
Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path?



Abstract
:1. Introduction
2. Insulin-Signaling Impairment and Neurodegeneration
2.1. Amyloidogenic Links between T2DM and AD
2.2. GSK-3β an Important Kinase for Insulin-Signaling Pathway and Phosphorylation of Tau Protein
3. Inflammation as One Common Mechanism for Insulin Dysregulation and Neurodegeneration
4. Oxidative Stress in Diabetes and Alzheimer’s Disease
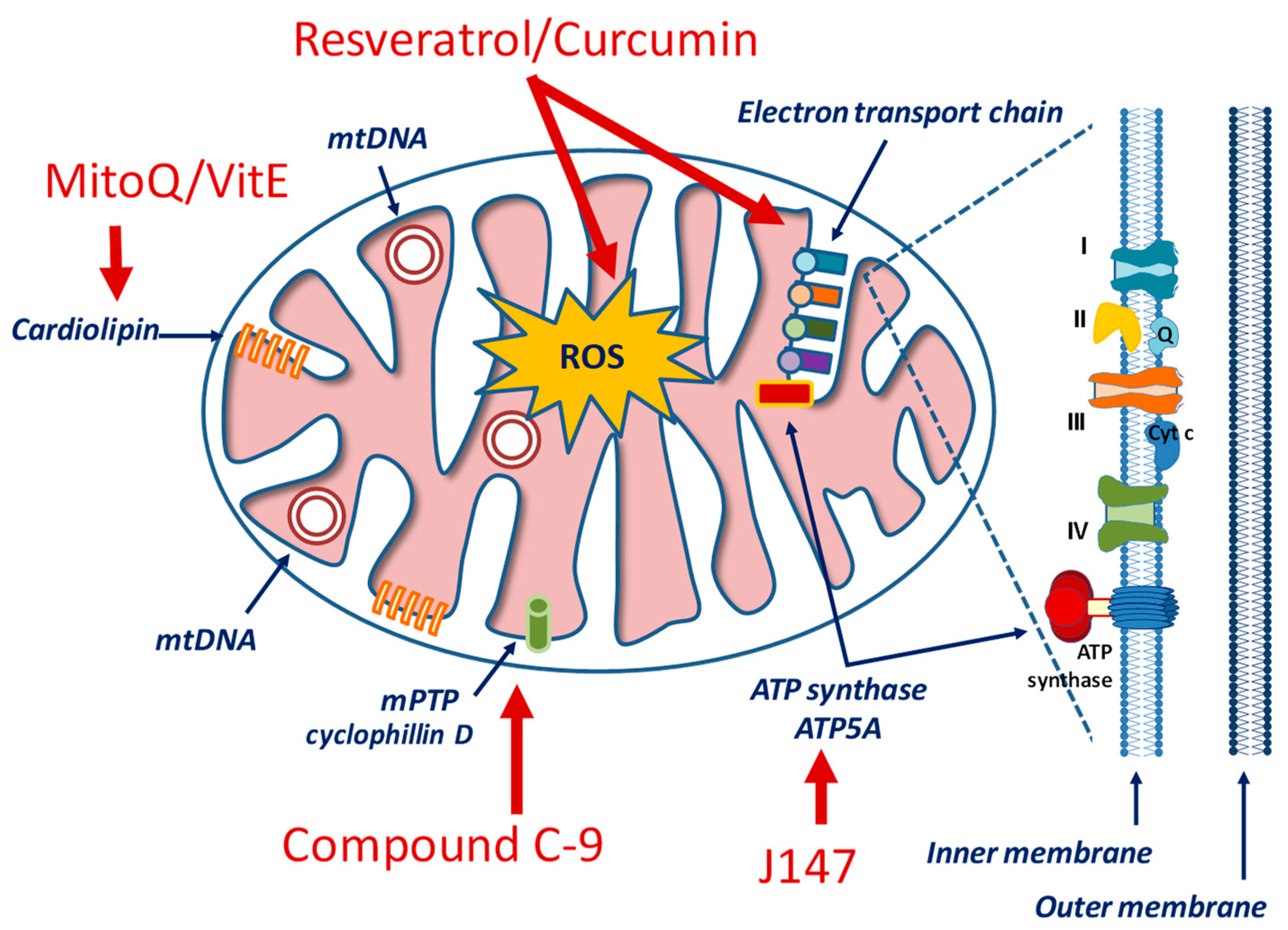
5. Could Mitochondrial Dysfunction Represent the Link between T2DM and AD?
6. Insights from Treatment Approaches for Both AD and T2DM
6.1. Ketogenic Diet (KD)
6.2. Antidiabetic Drugs
6.3. GSK-3β Inhibitors
6.4. Antioxidant Compounds
6.5. Mitochondria-Targeted Drugs
7. Therapeutic Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Umegaki, H. Type 2 diabetes as a risk factor for cognitive impairment: Current insights. Clin. Interv. Aging 2014, 9, 1011–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biessels, G.J.; Whitmer, R.A. Cognitive dysfunction in diabetes: How to implement emerging guidelines. Diabetologia 2019, 63, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF diabetes atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 2017, 66, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.K.; Vidoni, E.D.; Honea, R.A.; Burns, J.M. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol. Aging 2014, 35, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Xu, W.; Ou, Y.N.; Cao, X.P.; Tan, M.S.; Tan, L.; Yu, J.T. Diabetes mellitus and risks of cognitive impairment and dementia: A systematic review and meta-analysis of 144 prospective studies. Ageing Res. Rev. 2019, 55, 100944. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama study. Neurology 2010, 75, 764–770. [Google Scholar] [CrossRef]
- Crane, P.K.; Walker, R.; Hubbard, R.A.; Li, G.; Nathan, D.M.; Zheng, H.; Haneuse, S.; Craft, S.; Montine, T.J.; Kahn, S.E.; et al. Glucose levels and risk of dementia. N. Engl. J. Med. 2013, 369, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Sgarra, L.; Addabbo, F.; Potenza, M.A.; Montagnani, M. Determinants of evolving metabolic and cardiovascular benefit/risk profiles of rosiglitazone therapy during the natural history of diabetes: Molecular mechanisms in the context of integrated pathophysiology. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1171–E1182. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.Q.; Townsend, M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1792, 482–496. [Google Scholar] [CrossRef] [Green Version]
- Feinkohl, I.; Price, J.F.; Strachan, M.W.; Frier, B.M. The impact of diabetes on cognitive decline: Potential vascular, metabolic, and psychosocial risk factors. Alzheimers Res. Ther. 2015, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- S-Roriz-Filho, J.; Sa-Roriz, T.M.; Rosset, I.; Camozzato, A.L.; Santos, A.C.; Chaves, M.L.; Moriguti, J.C.; Roriz-Cruz, M. (Pre)diabetes, brain aging, and cognition. Biochim. Biophys. Acta 2009, 1792, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D. A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [Green Version]
- Anor, C.J.; O’Connor, S.; Saund, A.; Tang-Wai, D.F.; Keren, R.; Tartaglia, M.C. Neuropsychiatric symptoms in Alzheimer disease, vascular dementia, and mixed dementia. Neurodegener. Dis. 2017, 17, 127–134. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004457. [Google Scholar] [CrossRef]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373–379. [Google Scholar] [PubMed]
- Denver, P.; McClean, P.L. Distinguishing normal brain aging from the development of Alzheimer’s disease: Inflammation, insulin signaling and cognition. Neural Regen Res. 2018, 13, 1719–1730. [Google Scholar] [CrossRef]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications. J. Clin. Investig. 2013, 123, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.Y.; Wang, Z.J.; Holscher, C.; Yuan, L.; Zhang, J.; Sun, P.; Li, J.; Yang, W.; Wu, M.N.; Qi, J.S. Lixisenatide attenuates the detrimental effects of amyloid beta protein on spatial working memory and hippocampal neurons in rats. Behav. Brain Res. 2017, 318, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Zorzano, A.; Liesa, M.; Palacin, M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int. J. Biochem. Cell Bio. 2009, 41, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial dynamics in diabetes. Antioxid. Redox Signal. 2010, 14, 439–457. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Ravipati, A.; Nimmagadda, V.; Schubert, M.; Castellani, R.J.; Russell, J.W. Potential roles of PINK1 for increased PGC-1alpha-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion 2014, 18, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addabbo, F.; Montagnani, M.; Goligorsky, M.S. Mitochondria and reactive oxygen species. Hypertension 2009, 53, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, P.I.; Santos, M.S.; Seica, R.; Oliveira, C.R. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J. Neurol. Sci. 2007, 257, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Fang, D.; Swerdlow, R.H.; Yu, H.; Chen, J.X.; Yan, S.S. Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers Dis. 2016, 54, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Sheng, B.; Gong, K.; Niu, Y.; Liu, L.; Yan, Y.; Lu, G.; Zhang, L.; Hu, M.; Zhao, N.; Zhang, X.; et al. Inhibition of gamma-secretase activity reduces Abeta production, reduces oxidative stress, increases mitochondrial activity and leads to reduced vulnerability to apoptosis: Implications for the treatment of Alzheimer’s disease. Free Radic. Biol. Med. 2009, 46, 1362–1375. [Google Scholar] [CrossRef] [PubMed]
