
Na/H Exchange Regulatory Factor 1 Deficient Mice Show Evidence of Oxidative Stress and Altered Cisplatin Pharmacokinetics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. γ-Glutamyl Transferase Activity Assay
2.3. Histology and Immunohistochemistry (IHC)
2.4. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
2.5. Collection of Mouse Blood and Tissue for Glutathione (GSH), Glutathione Disulfide (GSSG), Cysteine (Cys), and Cystine (CySS)
2.6. HPLC Analysis of Kidney Cortex and Plasma for GSH, GSSG, Cys, and CySS
2.7. RNA Isolation and Quantitative Reverse-Transcriptase Polymerase Chain Reaction (PCR)
2.8. Protein Determination and Immunoblots
2.9. Inductively Coupled Plasma-Mass Spectrometry of Platinum
2.10. Statistical Analysis
3. Results
3.1. Kidneys of NHERF1 KO Mice Treated with Cisplatin Show Significantly Greater Damage when Compared to Those from Cisplatin Treated WT Mice at 24 h
3.2. Greater Renal Histologic Injury 24 h Post Cisplatin in NHERF1 KO Kidneys
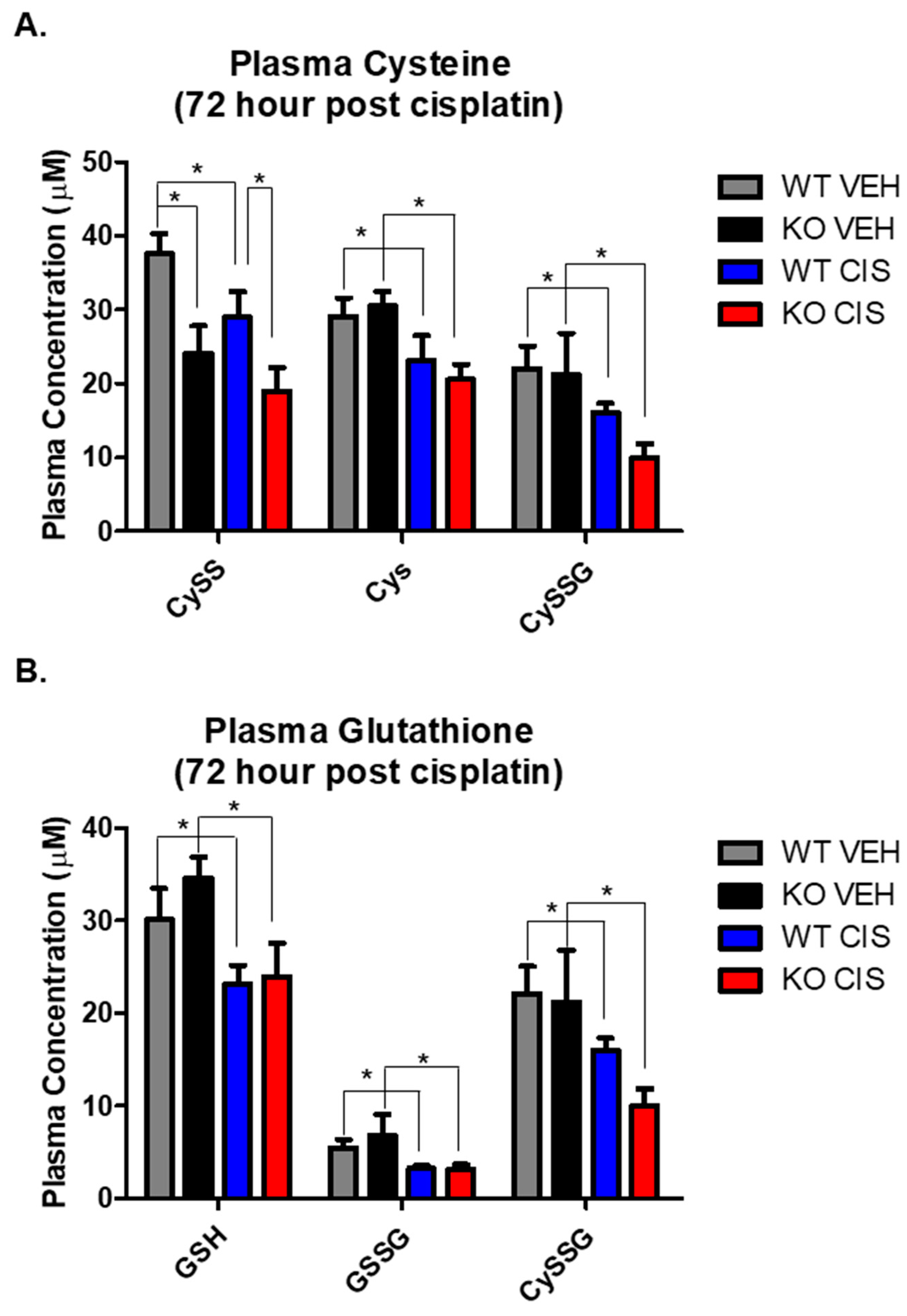
3.3. NHERF1 KO Kidneys Exhibit Altered Redox
3.4. Comparison of Genes Involved in Glutathione Synthesis between NHERF1 Deficient and WT Kidneys
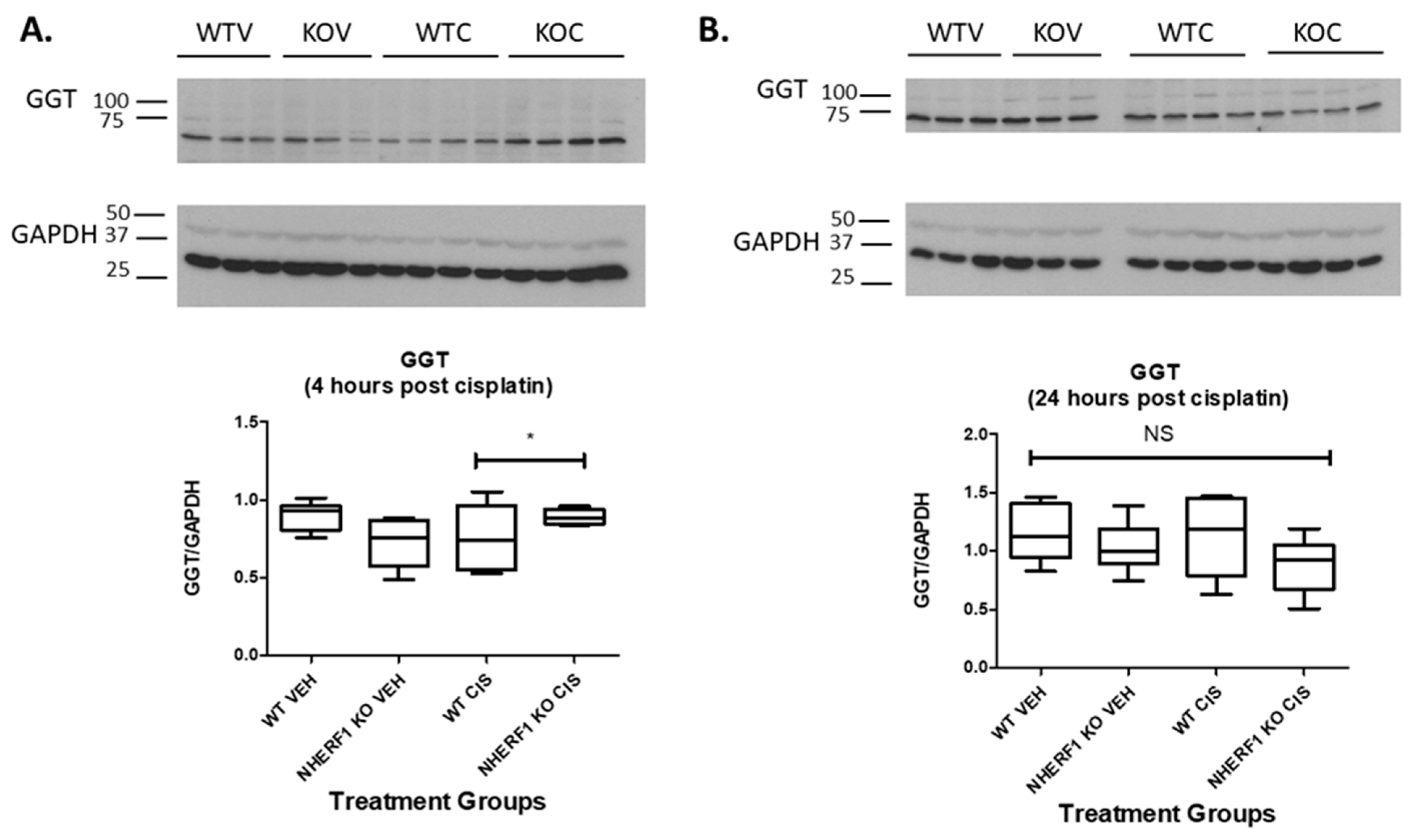
3.5. Cisplatin-Induced Changes in GGT Activity in WT and KO Kidneys
3.6. Cisplatin Treatment and NHERF1 Loss Affect GGT Protein Expression
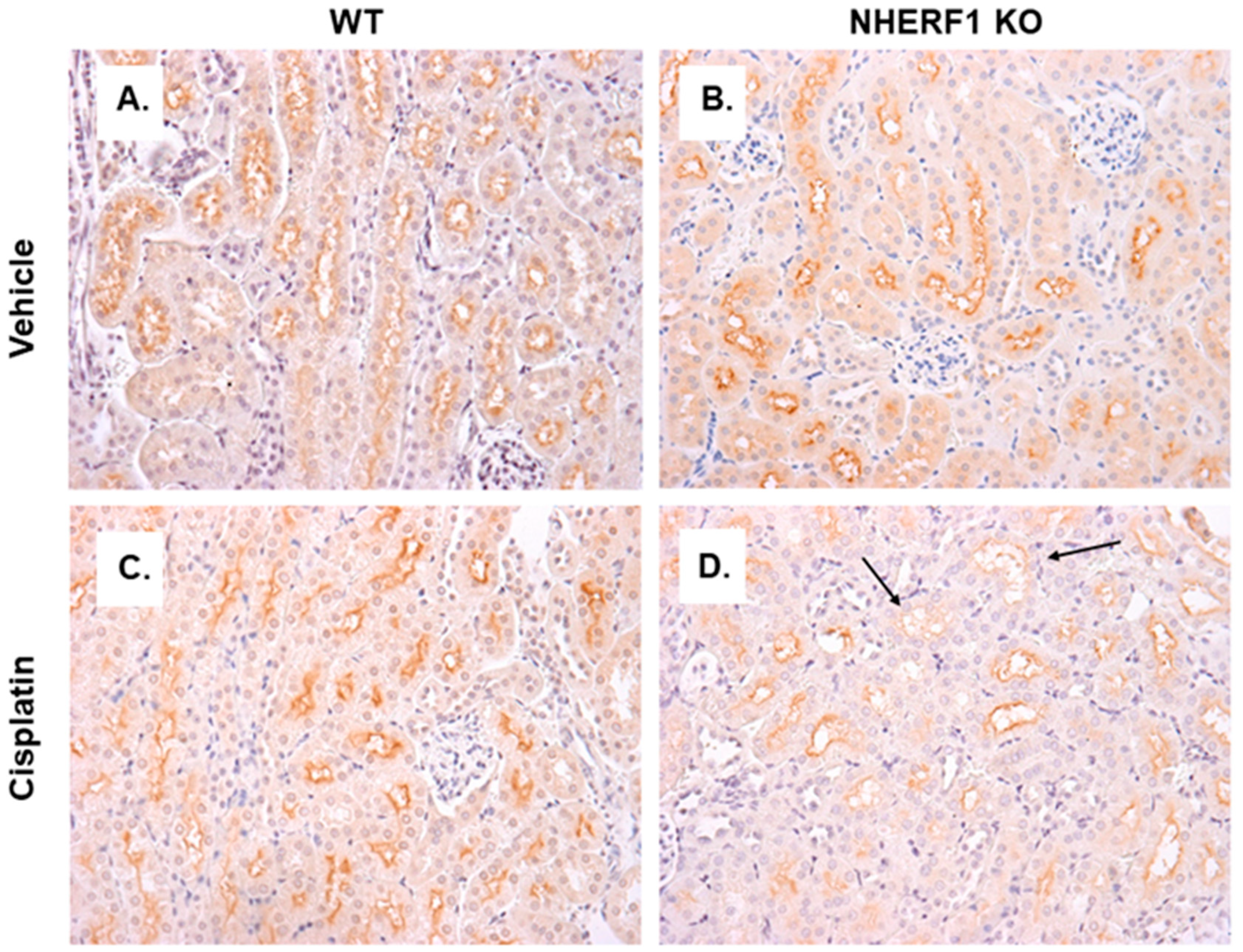
3.7. Differences in GGT Localization in NHERF1 KO and WT Kidneys
3.8. NHERF1 Expression Influences CCBL Protein Expression
3.9. CCBL Localization and Staining Pattern Is Altered in NHERF1 KO Kidneys Prior to Cisplatin Treatment
3.10. Pt Levels Are Decreased in NHERF1 KO Kidneys 72 h after Cisplatin Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alderden, R.A.; Hall, M.D.; Hambley, T.W. The Discovery and Development of Cisplatin. J. Chem. Educ. 2006, 83, 728. [Google Scholar] [CrossRef]
