
Two Birds One Stone: The Neuroprotective Effect of Antidiabetic Agents on Parkinson Disease—Focus on Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors

 , , , and
, , , and
Abstract
:1. Introduction
2. Mitochondria Biology and Oxidative Stress and Mitochondrial Dependent Cell Death
2.1. Mitochondrial Biology
2.2. Mitochondrial ROS
2.3. Mitochondria Dynamics, Autophagy, and Mitochondria Mediated Intrinsic Apoptosis
3. The Linkage between DM and PD
3.1. DM and PD Shared Common Pathogenesis- Mitochondrial Degeneration and Oxidative Damage
3.2. The Usage of Anti-Diabetes Agents in Slowing PD Progression
4. SGLT2 Inhibitors and PD
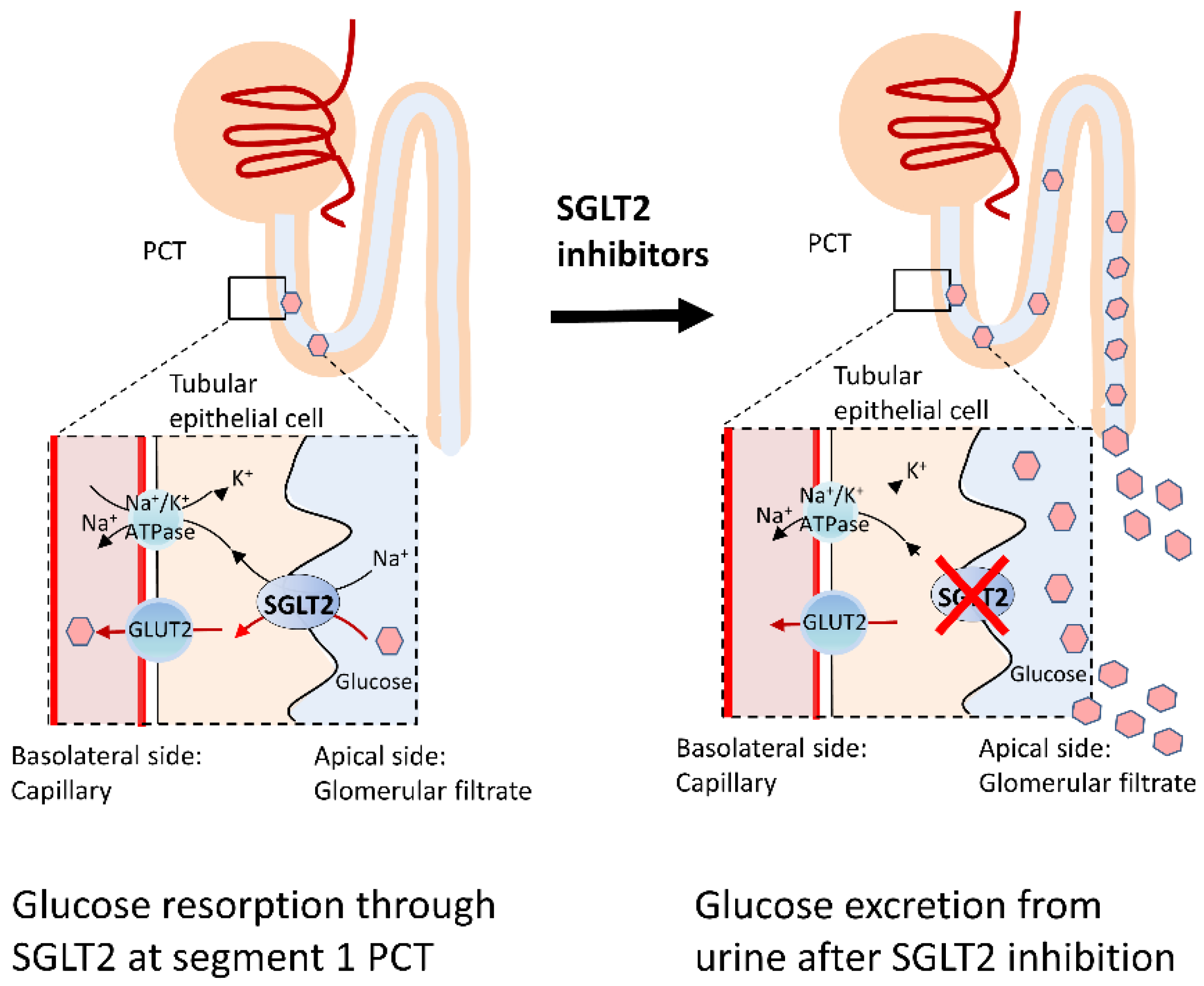
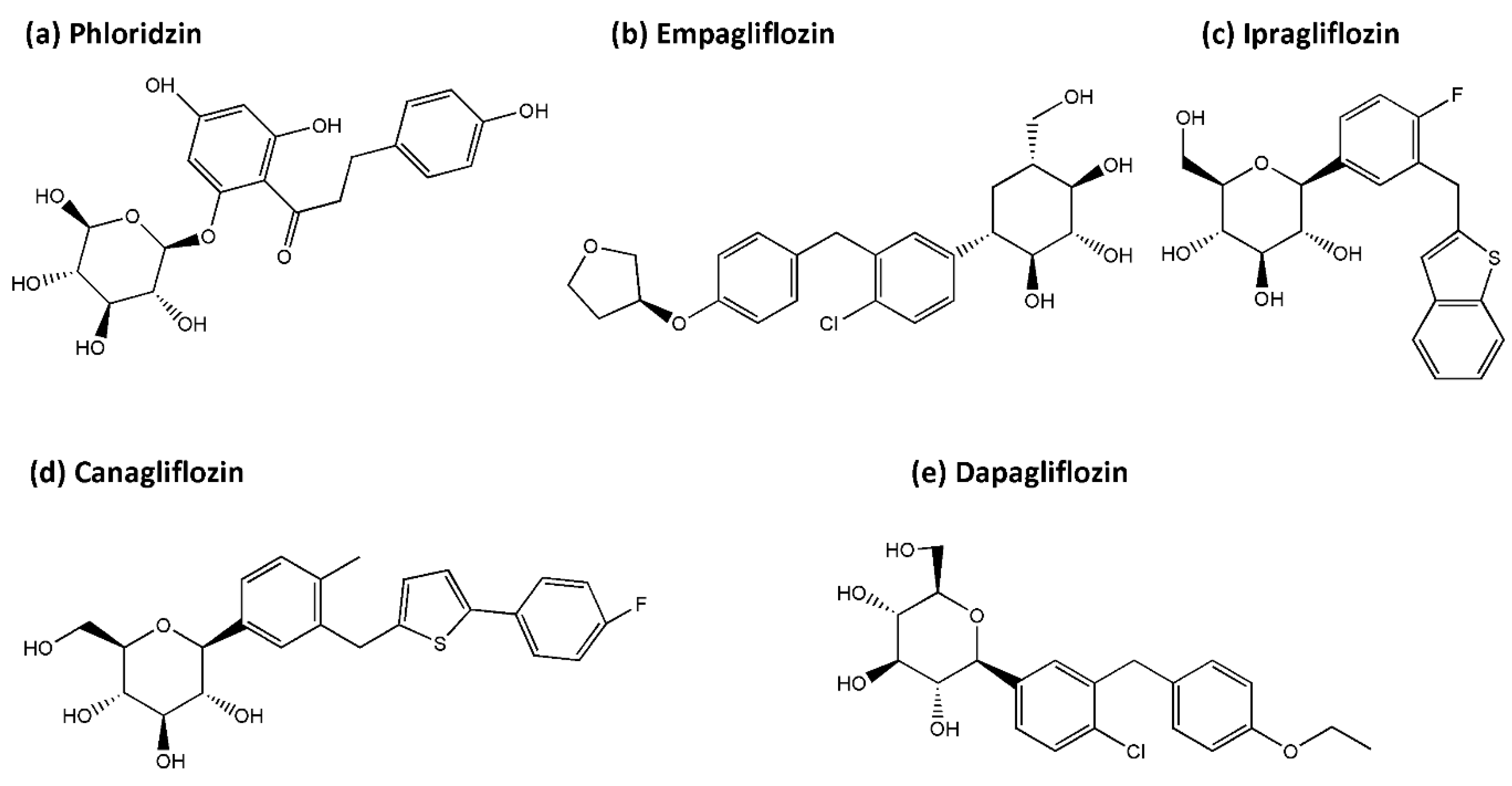
4.1. SGLT2 Inhibitors
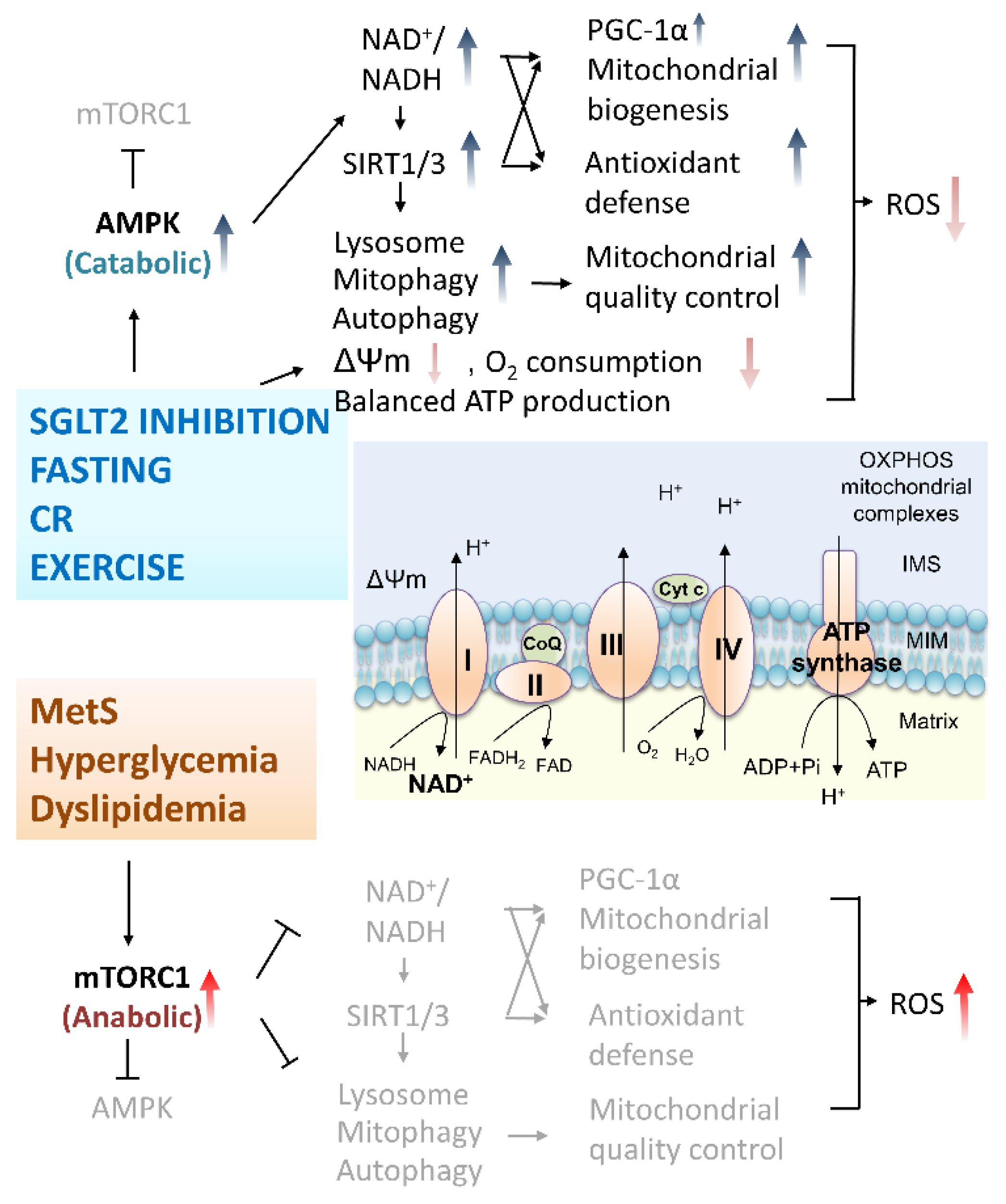
4.2. Protective Mechanisms of SGLT2 Inhibitors on PD and DM: Antioxidative Activities and Mitochondria Protection
4.3. Protective Evidence of SGLT2 Inhibitors for Complications of Metabolic Syndromes
4.4. Neuroprotection Provided by SGLT2 Inhibitors
4.5. Potential Usage of SGLT2 Inhibitor in PD Neuroprotective Strategy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SGLT2 Inhibitor/ Dose/Time | Model | Findings | Ref. |
|---|---|---|---|
| Empagliflozin/ 0.03% empagliflozin diet/10 wks | db/db mouse, obesity and T2DM model | Enhanced: BDNF in cerebral tissue, Reduced: cerebral oxidative stress, DNA oxidative damage Function: Slowed progression of cognitive impairment, | [175] Lin et al. |
| Empagliflozin/ 10 mg/kg/day/ 22 wks | APP/PS1xdb/db mice, Mixed AD and T2DM model | Reduced: brain atrophy, senile plaques, amyloid-β levels, Tau phosphorylation, hemorrhage density, microglia burden Function: Enhanced learning, memory | [176] Hierro-Bujalance et al. |
| Empagliflozin/ 10 mg/kg/day/ 10 wks | T2DM db/db and lean control female mice | Protects mice brain from severe T2DM-induced ultrastructural remodeling of the neurovascular unit | [185] Hayden et.al. |
| Empagliflozin/ 10 mg/kg/ 1 h and 24 h after reperfusion | Wistar rats hyperglycemic model + STZ (55 mg/kg)-induced I/R model | Reduced: cerebral infarct volume, neuroinflammation, oxidative stress, neuronal apoptosis Function: enhanced behavioral/ neurological functions | [177] Amin et al. |
| Empagliflozin/ 1 and 10 mg/kg/ 1 h and 24 h after reperfusion | Wistar rats, transient bilateral common carotid arteries occlusion induced I/R model | Enhanced: HIF-1α, VEGF Reduced: brain apoptotic cell death(↓caspase-3), infarct volume Function: Reduced motor dysfunction, neurological deficit * large dose with better neuroprotective effect | [186] Abdel-Latif et al. |
| Canagliflozin/ 10 mg/5mL/ vs. Galantamine/ 3 mg/5 mL/ 1st and 13th day | Wistar rats, Scopolamine hydrobromide (C17H21NO4·HBr) induced memory dysfunction model | ↓memory dysfunction (Y maze task) ↑hippocampus M1 mAChR | [179] Arafa et al. |
| Dapagliflozin/ 1 mg/kg/day/ vs. Vildagliptin/3 mg/kg/day/ 4 wks | Wistar rats, HFD model | Reduced: brain mt ROS production, ΔΨm change, mt swelling, neuroinflammation (↓p-NFκB, p65/ NFκB p65 ratio), neuronal apoptosis (↓Bax, Bcl2) Enhanced: brain mitochondrial function, p-IR and p-Akt/PKB ser473 Function: Both drugs: Enhanced insulin sensitivity, prevented cognitive decline. Only dapagliflozin improved hippocampal synaptic plasticity * the combination of two drugs has better effect than single therapies | [180] Sa-Nguanmoo et al. |
| Dapagliflozin/ 1 mg/kg/day/ 3 wks | Wistar rats, rotenone-induced PD model | Reduced: brain mt ROS production, α-synuclein expression, neuroinflammation (↓p-NFκB, p65/NFκB p65 ratio, TNF-α), striatal neuronal oxidative stress (↓DJ-1, Nrf2, HO-1, ↓GDNF, PI3K/AKT/GSK-3β), neuronal apoptosis (↓Bax, cleaved caspase 3) Preserved striatal dopaminergic neurons Function: Improved neurodegenerative aberrations/motor dysfunction | [181] Arab et al. |
| Dapagliflozin 75 and 150 mg/kg | Sprague–Dawley rats with pentylenetetrazol-induced seizures | ↓seizure activity (EEG SWP, RSS, TFMJ) * higher dose more effective | [182] Erdogan et al. |