- Flannery, P.J.; Trushina, E. Mitochondrial dysfunction in Alzheimer’s disease and progress in mitochondria-targeted therapeutics. Curr. Behav. Neurosci. Rep. 2019, 6, 88–102. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The link between type 2 diabetes and neurodegeneration: Roles for amyloid-beta, amylin, and tau Proteins. J. Alzheimers Dis. 2017, 59, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Huang, N.Q.; Yan, F.; Jin, H.; Zhou, S.Y.; Shi, J.S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3beta as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar]
- Lee, S.H.; Zabolotny, J.M.; Huang, H.; Lee, H.; Kim, Y.B. Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol. Metab. 2016, 5, 589–601. [Google Scholar] [CrossRef]
- Broughton, S.; Partridge, L. Insulin/IGF-like signalling, the central nervous system and aging. Biochem. J. 2009, 418, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Corp, E.S.; Woods, S.C.; Porte, D., Jr.; Dorsa, D.M.; Figlewicz, D.P.; Baskin, D.G. Localization of 125I-insulin binding sites in the rat hypothalamus by quantitative autoradiography. Neurosci. Lett. 1986, 70, 17–22. [Google Scholar] [CrossRef]
- Werther, G.A.; Hogg, A.; Oldfield, B.J.; McKinley, M.J.; Figdor, R.; Allen, A.M.; Mendelsohn, F.A. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 1987, 121, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, F.; Toth, C. Insulin and the brain. Curr. Diabetes Rev. 2013, 9, 102–116. [Google Scholar]
- Woods, S.C.; Seeley, R.J.; Baskin, D.G.; Schwartz, M.W. Insulin and the blood-brain barrier. Curr. Pharm. Des. 2003, 9, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.; Alves, C.J.; Neto, E.; Lamghari, M. Communication from the periphery to the hypothalamus through the blood-brain barrier: An in vitro platform. Int. J. Pharm. 2016, 499, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilliox, L.A.; Chadrasekaran, K.; Kwan, J.Y.; Russell, J.W. Diabetes and cognitive impairment. Curr. Diab. Rep. 2016, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, I.; Tweedie, D.; Li, Y.; Greig, N.H. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br. J. Pharmacol. 2012, 166, 1586–1599. [Google Scholar] [CrossRef] [Green Version]
- Akintola, A.A.; van Heemst, D. Insulin, aging, and the brain: Mechanisms and implications. Front. Endocrinol. 2015, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- de la Monte, S.M. Relationships between diabetes and cognitive impairment. Endocrinol. Metab. Clin. N. Am. 2014, 43, 245–267. [Google Scholar] [CrossRef] [Green Version]
- Burillo, J.; Marques, P.; Jimenez, B.; Gonzalez-Blanco, C.; Benito, M.; Guillen, C. Insulin resistance and diabetes mellitus in Alzheimer’s disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef]
- Koekkoek, P.S.; Kappelle, L.J.; van den Berg, E.; Rutten, G.E.; Biessels, G.J. Cognitive function in patients with diabetes mellitus: Guidance for daily care. Lancet Neurol. 2015, 14, 329–340. [Google Scholar] [CrossRef]
- He, J.T.; Zhao, X.; Xu, L.; Mao, C.Y. Vascular risk factors and Alzheimer’s disease: Blood-brain barrier disruption, metabolic syndromes, and molecular links. J. Alzheimers Dis. 2020, 73, 39–58. [Google Scholar] [CrossRef]
- Potenza, M.A.; Addabbo, F.; Montagnani, M. Vascular actions of insulin with implications for endothelial dysfunction. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E568–E577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potenza, M.A.; Nacci, C.; Gagliardi, S.; Montagnani, M. Cardiovascular complications in diabetes: Lessons from animal models. Curr. Med. Chem. 2011, 18, 1806–1819. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abner, E.L.; Nelson, P.T.; Kryscio, R.J.; Schmitt, F.A.; Fardo, D.W.; Woltjer, R.L.; Cairns, N.J.; Yu, L.; Dodge, H.H.; Xiong, C.; et al. Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement. 2016, 12, 882–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chornenkyy, Y.; Wang, W.X.; Wei, A.; Nelson, P.T. Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 2019, 29, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukazawa, R.; Hanyu, H.; Sato, T.; Shimizu, S.; Koyama, S.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Subgroups of Alzheimer’s disease associated with diabetes mellitus based on brain imaging. Dement. Geriatr. Cogn. Disord. 2013, 35, 280–290. [Google Scholar] [CrossRef]
- de Matos, A.M.; de Macedo, M.P.; Rauter, A.P. Bridging type 2 diabetes and Alzheimer’s disease: Assembling the puzzle pieces in the quest for the molecules with therapeutic and preventive potential. Med. Res. Rev. 2018, 38, 261–324. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Ann. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef]