- Eustace, P. History and development of cisplatin in the management of malignant disease. Cancer Nurs. 1980, 3, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H.; Devarajan, P. Cisplatin nephrotoxicity: Molecular mechanisms. Cancer Ther. 2003, 1, 47–61. [Google Scholar] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarimboli, G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014, 34, 547–550. [Google Scholar] [CrossRef]
- Nakamura, T.; Yonezawa, A.; Hashimoto, S.; Katsura, T.; Inui, K.-I. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem. Pharmacol. 2010, 80, 1762–1767. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a Nephrotoxin in Proximal Tubule Cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Fliedl, L. Controversial role of Gamma-Glutamyl Transferase activity in cisplatin nephrotoxicity. ALTEX 2014, 31, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Bushau-Sprinkle, A.M.; Lederer, E.D. New roles of the Na+/H+ exchange regulatory factor 1 scaffolding protein: A review. Am. J. Physiol. Physiol. 2020, 318, F804–F808. [Google Scholar] [CrossRef]
- Bushau-Sprinkle, A.; Barati, M.; Conklin, C.; Dupre, T.; Gagnon, K.; Khundmiri, S.J.; Clark, B.; Siskind, L.; Doll, M.A.; Rane, M.; et al. Loss of the Na+/H+ Exchange Regulatory Factor 1 Increases Susceptibility to Cisplatin-Induced Acute Kidney Injury. Am. J. Pathol. 2019, 189, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.; Song, J.M.; Park, M.Y.; Kang, S.K.; Kim, Y.K.; Jung, J.S. Cisplatin-induced apoptosis by translocation of endogenous Bax in mouse collecting duct cells11Abbreviations: ROS, reactive oxygen species; SAPK/JNK, stress-activated protein kinase/c-Jun NH2-terminal kinase; RT-PCR, reverse transcription-polymerase chain reaction; ECL, enhanced chemiluminescence; LDH, lactic dehydrogenase; DPPD, diphenyl-p-phenylene-diamine; DFO, deferoxamine; DMTU, dimethylthiourea; and BHA, butylated hydroxyanisole. Biochem. Pharmacol. 2001, 62, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Triaca, V.; Levine, J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: Apoptosis vs. necrosis. Am. J. Physiol. Physiol. 1996, 270, F700–F708. [Google Scholar] [CrossRef] [PubMed]