| Clinical trial data | |||
| SGLT2 inhibitors | (a systematic review and meta-analysis that included 5 clinical trials) EMPA-REG, CANVAS, DECLARE-TIMI 58, CREDENCE and VERTIS CV | * potential protective effect against hemorrhagic stroke * neutral effect on the risk of fatal stroke, non-fatal stroke, ischemic stroke or transient ischemic attack) | [183] Tsai et al. |
| dapagliflozin | T2DM patients | cognition decline (Recruiting) | NCT04304261 |
5. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
| AD | Alzheimer’s disease |
| ADP | Adenosine diphosphate |
| AGEs | advanced glycation end products |
| AMPK | 5’-adenosine monophosphate (AMP)-activated protein kinase |
| APAF1 | Apoptotic peptidase-activating factor 1 |
| ATP | Adenosine triphosphate |
| ATP13A2 | Atpase type 13 a 2 |
| BAK | Bcl2-antagonist/killer |
| BAX | Bcl-2-associated x protein |
| Ca2+ | Calcium |
| CoQ | Co-enzyme q |
| CR | Caloric restriction |
| CVD | Cardiovascular diseases |
| DJ-1 | Daisuke-junko-1 |
| DPP-4 | dipeptidyl peptidase-4 |
| ETC | Electric transport chain |
| FADH2 | flavin adenine dinucleotide |
| FBXO7 | F-box only protein 7 |
| GLP-1RA | glucagon-like peptide-1 |
| GPX | glutathione peroxidase |
| GSH | Glutathione |
| HO• | hydroxy radical |
| IMS | Intermembrane space |
| LRRK2 | Leucine rich repeat kinase 2 |
| MELAS | mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome |
| Mfn-1 and 2 | Mitofusins 1 and 2 |
| MIM | Mitochondrial inner membrane |
| ΔΨm | Mitochondrial membrane potential |
| MOM | Mitochondrial outer membrane |
| MOMP | Mitochondrial outer membrane permeabilization |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mtDNA | Mitochondrial DNA |
| mTOR/mTORC1 | Mammalian target of rapamycin/mammalian target of rapamycin complex I |
| nDNA | Nucleus DNA |
| NADH | nicotinamide adenine dinucleotide |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| Nrf2 | nuclear factor erythroid 2–related factor 2 |
| OPA1 | optic atrophy 1 |
| OXPHOS | Oxidative phosphorylation |
| PD | Parkinson disease |
| PGC | Peroxisome proliferator-activated receptors (PPAR) γ coactivator |
| PINK1 | Phosphatase and tensin homologue (PTEN)-induced putative kinase 1 |
| PPAR | Peroxisome proliferator-activated receptor |
| ROS | Reactive oxygen species |
| rRNA | Ribosomal rna |
| SGLT2 | sodium–glucose cotransporter 2 |
| SOD | Superoxide dismutase |
| SIRT-1 | Sirtuin-1 |
| TCA | Tricarboxylic acid |
| TNF-α | tumor necrosis factor α |
| TRX | Thioredoxin |
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Richardson, R.J. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology 1998, 50, 1346–1350. [Google Scholar] [CrossRef]
- Nonnekes, J.; Post, B.; Tetrud, J.W.; Langston, J.W.; Bloem, B.R. MPTP-induced parkinsonism: An historical case series. Lancet Neurol. 2018, 17, 300–301. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Ball, N.; Teo, W.P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.K.; Lin, K.J.; Lin, K.L.; Liou, C.W.; Chen, S.D.; Chuang, Y.C.; Wang, P.W.; Chuang, J.H.; Wang, T.J. When Friendship Turns Sour: Effective Communication between Mitochondria and Intracellular Organelles in Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 607392. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.B.; Hanss, Z.; Kruger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef] [Green Version]
- Billingsley, K.J.; Barbosa, I.A.; Bandrés-Ciga, S.; Quinn, J.P.; Bubb, V.J.; Deshpande, C.; Botia, J.A.; Reynolds, R.H.; Zhang, D.; Simpson, M.A.; et al. Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Parkinson’s Dis. 2019, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, G.; deSouza, R.M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase Mutations in Parkinson Disease. J. Parkinson’s Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Li, H.; Yan, H.; Zhang, P.; Chang, L.; Li, T. Risk of Parkinson Disease in Diabetes Mellitus: An Updated Meta-Analysis of Population-Based Cohort Studies. Medicine 2016, 95, e3549. [Google Scholar] [CrossRef]