- Wijesekara, N.; Ahrens, R.; Sabale, M.; Wu, L.; Ha, K.; Verdile, G.; Fraser, P.E. Amyloid-beta and islet amyloid pathologies link Alzheimer’s disease and type 2 diabetes in a transgenic model. FASEB J. 2017, 31, 5409–5418. [Google Scholar] [CrossRef] [Green Version]
- Westermark, G.T.; Westermark, P.; Berne, C.; Korsgren, O. Widespread amyloid deposition in transplanted human pancreatic islets. N. Engl. J. Med. 2008, 359, 977–979. [Google Scholar] [CrossRef]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawver, J.N.; Ghiwot, Y.; Koola, C.; Carrera, W.; Rodriguez-Rivera, J.; Hernandez, C.; Dineley, K.T.; Kong, Y.; Li, J.; Jhamandas, J.; et al. Islet Amyloid Polypeptide (IAPP): A second amyloid in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Eanes, E.D.; Wiley, C.A. Amyloid fibrils formed from a segment of the pancreatic islet amyloid protein. Biochem. Biophys. Res. Commun. 1988, 155, 608–614. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, O.; Hohn, A.; Grune, T.; Pfeiffer, A.F.; Rudovich, N. Insulin-degrading enzyme: New therapeutic target for diabetes and Alzheimer’s disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef]
- Zimbone, S.; Monaco, I.; Giani, F.; Pandini, G.; Copani, A.G.; Giuffrida, M.L.; Rizzarelli, E. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 2018, 17, e12684. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin resistance as a link between amyloid-beta and tau pathologies in Alzheimer’s disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; Van Bronswijk, W.; Martins, R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Kamat, P.K.; Rai, S.; Swarnkar, S.; Shukla, R.; Ali, S.; Najmi, A.K.; Nath, C. Okadaic acid-induced Tau phosphorylation in rat brain: Role of NMDA receptor. Neuroscience 2013, 238, 97–113. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6, F1000. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- Pap, M.; Cooper, G.M. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef] [Green Version]
- Krishnankutty, A.; Kimura, T.; Saito, T.; Aoyagi, K.; Asada, A.; Takahashi, S.I.; Ando, K.; Ohara-Imaizumi, M.; Ishiguro, K.; Hisanaga, S.I. In vivo regulation of glycogen synthase kinase 3beta activity in neurons and brains. Sci. Rep. 2017, 7, 8602. [Google Scholar] [CrossRef] [Green Version]
- Leroy, A.; Landrieu, I.; Huvent, I.; Legrand, D.; Codeville, B.; Wieruszeski, J.M.; Lippens, G. Spectroscopic studies of GSK3{beta} phosphorylation of the neuronal tau protein and its interaction with the N-terminal domain of apolipoprotein E. J. Biol. Chem. 2010, 285, 33435–33444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, S141–S144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Begum, A.N.; Jones, M.R.; Oh, M.S.; Beech, W.K.; Beech, B.H.; Yang, F.; Chen, P.; Ubeda, O.J.; Kim, P.C.; et al. GSK3 inhibitors show benefits in an Alzheimer’s Disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 2009, 33, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.L.; Cosseau, C.; Gardy, J.L.; Hancock, R.E. Complexities of targeting innate immunity to treat infection. Trends Immunol. 2007, 28, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Potenza, M.A.; Nacci, C.; De Salvia, M.A.; Sgarra, L.; Collino, M.; Montagnani, M. Targeting endothelial metaflammation to counteract diabesity cardiovascular risk: Current and perspective therapeutic options. Pharmacol. Res. 2017, 120, 226–241. [Google Scholar] [CrossRef]
- Akash, M.S.; Rehman, K.; Chen, S. Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J. Cell Biochem. 2013, 114, 525–531. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Banks, W.A. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr. Pharm. Des. 2005, 11, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Gayral, S.; Garnotel, R.; Castaing-Berthou, A.; Blaise, S.; Fougerat, A.; Berge, E.; Montheil, A.; Malet, N.; Wymann, M.P.; Maurice, P.; et al. Elastin-derived peptides potentiate atherosclerosis through the immune Neu1-PI3Kgamma pathway. Cardiovasc. Res. 2014, 102, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolov, A.; Tsinlikov, I.; Nicoloff, G.; Tsinlikova, I.; Blazhev, A.; Garev, A. Abnormal levels of age-elastin derived peptides in sera of diabetic patients with arterial hypertension. Cent. Eur. J. Immunol. 2014, 39, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Degan, D.; Ornello, R.; Tiseo, C.; Carolei, A.; Sacco, S.; Pistoia, F. The Role of Inflammation in Neurological Disorders. Curr. Pharm. Des. 2018, 24, 1485–1501. [Google Scholar] [CrossRef] [PubMed]
- Blaise, S.; Romier, B.; Kawecki, C.; Ghirardi, M.; Rabenoelina, F.; Baud, S.; Duca, L.; Maurice, P.; Heinz, A.; Schmelzer, C.E.; et al. Elastin-derived peptides are new regulators of insulin resistance development in mice. Diabetes 2013, 62, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and inflammatory adaptation of reactive astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef] [PubMed]