- Bushau-Sprinkle, A.; Barati, M.T.; Gagnon, K.B.; Khundmiri, S.J.; Kitterman, K.; Hill, B.G.; Sherwood, A.; Merchant, M.; Rai, S.N.; Srivastava, S.; et al. NHERF1 Loss Upregulates Enzymes of the Pentose Phosphate Pathway in Kidney Cortex. Antioxidants 2020, 9, 862. [Google Scholar] [CrossRef]
- Barati, M.T.; Ketchem, C.J.; Merchant, M.L.; Kusiak, W.B.; Jose, P.A.; Weinman, E.J.; Leblanc, A.J.; Lederer, E.D.; Khundmiri, S.J. Loss of NHERF-1 expression prevents dopamine-mediated Na-K-ATPase regulation in renal proximal tubule cells from rat models of hypertension: Aged F344 rats and spontaneously hypertensive rats. Am. J. Physiol. Physiol. 2017, 313, C197–C206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenolikar, S.; Voltz, J.W.; Minkoff, C.M.; Wade, J.B.; Weinman, E.J. Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc. Natl. Acad. Sci. USA 2002, 99, 11470–11475. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Liang, Y. Measuring the poise of thiol/disulfide couples in vivo. Free. Radic. Biol. Med. 2009, 47, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Watson, W.H.; Burke, T.J.; Zelko, I.N.; Torres-González, E.; Ritzenthaler, J.D.; Roman, J. Differential Regulation of the Extracellular Cysteine/Cystine Redox State (EhCySS) by Lung Fibroblasts from Young and Old Mice. Oxidative Med. Cell. Longev. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sadighi-Moghaddam, B.; Salek Farrokhi, A.; Namdar Ahmadabad, H.; Barati, M.; Moazzeni, S.M. Mesenchymal Stem Cell Therapy Prevents Abortion in CBA/J × DBA/2 Mating. Reprod. Sci. 2017, 25, 1261–1269. [Google Scholar] [CrossRef]
- Lash, L.H. Renal glutathione transport: Identification of carriers, physiological functions, and controversies. BioFactors 2009, 35, 500–508. [Google Scholar] [CrossRef]