- De Pablo-Fernandez, E.; Goldacre, R.; Pakpoor, J.; Noyce, A.J.; Warner, T.T. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 2018, 91, e139–e142. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-W.; Hsieh, T.-F.; Li, C.-I.; Liu, C.-S.; Lin, W.-Y.; Chiang, J.-H.; Li, T.-C.; Lin, C.-C. Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Medicine 2017, 96, e5921. [Google Scholar] [CrossRef]
- Renaud, J.; Bassareo, V.; Beaulieu, J.; Pinna, A.; Schlich, M.; Lavoie, C.; Murtas, D.; Simola, N.; Martinoli, M.-G. Dopaminergic neurodegeneration in a rat model of long-term hyperglycemia: Preferential degeneration of the nigrostriatal motor pathway. Neurobiol. Aging 2018, 69, 117–128. [Google Scholar] [CrossRef]
- Sandyk, R. The relationship between diabetes mellitus and Parkinson’s disease. Int. J. Neurosci. 1993, 69, 125–130. [Google Scholar] [CrossRef]
- Cheong, J.L.Y.; de Pablo-Fernandez, E.; Foltynie, T.; Noyce, A.J. The Association between Type 2 Diabetes Mellitus and Parkinson’s Disease. J. Parkinson’s Dis. 2020, 10, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olgar, Y.; Tuncay, E.; Degirmenci, S.; Billur, D.; Dhingra, R.; Kirshenbaum, L.; Turan, B. Ageing-associated increase in SGLT2 disrupts mitochondrial/sarcoplasmic reticulum Ca(2+) homeostasis and promotes cardiac dysfunction. J. Cell. Mol. Med. 2020, 24, 8567–8578. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.J.H.; Foster-Davies, H.; Stephens, J.W. A systematic review examining the effects of sodium-glucose cotransporter-2 inhibitors (SGLT2is) on biomarkers of inflammation and oxidative stress. Diabetes Res. Clin. Pract. 2020, 168, 108368. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ni, Y.Q.; Zhan, J.K.; Liu, Y.S. The Role of SGLT2 Inhibitors in Vascular Aging. Aging Dis. 2021, 12, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.; Chau, Y.-Y.; Ng, H.-Y.; Chen, C.-H.; Wang, P.-W.; Liou, C.-W.; Lin, T.-K.; Chen, J.-B. Empagliflozin Protects HK-2 Cells from High Glucose-Mediated Injuries via a Mitochondrial Mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuszak, A.J.; Espey, M.G.; Falk, M.J.; Holmbeck, M.A.; Manfredi, G.; Shadel, G.S.; Vernon, H.J.; Zolkipli-Cunningham, Z. Nutritional Interventions for Mitochondrial OXPHOS Deficiencies: Mechanisms and Model Systems. Annu. Rev. Pathol. 2018, 13, 163–191. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Lyngsie, G.; Krumina, L.; Tunlid, A.; Persson, P. Generation of hydroxyl radicals from reactions between a dimethoxyhydroquinone and iron oxide nanoparticles. Sci. Rep. 2018, 8, 10834. [Google Scholar] [CrossRef] [Green Version]
- Pham, A.N.; Xing, G.; Miller, C.J.; Waite, T.D. Fenton-like copper redox chemistry revisited: Hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J. Catal. 2013, 301, 54–64. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chuang, J.H.; Wang, P.W.; Lin, T.K.; Wu, M.T.; Hsu, W.M.; Chuang, H.C. 5-aza-2’-Deoxycytidine Induces a RIG-I-Related Innate Immune Response by Modulating Mitochondria Stress in Neuroblastoma. Cells 2020, 9, 1920. [Google Scholar] [CrossRef]
- Han, W.; Fessel, J.P.; Sherrill, T.; Kocurek, E.G.; Yull, F.E.; Blackwell, T.S. Enhanced Expression of Catalase in Mitochondria Modulates NF-κB-Dependent Lung Inflammation through Alteration of Metabolic Activity in Macrophages. J. Immunol. 2020, 205, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Man, M.Q.; Abuabara, K.; Wakefield, J.S.; Sheu, H.M.; Tsai, J.C.; Lee, C.H.; Elias, P.M. By protecting against cutaneous inflammation, epidermal pigmentation provided an additional advantage for ancestral humans. Evol. Appl. 2019, 12, 1960–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Hoitzing, H.; Johnston, I.G.; Jones, N.S. What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. BioEssays 2015, 37, 687–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, T.B.; Sanchez-Guerrero, A.