- Szychowski, K.A.; Gminski, J. The elastin-derived peptide VGVAPG does not activate the inflammatory process in mouse cortical astrocytes in vitro. Neurotox. Res. 2020, 37, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Park, Y.; Zhang, H.; Xu, X.; Laine, G.A.; Dellsperger, K.C.; Zhang, C. Feed-forward signaling of TNF-alpha and NF-kappaB via IKK-beta pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1850–H1858. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D. PKR-dependent inflammatory signals. Sci. Signal. 2012, 5, pe47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Salvioli, S.; Capri, M.; Valensin, S.; Tieri, P.; Monti, D.; Ottaviani, E.; Franceschi, C. Inflamm-aging, cytokines and aging: State of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr. Pharm. Des. 2006, 12, 3161–3171. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Ledo, J.H.; Barbosa, G.; Sobrinho, M.; Diniz, L.; Fonseca, A.C.; Gomes, F.; Romao, L.; Lima, F.R.; Palhano, F.L.; et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. 2013, 4, e789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.M.; Bowers, W.J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal. 2010, 22, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Ruan, L.; Kang, Z.; Pei, G.; Le, Y. Amyloid deposition and inflammation in APPswe/PS1dE9 mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 531–540. [Google Scholar] [CrossRef]
- Potenza, M.A.; Nacci, C.; Carratu, M.R. Alzheimer’s disease murine models: Focus on late-onset disease. Drugs Future 2020, 45, 109–117. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermejo, P.; Martin-Aragon, S.; Benedi, J.; Susin, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer’s disease. Immunol. Lett. 2008, 117, 198–202. [Google Scholar] [CrossRef]
- Tan, Z.S.; Beiser, A.S.; Vasan, R.S.; Roubenoff, R.; Dinarello, C.A.; Harris, T.B.; Benjamin, E.J.; Au, R.; Kiel, D.P.; Wolf, P.A.; et al. Inflammatory markers and the risk of Alzheimer disease: The Framingham Study. Neurology 2007, 68, 1902–1908. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, F.; Klann, E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 2004, 3, 431–443. [Google Scholar] [CrossRef]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.P.; Zhu, X.; Perry, G.; Smith, M.A. Oxidative stress in diabetes and Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Wijesekara, N.; Goncalves, R.A.; De Felice, F.G.; Fraser, P.E. Impaired peripheral glucose homeostasis and Alzheimer’s disease. Neuropharmacology 2018, 136, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Nikooyeh, B.; Neyestani, T.R. Oxidative stress, type 2 diabetes and vitamin D: Past, present and future. Diabetes Metab. Res. Rev. 2015, 32, 260–267. [Google Scholar] [CrossRef]
- Ceretta, L.B.; Reus, G.Z.; Abelaira, H.M.; Ribeiro, K.F.; Zappellini, G.; Felisbino, F.F.; Steckert, A.V.; Dal-Pizzol, F.; Quevedo, J. Increased oxidative stress and imbalance in antioxidant enzymes in the brains of alloxan-induced diabetic rats. Exp. Diabetes Res. 2012, 2012, 302682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Nolting, R.; Arnal, E.; Barcia, J.M.; Miranda, M.; Romero, F.J. Protection by DHA of early hippocampal changes in diabetes: Possible role of CREB and NF-kappaB. Neurochem. Res. 2012, 37, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative stress and beta-amyloid protein in Alzheimer’s disease. Neuromolecular Med. 2011, 13, 223–250. [Google Scholar] [CrossRef]
- Jo, D.G.; Arumugam, T.V.; Woo, H.N.; Park, J.S.; Tang, S.C.; Mughal, M.; Hyun, D.H.; Park, J.H.; Choi, Y.H.; Gwon, A.R.; et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Oda, A.; Tamaoka, A.; Araki, W. Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J. Neurosci. Res. 2010, 88, 1137–1145. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Monteleone, D.; Tabaton, M. Amyloid-beta production: Major link between oxidative stress and BACE1. Neurotox. Res. 2012, 22, 208–219. [Google Scholar] [CrossRef]
- Filosto, M.; Scarpelli, M.; Cotelli, M.S.; Vielmi, V.; Todeschini, A.; Gregorelli, V.; Tonin, P.; Tomelleri, G.; Padovani, A. The role of mitochondria in neurodegenerative diseases. J. Neurol. 2011, 258, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Sohn, P.D.; Cho, S.H.; Swanson, R.A.; Gan, L. Sirtuins in neurodegenerative diseases: An update on potential mechanisms. Front. Aging Neurosci. 2013, 5, 53. [Google Scholar] [CrossRef] [Green Version]