- Brozovic, A.; Ambriović-Ristov, A.; Osmak, M.; Ambriovic-Ristov, A. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Sikalidis, A.; Mazor, K.M.; Lee, J.-I.; Roman, H.B.; Hirschberger, L.L.; Stipanuk, M.H. Upregulation of capacity for glutathione synthesis in response to amino acid deprivation: Regulation of glutamate-cysteine ligase subunits. Amino Acids 2014, 46, 1285–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleena, M.A.; Vadhiraha, B.M.; Fernandes, D.H.; Prabhu, R.; Nalini, K. Evaluation of urinary tubular enzymes for the detection of early kidney injury due to cisplatin chemotherapy. Int. J. Biol. Med. Res. 2012, 3, 2241–2246. [Google Scholar]
- Giral, H.; Cranston, D.; Lanzano, L.; Caldas, Y.; Sutherland, E.; Rachelson, J.; Dobrinskikh, E.; Weinman, E.J.; Doctor, R.B.; Gratton, E.; et al. NHE3 Regulatory Factor 1 (NHERF1) Modulates Intestinal Sodium-dependent Phosphate Transporter (NaPi-2b) Expression in Apical Microvilli*. J. Biol. Chem. 2012, 287, 35047–35056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, R.; Biswas, R.; Steplock, D.; Shenolikar, S.; Weinman, E. Role of NHERF and scaffolding proteins in proximal tubule transport. Urol. Res. 2010, 38, 257–262. [Google Scholar] [CrossRef]
- Tokunaga, J.; Kobayashi, M.; Kitagawa, A.; Nakamura, C.; Arimori, K.; Nakano, M. Protective effects of betamipron on renal toxicity during repeated cisplatin administration in rats and protective mechanism. Ren. Fail. 1998, 20, 27–38. [Google Scholar] [CrossRef]
- Hajian, S.; Rafieian-Kopaei, M.; Nasri, H. Renoprotective effects of antioxidants against cisplatin nephrotoxicity. J. Nephropharmacology 2014, 3, 39–42. [Google Scholar]
- Verma, P.K.; Raina, R.; Sultana, M.; Singh, M.; Kumar, P. Total antioxidant and oxidant status of plasma and renal tissue of cisplatin-induced nephrotoxic rats: Protection by floral extracts of Calendula officinalis Linn. Ren. Fail. 2015, 38, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Long, Y.C. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci. Rep. 2016, 6, 30033. [Google Scholar] [CrossRef]