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; López-Doménech, G.; Halff, E.F.; Covill-Cooke, C.; Ivankovic, D.; Melandri, D.; Arancibia-Cárcamo, I.L.; Burden, J.J.; Lowe, A.R.; Kittler, J.T. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 2019, 10, 4399. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, C.; Ott, M.; Buttner, S. Mitochondria orchestrate proteostatic and metabolic stress responses. EMBO Rep. 2019, 20, e47865. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Suh, D.H.; Kim, M.-K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front. Oncol. 2013, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, M.Z.; Tabassum, H.; Parvez, S. Mitochondrial permeability transition pore: A promising target for the treatment of Parkinson’s disease. Protoplasma 2017, 254, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Shevtsova, E.F.; Maltsev, A.V.; Vinogradova, D.V.; Shevtsov, P.N.; Bachurin, S.O. Mitochondria as a promising target for developing novel agents for treating Alzheimer’s disease. Med. Res. Rev. 2021, 41, 803–827. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef]
- Wacquier, B.; Combettes, L.; Dupont, G. Dual dynamics of mitochondrial permeability transition pore opening. Sci. Rep. 2020, 10, 3924. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef]
- Lee, Y.; Chiou, Y.J.; Hung, C.F.; Chang, Y.Y.; Chen, Y.F.; Lin, T.K.; Wang, L.J. A dyadic study of psychological well-being of individuals with Parkinson’s disease and their caregivers. Sci. Rep. 2021, 11, 957. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, R.S. Progression and Prognosis in Parkinson’s Disease. J. Nerv. Ment. Dis. 1960, 130, 556–566. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schernhammer, E.; Hansen, J.; Rugbjerg, K.; Wermuth, L.; Ritz, B. Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care 2011, 34, 1102–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Park, Y.; Huang, X.; Hollenbeck, A.; Blair, A.; Schatzkin, A.; Chen, H. Diabetes and risk of Parkinson’s disease. Diabetes Care 2011, 34, 910–915. [Google Scholar] [CrossRef] [Green Version]
- Pagano, G.; Polychronis, S.; Wilson, H.; Giordano, B.; Ferrara, N.; Niccolini, F.; Politis, M. Diabetes mellitus and Parkinson disease. Neurology 2018, 90, e1654–e1662. [Google Scholar] [CrossRef]
- Ong, M.; Foo, H.; Chander, R.J.; Wen, M.-C.; Au, W.L.; Sitoh, Y.Y.; Tan, L.; Kandiah, N. Influence of diabetes mellitus on longitudinal atrophy and cognition in Parkinson’s disease. J. Neurol. Sci. 2017, 377, 122–126. [Google Scholar] [CrossRef]
- Marques, A.; Dutheil, F.; Durand, E.; Rieu, I.; Mulliez, A.; Fantini, M.L.; Boirie, Y.; Durif, F. Glucose dysregulation in Parkinson’s disease: Too much glucose or not enough insulin? Parkinsonism Relat. Disord. 2018, 55, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012, 2012, 384017. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the Brain: Its Pathophysiological Implications for States Related with Central Insulin Resistance, Type 2 Diabetes and Alzheimer’s Disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Young, W.S. Periventricular hypothalamic cells in the rat brain contain insulin mRNA. Neuropeptides 1986, 8, 93–97. [Google Scholar] [CrossRef]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Dorn, A.; Rinne, A.; Bernstein, H.G.; Hahn, H.J.; Ziegler, M. Insulin and C-peptide in human brain neurons (insulin/C-peptide/brain peptides/immunohistochemistry/radioimmunoassay). J. Fur Hirnforsch. 1983, 24, 495–499. [Google Scholar]
- Pagotto, U. Where does insulin resistance start? The brain. Diabetes Care 2009, 32 (Suppl. 2), S174–S177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Könner, A.C.; Brüning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef]