- Herskovits, A.Z.; Guarente, L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013, 23, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesko, H.; Wencel, P.; Strosznajder, R.P.; Strosznajder, J.B. Sirtuins and their roles in brain aging and neurodegenerative disorders. Neurochem, Res. 2016, 42, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Y.; Gan, X.; Fang, D.; Zhong, C.; Wu, L.; Hu, G.; Sosunov, A.A.; McKhann, G.M.; Yu, H.; et al. Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes 2015, 64, 1728–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Schapira, A.H. Mitochondria and degenerative disorders. Am. J. Med. Genet. 2001, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef] [Green Version]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.S. Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid. Redox. Signal. 2012, 16, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Guo, L.; Lu, L.; Sun, H.; Shao, M.; Beck, S.J.; Li, L.; Ramachandran, J.; Du, Y.; Du, H. Synaptosomal mitochondrial dysfunction in 5xFAD mouse model of Alzheimer’s disease. PLoS ONE 2016, 11, e0150441. [Google Scholar] [CrossRef] [Green Version]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Parks, J.K.; Smith, T.S.; Trimmer, P.A.; Bennett, J.P., Jr.; Parker, W.D., Jr. Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 2001, 76, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Beal, F.M. Portal to Alzheimer’s disease. Nat. Med. 2008, 14, 1020–1021. [Google Scholar] [CrossRef] [Green Version]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Giuffrida Stella, A.M.; Bates, T.E.; Clark, J.B. Mitochondrial involvement in brain function and dysfunction: Relevance to aging, neurodegenerative disorders and longevity. Neurochem. Res. 2001, 26, 739–764. [Google Scholar] [CrossRef]
- Ke, Y.D.; Delerue, F.; Gladbach, A.; Gotz, J.; Ittner, L.M. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 2009, 4, e7917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Burdo, J.R.; Chen, Q.; Calcutt, N.A.; Schubert, D. The pathological interaction between diabetes and presymptomatic Alzheimer’s disease. Neurobiol. Aging 2009, 30, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Galloway, C.A.; Lee, H.; Nejjar, S.; Jhun, B.S.; Yu, T.; Hsu, W.; Yoon, Y. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes 2012, 61, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Stefanova, N.A.; Ershov, N.I.; Maksimova, K.Y.; Muraleva, N.A.; Tyumentsev, M.A.; Kolosova, N.G. The rat prefrontal-cortex transcriptome: Effects of aging and sporadic Alzheimer’s disease-like pathology. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Tyumentsev, M.A.; Stefanova, N.A.; Muraleva, N.A.; Rumyantseva, Y.V.; Kiseleva, E.; Vavilin, V.A.; Kolosova, N.G. Mitochondrial Dysfunction as a Predictor and Driver of Alzheimer’s Disease-Like Pathology in OXYS Rats. J. Alzheimers Dis. 2018, 63, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xiao, M.; Chang, L.; Yan, L.J. Role of insulin resistance in Alzheimer’s disease. Metab. Brain Dis. 2015, 30, 839–851. [Google Scholar] [CrossRef]
- Kim, D.I.; Lee, K.H.; Oh, J.Y.; Kim, J.S.; Han, H.J. Relationship between beta-amyloid and mitochondrial dynamics. Cell Mol. Neurobiol. 2017, 37, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.A.; Li, C.; Soleimanpour, S.A. Mitochondrial regulation of beta-cell function: Maintaining the momentum for insulin release. Mol. Aspects Med. 2015, 42, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civitarese, A.E.; MacLean, P.S.; Carling, S.; Kerr-Bayles, L.; McMillan, R.P.; Pierce, A.; Becker, T.C.; Moro, C.; Finlayson, J.; Lefort, N.; et al. Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metab. 2011, 11, 412–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Shulman, G.I. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005, 2, e233. [Google Scholar] [CrossRef] [PubMed]
- Befroy, D.E.; Petersen, K.F.; Dufour, S.; Mason, G.F.; de Graaf, R.A.; Rothman, D.L.; Shulman, G.I. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 2007, 56, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archuleta, T.L.; Lemieux, A.M.; Saengsirisuwan, V.; Teachey, M.K.; Lindborg, K.A.; Kim, J.S.; Henriksen, E.J. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: Role of p38 MAPK. Free Radic. Biol. Med. 2009, 47, 1486–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Nucleus-encoded regulators of mitochondrial function: Integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim. Biophys. Acta 2012, 1819, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Madsen-Bouterse, S.A.; Zhong, Q.; Mohammad, G.; Ho, Y.S.; Kowluru, R.A. Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radic Res. 2010, 44, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.Y.; Tomizawa, K. Functional loss of Cdkal1, a novel tRNA modification enzyme, causes the development of type 2 diabetes. Endocr. J. 2011, 58, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Coskun, P.E.; Wyrembak, J.; Derbereva, O.; Melkonian, G.; Doran, E.; Lott, I.T.; Head, E.; Cotman, C.W.; Wallace, D.C. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. J. Alzheimers Dis. 2010, 20, S293–S310. [Google Scholar] [CrossRef] [Green Version]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 2011, 17, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef]
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen claims aducanumab turnaround. Nat. Rev. Neurol. 2020, 16, 63–64. [Google Scholar] [CrossRef]
- Atri, A. Current and future treatments in Alzheimer’s disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef]