- Filomeni, G.; Aquilano, K.; Civitareale, P.; Rotilio, G.; Ciriolo, M.R. Activation of c-Jun-N-terminal kinase is required for apoptosis triggered by glutathione disulfide in neuroblastoma cells. Free. Radic. Biol. Med. 2005, 39, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 mitogen-activated protein kinase pathway. FASEB J. 2002, 17, 64–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Zhuang, L.; Olszewski, K.; Gan, B. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edge sword in cellular redox regulation. Genes Dis. 2020. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Hanigan, M.H. Inhibition of γ-Glutamyl Transpeptidase or Cysteine S-Conjugate β-Lyase Activity Blocks the Nephrotoxicity of Cisplatin in Mice. J. Pharmacol. Exp. Ther. 2002, 300, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H.; Gallagher, B.C.; Taylor, P.T.; Large, M.K. Inhibition of gamma-glutamyl transpeptidase activity by acivicin in vivo protects the kidney from cisplatin-induced toxicity. Cancer Res. 1994, 54, 5925–5929. [Google Scholar]
- Hanigan, M.H.; Lykissa, E.D.; Townsend, D.M.; Ou, C.-N.; Barrios, R.; Lieberman, M.W. γ-Glutamyl Transpeptidase-Deficient Mice Are Resistant to the Nephrotoxic Effects of Cisplatin. Am. J. Pathol. 2001, 159, 1889–1894. [Google Scholar] [CrossRef]
- Paolicchi, A.; Sotiropuolou, M.; Perego, P.; Daubeuf, S.; Visvikis, A.; Lorenzini, E.; Franzini, M.; Romiti, N.; Chieli, E.; Leone, R.; et al. γ-Glutamyl transpeptidase catalyses the extracellular detoxification of cisplatin in a human cell line derived from the proximal convoluted tubule of the kidney. Eur. J. Cancer 2003, 39, 996–1003. [Google Scholar] [CrossRef]
- Almaghrabi, O.A. Molecular and biochemical investigations on the effect of quercetin on oxidative stress induced by cisplatin in rat kidney. Saudi J. Biol. Sci. 2015, 22, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Perše, M.; Večerić-Haler, Ž. Cisplatin-Induced Rodent Model of Kidney Injury: Characteristics and Challenges. BioMed Res. Int. 2018, 2018, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Dobyan, D.C.; Levi, J.; Jacobs, C.; Kosek, J.; Weiner, M.W. Mechanism of cis-platinum nephrotoxicity: II. Morphologic ob-servations. J. Pharmacol. Exp. Ther. 1980, 213, 551–556. [Google Scholar]
