress - Maintained ATP generation - Stabilized mitochondrial membrane - Prevented loss of dopaminergic neurons - Activated pro -survival astrocytes - Ameliorated motor dysfunction | Somayajulu-Nitu 2009 [60] | |
| In-Vivo (Mice) | 6 mg/kg/day | Oral | Muthukumaran 2014 [61] | |||
| In-Vivo (Rats) | 6 mg/kg/day | Oral | Muthukumaran 2014 [62] | |||
| In-Vivo (Mice) | 3 mg/kg/day | Oral | Sikorska 2014 [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wear, D.; Vegh, C.; Sandhu, J.K.; Sikorska, M.; Cohen, J.; Pandey, S. Ubisol-Q10, a Nanomicellar and Water-Dispersible Formulation of Coenzyme-Q10 as a Potential Treatment for Alzheimer’s and Parkinson’s Disease. Antioxidants 2021, 10, 764. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050764
Wear D, Vegh C, Sandhu JK, Sikorska M, Cohen J, Pandey S. Ubisol-Q10, a Nanomicellar and Water-Dispersible Formulation of Coenzyme-Q10 as a Potential Treatment for Alzheimer’s and Parkinson’s Disease. Antioxidants. 2021; 10(5):764. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050764
Chicago/Turabian StyleWear, Darcy, Caleb Vegh, Jagdeep K. Sandhu, Marianna Sikorska, Jerome Cohen, and Siyaram Pandey. 2021. "Ubisol-Q10, a Nanomicellar and Water-Dispersible Formulation of Coenzyme-Q10 as a Potential Treatment for Alzheimer’s and Parkinson’s Disease" Antioxidants 10, no. 5: 764. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050764