- Husna Ibrahim, N.; Yahaya, M.F.; Mohamed, W.; Teoh, S.L.; Hui, C.K.; Kumar, J. Pharmacotherapy of Alzheimer’s disease: Seeking clarity in a time of uncertainty. Front. Pharmacol. 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ulamek-Koziol, M.; Czuczwar, S.J. Ketogenic diet in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [Green Version]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 diabetes mellitus and Alzheimer’s disease: Role of insulin signaling and therapeutic implications. Int. J. Mol. Sci. 2016, 19, 3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Haj-ali, V.; Mohaddes, G.; Babri, S.H. Intracerebroventricular insulin improves spatial learning and memory in male Wistar rats. Behav. Neurosci. 2009, 123, 1309–1314. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Aleman, I. The many faces of insulin-like peptide signaling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Brooks, S.J.; Kullberg, J.; Burgos, J.; Kempton, M.J.; Nordenskjold, R.; Nylander, R.; Kilander, L.; Craft, S.; Larsson, E.M.; et al. Impaired insulin sensitivity as indexed by the HOMA score is associated with deficits in verbal fluency and temporal lobe gray matter volume in the elderly. Diabetes Care 2012, 35, 488–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, C.; Frey, W.H., II; Schioth, H.B.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp. Gerontol. 2011, 46, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: A pilot clinical trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freiherr, J.; Hallschmid, M.; Frey, W.H., II; Brunner, Y.F.; Chapman, C.D.; Holscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reger, M.A.; Watson, G.S.; Frey, W.H., II; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Drucker, D.J.; Sherman, S.I.; Gorelick, F.S.; Bergenstal, R.M.; Sherwin, R.S.; Buse, J.B. Incretin-based therapies for the treatment of type 2 diabetes: Evaluation of the risks and benefits. Diabetes Care 2010, 33, 428–433. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wang, L.X.; Chen, Z.; Liu, L.B. Liraglutide prevents beta-amyloid-induced neurotoxicity in SH-SY5Y cells via a PI3K-dependent signaling pathway. Neurol. Res. 2016, 38, 313–319. [Google Scholar] [CrossRef]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: Study protocol for a randomised controlled trial (ELAD study). Trials 2019, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Landreth, G. Therapeutic use of agonists of the nuclear receptor PPARgamma in Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 159–164. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risner, M.E.; Saunders, A.M.; Altman, J.F.; Ormandy, G.C.; Craft, S.; Foley, I.M.; Zvartau-Hind, M.E.; Hosford, D.A.; Roses, A.D. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharm. J. 2006, 6, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglietto-Vargas, D.; Shi, J.; Yaeger, D.M.; Ager, R.; LaFerla, F.M. Diabetes and Alzheimer’s disease crosstalk. Neurosci. Biobehav. Rev. 2016, 64, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Donovan, C.; Saunders, A.M.; Irizarry, M.; Jeter, B.; Zvartau-Hind, M.; van Dyck, C.H.; et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: Two phase 3 studies. Curr. Alzheimer Res. 2011, 8, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Tzimopoulou, S.; Cunningham, V.J.; Nichols, T.E.; Searle, G.; Bird, N.P.; Mistry, P.; Dixon, I.J.; Hallett, W.A.; Whitcher, B.; Brown, A.P.; et al. A multi-center randomized proof-of-concept clinical trial applying [(1)(8)F]FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Wallach, J.D.; Wang, K.; Zhang, A.D.; Cheng, D.; Grossetta Nardini, H.K.; Lin, H.; Bracken, M.B.; Desai, M.; Krumholz, H.M.; Ross, J.S. Updating insights into rosiglitazone and cardiovascular risk through shared data: Individual patient and summary level meta-analyses. BMJ 2020, 368, l7078. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Li, X.; Blanchard, J.; Li, Y.; Iqbal, K.; Liu, F.; Gong, C.X. Insulin sensitizers improve learning and attenuate tau hyperphosphorylation and neuroinflammation in 3xTg-AD mice. J. Neural Transm. 2015, 122, 593–606. [Google Scholar] [CrossRef]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Hanyu, H.; Sato, T.; Kiuchi, A.; Sakurai, H.; Iwamoto, T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J. Am. Geriatr. Soc. 2009, 57, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H.; Sato, T.; Sakurai, H.; Iwamoto, T. The role of tumor necrosis factor-alpha in cognitive improvement after peroxisome proliferator-activator receptor gamma agonist pioglitazone treatment in Alzheimer’s disease. J. Am. Geriatr. Soc. 2010, 58, 1000–1001. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Rosenblat, J.D.; Brietzke, E.; Park, C.; Lee, Y.; Musial, N.; Pan, Z.; Mansur, R.B.; McIntyre, R.S. Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: A systematic review and network meta-analysis. Diabetes Obes. Metab. 2018, 20, 2467–2471. [Google Scholar] [CrossRef]