- Boden, G. Fatty acid—induced inflammation and insulin resistance in skeletal muscle and liver. Curr. Diabetes Rep. 2006, 6, 177–181. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Fiory, F.; Perruolo, G.; Cimmino, I.; Cabaro, S.; Pignalosa, F.C.; Miele, C.; Beguinot, F.; Formisano, P.; Oriente, F. The Relevance of Insulin Action in the Dopaminergic System. Front. Neurosci. 2019, 13, 868. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamada, T.; Tooyama, I.; Moroo, I.; Kimura, H.; Yamamoto, T.; Okada, H. Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 1996, 204, 201–204. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Chou, S.-Y.; Chan, L.; Chung, C.-C.; Chiu, J.-Y.; Hsieh, Y.-C.; Hong, C.-T. Altered Insulin Receptor Substrate 1 Phosphorylation in Blood Neuron-Derived Extracellular Vesicles from Patients with Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 1490. [Google Scholar] [CrossRef]
- Gao, S.; Duan, C.; Gao, G.; Wang, X.; Yang, H. Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int. J. Biochem. Cell Biol. 2015, 64, 25–33. [Google Scholar] [CrossRef]
- Hölscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Vidoni, E.D.; Perea, R.D.; Rada, R.; Johnson, D.K.; Lyons, K.; Pahwa, R.; Burns, J.M.; Honea, R.A. Insulin resistance and gray matter volume in neurodegenerative disease. Neuroscience 2014, 270, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Hogg, E.; Athreya, K.; Basile, C.; Tan, E.E.; Kaminski, J.; Tagliati, M. High Prevalence of Undiagnosed Insulin Resistance in Non-Diabetic Subjects with Parkinson’s Disease. J. Parkinson’s Dis. 2018, 8, 259–265. [Google Scholar] [CrossRef]
- Markaki, I.; Ntetsika, T.; Sorjonen, K.; Svenningsson, P.; BioPark Study Group. Euglycemia Indicates Favorable Motor Outcome in Parkinson’s Disease. Mov. Disord. 2021, 36, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, N.; Xue, W.; Long, Q.; Ownby, J.G.; Olson, D.E.; Barb, D.; Rhee, M.K.; Mohan, A.V.; Watson-Williams, P.I.; Jackson, S.L.; et al. Aging is associated with increased HbA1c levels, independently of glucose levels and insulin resistance, and also with decreased HbA1c diagnostic specificity. Diabet. Med. 2014, 31, 927–935. [Google Scholar] [CrossRef]
- Brunerova, L.; Potockova, J.; Horacek, J.; Suchy, J.; Andel, M. Central Dopaminergic Activity Influences Metabolic Parameters in Healthy Men. Neuroendocrinology 2013, 97, 132–138. [Google Scholar] [CrossRef]
- Glei, D.A.; Goldman, N.; Lin, Y.H.; Weinstein, M. Age-Related Changes in Biomarkers: Longitudinal Data from a Population-Based Sample. Res. Aging 2011, 33, 312–326. [Google Scholar] [CrossRef] [Green Version]
- De Mello, N.P.; Orellana, A.M.; Mazucanti, C.H.; de Morais Lima, G.; Scavone, C.; Kawamoto, E.M. Insulin and Autophagy in Neurodegeneration. Front. Neurosci. 2019, 13, 491. [Google Scholar] [CrossRef] [Green Version]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Lin, K.L.; Chen, S.D.; Lin, K.J.; Liou, C.W.; Chuang, Y.C.; Wang, P.W.; Chuang, J.H.; Lin, T.K. Quality Matters? The Involvement of Mitochondrial Quality Control in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 9, 636295. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, B. Neuroprotective and Anti-inflammatory Activities of Ketogenic Diet on MPTP-induced Neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Yang, X.; An, L.; Gao, B.; Liu, X.; Liu, S. Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 2009, 1286, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Miyake, Y.; Sasaki, S.; Murakami, K.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; et al. Dietary patterns and risk of Parkinson’s disease: A case-control study in Japan. Eur. J. Neurol. 2012, 19, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; et al. Dietary glycemic index is inversely associated with the risk of Parkinson’s disease: A case–control study in Japan. Nutrition 2010, 26, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial Dysfunction and β-Cell Failure in Type 2 Diabetes Mellitus. Exp. Diabetes Res. 2012, 2012, 703538. [Google Scholar] [CrossRef] [Green Version]