- Zhang, L.; Hanigan, M.H. Role of CysteineS-Conjugate β-Lyase in the Metabolism of Cisplatin. J. Pharmacol. Exp. Ther. 2003, 306, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, E.M.; Mieyal, J.J. Protein-Thiol Oxidation and Cell Death: Regulatory Role of Glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Hanigan, M.H. Gamma-Glutamyl Transpeptidase. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar] [CrossRef] [Green Version]
- Crook, T.R.; Souhami, R.L.; Whyman, G.D.; McLean, A. Glutathione depletion as a determinant of sensitivity of human leukemia cells to cyclophosphamide. Cancer Res. 1986, 46, 5035–5038. [Google Scholar] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bushau-Sprinkle, A.M.; Barati, M.T.; Zheng, Y.; Watson, W.H.; Gagnon, K.B.; Khundmiri, S.J.; Kitterman, K.T.; Clark, B.J.; Siskind, L.J.; Doll, M.A.; et al. Na/H Exchange Regulatory Factor 1 Deficient Mice Show Evidence of Oxidative Stress and Altered Cisplatin Pharmacokinetics. Antioxidants 2021, 10, 1036. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10071036
Bushau-Sprinkle AM, Barati MT, Zheng Y, Watson WH, Gagnon KB, Khundmiri SJ, Kitterman KT, Clark BJ, Siskind LJ, Doll MA, et al. Na/H Exchange Regulatory Factor 1 Deficient Mice Show Evidence of Oxidative Stress and Altered Cisplatin Pharmacokinetics. Antioxidants. 2021; 10(7):1036. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10071036
Chicago/Turabian StyleBushau-Sprinkle, Adrienne M., Michelle T. Barati, Yuxuan Zheng, Walter H. Watson, Kenneth B. Gagnon, Syed Jalal Khundmiri, Kathleen T. Kitterman, Barbara J. Clark, Leah J. Siskind, Mark A. Doll, and et al. 2021. "Na/H Exchange Regulatory Factor 1 Deficient Mice Show Evidence of Oxidative Stress and Altered Cisplatin Pharmacokinetics" Antioxidants 10, no. 7: 1036. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10071036