- Hildreth, K.L.; Van Pelt, R.E.; Moreau, K.L.; Grigsby, J.; Hoth, K.F.; Pelak, V.; Anderson, C.A.; Parnes, B.; Kittelson, J.; Wolfe, P.; et al. Effects of pioglitazone or exercise in older adults with mild cognitive impairment and insulin resistance: A pilot study. Dement. Geriatr. Cogn. Dis. Extra 2015, 5, 51–63. [Google Scholar] [CrossRef]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, Y.P.; Miao, R.J.; Lin, N.N.; Gao, L. Risk factors for dementia with type 2 diabetes mellitus among elderly people in China. Age Ageing 2013, 42, 398–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolosa, E.; Litvan, I.; Hoglinger, G.U.; Burn, D.; Lees, A.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; Del Ser, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014, 29, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.M.; Fuertes, A.; Orozco, L.; del Monte-Millan, M.; Delgado, E.; Medina, M. Evidence for irreversible inhibition of glycogen synthase kinase-3beta by tideglusib. J. Biol. Chem. 2012, 287, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sereno, L.; Coma, M.; Rodriguez, M.; Sanchez-Ferrer, P.; Sanchez, M.B.; Gich, I.; Agullo, J.M.; Perez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Luna-Medina, R.; Cortes-Canteli, M.; Sanchez-Galiano, S.; Morales-Garcia, J.A.; Martinez, A.; Santos, A.; Perez-Castillo, A. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: Potential therapeutic role in brain disorders. J. Neurosci. 2007, 27, 5766–5776. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-mediated activation of GSK3beta promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 2017, 57, 75–83. [Google Scholar] [CrossRef]
- Darenskaya, M.A.; Kolesnikova, L.I.; Kolesnikov, S.I. Oxidative stress: Pathogenetic role in diabetes mellitus and its complications and therapeutic approaches to correction. Bull. Exp. Biol. Med. 2021, 171, 179–189. [Google Scholar] [CrossRef]
- Fracassi, A.; Marcatti, M.; Zolochevska, O.; Tabor, N.; Woltjer, R.; Moreno, S.; Taglialatela, G. Oxidative damage and antioxidant response in frontal cortex of demented and nondemented individuals with Alzheimer’s neuropathology. J. Neurosci. 2021, 41, 538–554. [Google Scholar] [CrossRef]
- Reddy, V.P.; Beyaz, A. Inhibitors of the Maillard reaction and AGE breakers as therapeutics for multiple diseases. Drug Discov. Today 2006, 11, 646–654. [Google Scholar] [CrossRef]
- Zheng, Q.; Kebede, M.T.; Kemeh, M.M.; Islam, S.; Lee, B.; Bleck, S.D.; Wurfl, L.A.; Lazo, N.D. Inhibition of the self-assembly of abeta and of tau by polyphenols: Mechanistic studies. Molecules 2019, 24, 2316. [Google Scholar] [CrossRef] [Green Version]
- Ladiwala, A.R.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Abeta into off-pathway conformers. J. Biol. Chem. 2010, 285, 24228–24237. [Google Scholar] [CrossRef] [Green Version]
- Sarubbo, F.; Esteban, S.; Miralles, A.; Moranta, D. Effects of resveratrol and other polyphenols on Sirt1: Relevance to brain function during aging. Curr. Neuropharmacol. 2018, 16, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective mechanisms of resveratrol in Alzheimer’s disease: Role of SIRT1. Oxid Med. Cell Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.J.T.; Bagit, A.; MacPherson, R.E.K. Resveratrol, metabolic dysregulation, and Alzheimer’s disease: Considerations for neurogenerative disease. Int J. Mol. Sci 2021, 22, 4628. [Google Scholar] [CrossRef] [PubMed]
- Lolicato, F.; Raudino, A.; Milardi, D.; La Rosa, C. Resveratrol interferes with the aggregation of membrane-bound human-IAPP: A molecular dynamics study. Eur. J. Med. Chem. 2015, 92, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Khazaei, M.; Karimi, J.; Sheikh, N.; Goodarzi, M.T.; Saidijam, M.; Khodadadi, I.; Moridi, H. Effects of resveratrol on receptor for advanced glycation end products (RAGE) expression and oxidative stress in the liver of rats with type 2 diabetes. Phytother. Res. 2016, 30, 66–71. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Santamaria, B.; Mas-Gutierrez, J.A.; Rada, P.; Fernandez-Millan, E.; Pardo, V.; Alvarez, C.; Cuadrado, A.; Ros, M.; Serrano, M.; et al. Resveratrol treatment restores peripheral insulin sensitivity in diabetic mice in a sirt1-independent manner. Mol. Nutr. Food Res. 2015, 59, 1431–1442. [Google Scholar] [CrossRef]
- Ozturk, E.; Arslan, A.K.K.; Yerer, M.B.; Bishayee, A. Resveratrol and diabetes: A critical review of clinical studies. Biomed. Pharm. 2017, 95, 230–234. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.J.T.; Frendo-Cumbo, S.; MacPherson, R.E.K. Resveratrol and metformin recover prefrontal cortex AMPK activation in diet-induced obese mice but reduce BDNF and synaptophysin protein content. J. Alzheimers Dis. 2019, 71, 945–956. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflammation 2017, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.C.; Zheng, B.W.; Guo, Y.; Zhao, J.; Zhao, J.Y.; Ma, X.W.; Jiang, Z.F. Antioxidative and neuroprotective effects of curcumin in an Alzheimer’s disease rat model co-treated with intracerebroventricular streptozotocin and subcutaneous D-galactose. J. Alzheimers Dis. 2016, 52, 899–911. [Google Scholar] [CrossRef]