- Esteghamati, A.; Eskandari, D.; Mirmiranpour, H.; Noshad, S.; Mousavizadeh, M.; Hedayati, M.; Nakhjavani, M. Effects of metformin on markers of oxidative stress and antioxidant reserve in patients with newly diagnosed type 2 diabetes: A randomized clinical trial. Clin. Nutr. 2013, 32, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Coarfa, C.; Qin, X.; Bonnen, P.E.; Milosavljevic, A.; Versalovic, J.; Aagaard, K. mtDNA haplogroup and single nucleotide polymorphisms structure human microbiome communities. BMC Genom. 2014, 15, 257. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante, G.C.; Magalhaes, L.; Ribeiro-Dos-Santos, A.; Vidal, A.F. Mitochondrial Epigenetics: Non-Coding RNAs as a Novel Layer of Complexity. Int. J. Mol. Sci. 2020, 21, 1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, M.-Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [Green Version]
- Kumari, T.; Vachher, M.; Bansal, S.; Bamezai, R.N.K.; Kumar, B. Meta-analysis of mitochondrial T16189C polymorphism for cancer and Type 2 diabetes risk. Clin. Chim. Acta 2018, 482, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.W.; Lin, T.K.; Huang, F.M.; Chen, T.L.; Lee, C.F.; Chuang, Y.C.; Tan, T.Y.; Chang, K.C.; Wei, Y.H. Association of the mitochondrial DNA 16189 T to C variant with lacunar cerebral infarction: Evidence from a hospital-based case-control study. Ann. N. Y. Acad. Sci. 2004, 1011, 317–324. [Google Scholar] [CrossRef]
- Chen, Y.N.; Liou, C.W.; Huang, C.C.; Lin, T.K.; Wei, Y.H. Maternally inherited diabetes and deafness (MIDD) syndrome: A clinical and molecular genetic study of a Taiwanese family. Chang Gung Med. J. 2004, 27, 66–73. [Google Scholar] [PubMed]
- Liou, C.W.; Huang, C.C.; Lee, C.F.; Lin, T.K.; Wei, Y.H. Low antioxidant content and mutation load in mitochondrial DNA A3243G mutation-related diabetes mellitus. J. Med. Assoc. 2003, 102, 527–533. [Google Scholar]
- Liou, C.W.; Huang, C.C.; Lin, T.K.; Tsai, J.L.; Wei, Y.H. Correction of pancreatic beta-cell dysfunction with coenzyme Q(10) in a patient with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome and diabetes mellitus. Eur. Neurol. 2000, 43, 54–55. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, M.T.; Schaefer, A.M.; Blakely, E.L.; McFarland, R.; Chinnery, P.F.; Turnbull, D.M.; Taylor, R.W. Noninvasive diagnosis of the 3243A>G mitochondrial DNA mutation using urinary epithelial cells. Eur. J. Hum. Genet. 2004, 12, 778–781. [Google Scholar] [CrossRef]
- Ikeda, T.; Osaka, H.; Shimbo, H.; Tajika, M.; Yamazaki, M.; Ueda, A.; Murayama, K.; Yamagata, T. Mitochondrial DNA 3243A>T mutation in a patient with MELAS syndrome. Hum. Genome Var. 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.W.; Lin, T.K.; Chen, J.B.; Tiao, M.M.; Weng, S.W.; Chen, S.D.; Chuang, Y.C.; Chuang, J.H.; Wang, P.W. Association between a common mitochondrial DNA D-loop polycytosine variant and alteration of mitochondrial copy number in human peripheral blood cells. J. Med. Genet. 2010, 47, 723–728. [Google Scholar] [CrossRef]
- Wang, P.W.; Lin, T.K.; Weng, S.W.; Liou, C.W. Mitochondrial DNA variants in the pathogenesis of type 2 diabetes—Relevance of asian population studies. Rev. Diabet. Stud. 2009, 6, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Tiao, M.M.; Lin, T.K.; Liou, C.W.; Wang, P.W.; Chen, J.B.; Kuo, F.Y.; Huang, C.C.; Chou, Y.M.; Chuang, J.H. Early transcriptional deregulation of hepatic mitochondrial biogenesis and its consequent effects on murine cholestatic liver injury. Apoptosis 2009, 14, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.W.; Lin, T.K.; Huei Weng, H.; Lee, C.F.; Chen, T.L.; Wei, Y.H.; Chen, S.D.; Chuang, Y.C.; Weng, S.W.; Wang, P.W. A common mitochondrial DNA variant and increased body mass index as associated factors for development of type 2 diabetes: Additive effects of genetic and environmental factors. J. Clin. Endocrinol. Metab. 2007, 92, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.K.; Chen, S.D.; Wang, P.W.; Wei, Y.H.; Lee, C.F.; Chen, T.L.; Chuang, Y.C.; Tan, T.Y.; Chang, K.C.; Liou, C.W. Increased oxidative damage with altered antioxidative status in type 2 diabetic patients harboring the 16189 T to C variant of mitochondrial DNA. Ann. N. Y. Acad. Sci. 2005, 1042, 64–69. [Google Scholar] [CrossRef]