- Thapa, A.; Jett, S.D.; Chi, E.Y. Curcumin attenuates amyloid-beta aggregate toxicity and modulates amyloid-beta aggregation pathway. ACS Chem. Neurosci. 2016, 7, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Bustanji, Y.; Taha, M.O.; Almasri, I.M.; Al-Ghussein, M.A.; Mohammad, M.K.; Alkhatib, H.S. Inhibition of glycogen synthase kinase by curcumin: Investigation by simulated molecular docking and subsequent in vitro/in vivo evaluation. J. Enzyme Inhib. Med. Chem. 2009, 24, 771–778. [Google Scholar] [CrossRef]
- Goozee, K.G.; Shah, T.M.; Sohrabi, H.R.; Rainey-Smith, S.R.; Brown, B.; Verdile, G.; Martins, R.N. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br. J. Nutr. 2016, 115, 449–465. [Google Scholar] [CrossRef]
- Rainey-Smith, S.R.; Brown, B.M.; Sohrabi, H.R.; Shah, T.; Goozee, K.G.; Gupta, V.B.; Martins, R.N. Curcumin and cognition: A randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br. J. Nutr. 2016, 115, 2106–2113. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzillotta, C.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. Targeting mitochondria in Alzheimer disease: Rationale and perspectives. CNS Drugs 2019, 33, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Geissler, A. Versatility of the mitochondrial protein import machinery. Nat. Rev. Mol. Cell Biol. 2001, 2, 339–349. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-targeted drugs. Curr. Mol. Pharmacol. 2018, 12, 202–214. [Google Scholar] [CrossRef]
- Goldberg, J.; Currais, A.; Prior, M.; Fischer, W.; Chiruta, C.; Ratliff, E.; Daugherty, D.; Dargusch, R.; Finley, K.; Esparza-Molto, P.B.; et al. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 2018, 17, e12715. [Google Scholar] [CrossRef]
- Zhang, S.; Hedskog, L.; Petersen, C.A.; Winblad, B.; Ankarcrona, M. Dimebon (latrepirdine) enhances mitochondrial function and protects neuronal cells from death. J. Alzheimers Dis. 2010, 21, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of mitochondrial dysfunction in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Shan, X.; Jones, B.A.; Zhao, G.; Woodward, M.L.; Li, X.; Zhu, Y.; McEachern, E.J.; Silverman, M.A.; Watson, N.V.; et al. Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol. Neurodegener. 2014, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covey, M.V.; Murphy, M.P.; Hobbs, C.E.; Smith, R.A.; Oorschot, D.E. Effect of the mitochondrial antioxidant, Mito Vitamin E, on hypoxic-ischemic striatal injury in neonatal rats: A dose-response and stereological study. Exp. Neurol. 2006, 199, 513–519. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 ameliorates synaptic depression, abeta deposition, and cognitive impairment in an Alzheimer’s disease model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.M.; Park, J.; Kim, S.H.; Jung, Y.K. Emerging perspectives on mitochondrial dysfunction and inflammation in Alzheimer’s disease. BMB Rep. 2020, 53, 35–46. [Google Scholar] [CrossRef]
- Valasani, K.R.; Sun, Q.; Fang, D.; Zhang, Z.; Yu, Q.; Guo, Y.; Li, J.; Roy, A.; ShiDu Yan, S. Identification of a small molecule cyclophilin D inhibitor for rescuing abeta-mediated mitochondrial dysfunction. ACS Med. Chem. Lett. 2016, 7, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| T2DM | Drugs | AD |
|---|---|---|
| ↑ glucose uptake and regulation | INSULIN | ↑ deactivation of GSK-3β ↑ Aβ clearance |
| ↑ insulin secretion | GLP1-RA (Exenatide-4, liraglutide, lixisenatide) | ↑ deactivation of GSK-3β ↓ neuronal Tau hyperphoshorylation |
| ↑ insulin sensitivity, ↑ transcription of insulin sensitive genes | TZDs (rosiglitazone, pioglitazone) | ↓ Aβ generation and deposition |
| ↑ insulin sensitivity | GSK-3β INHIBITORS (Tideglusib, NP12, TDZD-8) | ↑ deactivation of GSK-3β ↓ Tau hyperphosh ↓ Aβ deposition |
| ↓ RAGE expression ↑ glucose uptake ↑ insulin sensitivity | RESVERATROL | ↑ AMPK ↓ mTOR ↑autophagy ↓ Aβ deposition |
| ↑ insulin sensitivity ↓ glucose plasma levels | CURCUMIN | ↓ IAPP amyloid fibrils ↑ deactivation of GSK-3β |
| ↑ insulin sensitivity | ZERUMBONE | AChE inhibitor |
| ↓ intestinal glucose absorption | CAPSAICIN | ↓ RAGE activation ↓ blood–brain Aβ |
| ↓ free radicals | LYCOPENE | ↓ free radicals |
| J147 | ↑ AMPK ↓ mTOR ↑autophagy ↓ Aβ deposition | |
| THIAMET G | O-GlcNAcase inhibitor ↓ Aβ and Tau pathology | |
| MITOCHONDRIA ANTIOXIDANTS (MitoQ and MitoVitE) | ↓ free radicals ↓ Aβ neurotoxicity | |
| RAPAMYCIN | ↓ mTOR ↑autophagy/mitophagy | |
| LATREPIRDINE | ↑autophagy/mitophagy ↓Aβ toxicity | |
| NICOTINAMIDE | ↑ autophagy ↓Aβ and Tau pathology. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potenza, M.A.; Sgarra, L.; Desantis, V.; Nacci, C.; Montagnani, M. Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path? Antioxidants 2021, 10, 1257. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081257
Potenza MA, Sgarra L, Desantis V, Nacci C, Montagnani M. Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path? Antioxidants. 2021; 10(8):1257. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081257
Chicago/Turabian StylePotenza, Maria Assunta, Luca Sgarra, Vanessa Desantis, Carmela Nacci, and Monica Montagnani. 2021. "Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path?" Antioxidants 10, no. 8: 1257. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081257