- Van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef]
- Tsai, M.H.; Kuo, C.W.; Lin, T.K.; Ho, C.J.; Wang, P.W.; Chuang, J.H.; Liou, C.W. Ischemic Stroke Risk Associated with Mitochondrial Haplogroup F in the Asian Population. Cells 2020, 9, 1885. [Google Scholar] [CrossRef]
- Liou, C.W.; Chuang, J.H.; Chen, J.B.; Tiao, M.M.; Wang, P.W.; Huang, S.T.; Huang, T.L.; Lee, W.C.; Weng, S.W.; Huang, P.H.; et al. Mitochondrial DNA variants as genetic risk factors for Parkinson disease. Eur. J. Neurol. 2016, 23, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Li, T.; Wang, Z.F.; Huang, S.S.; Shao, Z.Q.; Wang, K.; Zhong, H.Q.; Chen, S.F.; Zhang, X.; Zhu, J.H. Mitochondrial DNA variants modulate genetic susceptibility to Parkinson’s disease in Han Chinese. Neurobiol. Dis. 2018, 114, 17–23. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Limousin, P.; Lees, A.; Foltynie, T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain 2013, 136, 374–384. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zeng, F.; Jin, W.-S.; Zhu, C.; Wang, Q.-H.; Bu, X.-L.; Luo, H.-B.; Zou, H.-Q.; Pu, J.; Zhou, Z.-H.; et al. Comorbidity burden of patients with Parkinson’s disease and Parkinsonism between 2003 and 2012: A multicentre, nationwide, retrospective study in China. Sci. Rep. 2017, 7, 1671. [Google Scholar] [CrossRef] [Green Version]
- Sergi, D.; Renaud, J.; Simola, N.; Martinoli, M.G. Diabetes, a Contemporary Risk for Parkinson’s Disease: Epidemiological and Cellular Evidences. Front. Aging Neurosci. 2019, 11, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlqvist, M.L.; Lee, M.S.; Hsu, C.C.; Chuang, S.Y.; Lee, J.T.; Tsai, H.N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 2012, 18, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Shaikh, M.F.; Othman, I. Emerging neuroprotective effect of metformin in Parkinson’s disease: A molecular crosstalk. Pharmacol. Res. 2020, 152, 104593. [Google Scholar] [CrossRef]
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Carta, A.R.; Frau, L.; Pisanu, A.; Wardas, J.; Spiga, S.; Carboni, E. Rosiglitazone decreases peroxisome proliferator receptor-γ levels in microglia and inhibits TNF-α production: New evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 2011, 194, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, M.; Marinova-Mutafchieva, L.; Broom, L.; Davis, J.B.; Virley, D.; Medhurst, A.D.; Dexter, D.T. Full and partial peroxisome proliferation-activated receptor-gamma agonists, but not delta agonist, rescue of dopaminergic neurons in the 6-OHDA parkinsonian model is associated with inhibition of microglial activation and MMP expression. J. Neuroimmunol. 2012, 246, 69–77. [Google Scholar] [CrossRef]
- Brauer, R.; Bhaskaran, K.; Chaturvedi, N.; Dexter, D.T.; Smeeth, L.; Douglas, I. Glitazone Treatment and Incidence of Parkinson’s Disease among People with Diabetes: A Retrospective Cohort Study. PLoS Med. 2015, 12, e1001854. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Pu, J.; Chen, Y.; Zhang, B. Decreased risk of Parkinson’s disease in diabetic patients with thiazolidinediones therapy: An exploratory meta-analysis. PLoS ONE 2019, 14, e0224236. [Google Scholar] [CrossRef] [Green Version]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: Mechanisms of action. Drug Discov. Today 2016, 21, 802–818. [Google Scholar] [CrossRef] [Green Version]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145, 98–120. [Google Scholar] [CrossRef]
- Grieco, M.; Giorgi, A.; Gentile, M.C.; d’Erme, M.; Morano, S.; Maras, B.; Filardi, T. Glucagon-Like Peptide-1: A Focus on Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1112. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-K.; Lin, K.-J.; Lin, H.-Y.; Lin, K.-L.; Lan, M.-Y.; Wang, P.-W.; Wang, T.-J.; Wang, F.-S.; Tsai, P.-C.; Liou, C.-W.; et al. Glucagon-Like Peptide-1 Receptor Agonist Ameliorates 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Neurotoxicity Through Enhancing Mitophagy Flux and Reducing α-Synuclein and Oxidative Stress. Front. Mol. Neurosci. 2021, 14, 697440. [Google Scholar] [CrossRef]
- Badawi, G.A.; Abd El Fattah, M.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and Liraglutide Modulate L-dopa Effect and Attenuate Dyskinetic Movements in Rotenone-Lesioned Rats. Neurotox. Res. 2019, 35, 635–653. [Google Scholar] [CrossRef]
- Jeong, S.H.; Chung, S.J.; Yoo, H.S.; Hong, N.; Jung, J.H.; Baik, K.; Lee, Y.H.; Sohn, Y.H.; Lee, P.H. Beneficial effects of dipeptidyl peptidase-4 inhibitors in diabetic Parkinson’s disease. Brain 2021, 144, 1127–1137. [Google Scholar] [CrossRef]
- Brauer, R.; Wei, L.; Ma, T.; Athauda, D.; Girges, C.; Vijiaratnam, N.; Auld, G.; Whittlesea, C.; Wong, I.; Foltynie, T. Diabetes medications and risk of Parkinson’s disease: A cohort study of patients with diabetes. Brain 2020, 143, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Chao, E.C.; Henry, R.R. SGLT2 inhibition—A novel strategy for diabetes treatment. Nat. Rev. Drug Discov. 2010, 9, 551–559. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Sjöström, C.D.; Greasley, P.J.; Cain, V.; Boulton, D.W.; Heerspink, H.J.L. Effects of the sodium-glucose co-transporter-2 inhibitor dapagliflozin on estimated plasma volume in patients with type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 2667–2673. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Kosiborod, M.; Cavender, M.A.; Fu, A.Z.; Wilding, J.P.; Khunti, K.; Holl, R.W.; Norhammar, A.; Birkeland, K.I.; Jørgensen, M.E.; Thuresson, M.; et al. Lower Risk of Heart Failure and Death in Patients Initiated on Sodium-Glucose Cotransporter-2 Inhibitors Versus Other Glucose-Lowering Drugs: The CVD-REAL Study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation 2017, 136, 249–259. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Atkin, S.L.; Butler, A.E.; Sahebkar, A. Sodium-glucose cotransporter inhibitors and oxidative stress: An update. J. Cell. Physiol. 2019, 234, 3231–3237. [Google Scholar] [CrossRef]
- Esterline, R.L.; Vaag, A.; Oscarsson, J.; Vora, J. MECHANISMS IN ENDOCRINOLOGY: SGLT2 inhibitors: Clinical benefits by restoration of normal diurnal metabolism? Eur. J. Endocrinol. 2018, 178, R113–R125. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Desilva, S.; Abbruscato, T. The role of glucose transporters in brain disease: Diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 2012, 13, 12629–12655. [Google Scholar] [CrossRef]
- Camargo Maluf, F.; Feder, D.; Alves de Siqueira Carvalho, A. Analysis of the Relationship between Type II Diabetes Mellitus and Parkinson’s Disease: A Systematic Review. Parkinson’s Dis. 2019, 2019, 4951379. [Google Scholar] [CrossRef] [Green Version]
- De Lazzari, F.; Bubacco, L.; Whitworth, A.J.; Bisaglia, M. Superoxide Radical Dismutation as New Therapeutic Strategy in Parkinson’s Disease. Aging Dis. 2018, 9, 716–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2020, 6, 186. [Google Scholar] [CrossRef]
- Lopaschuk, G.D. Fatty Acid Oxidation and Its Relation with Insulin Resistance and Associated Disorders. Ann. Nutr. Metab. 2016, 68 (Suppl. 3), 15–20. [Google Scholar] [CrossRef]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Yuan, H.-X.; Guan, K.-L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.-F.; Chen, Y.-L.; Chiou, T.T.-Y.; Chu, T.-H.; Li, L.-C.; Ng, H.-Y.; Lee, W.-C.; Lee, C.-T. Emergence of SGLT2 Inhibitors as Powerful Antioxidants in Human Diseases. Antioxidants 2021, 10, 1166. [Google Scholar] [CrossRef]
- Feng, R.; Dong, L.; Wang, L.; Xu, Y.; Lu, H.; Zhang, J. Development of sodium glucose co-transporter 2 (SGLT2) inhibitors with novel structure by molecular docking and dynamics simulation. J. Mol. Modeling 2019, 25, 175. [Google Scholar] [CrossRef]
- Cai, W.; Jiang, L.; Xie, Y.; Liu, Y.; Liu, W.; Zhao, G. Design of SGLT2 Inhibitors for the Treatment of Type 2 Diabetes: A History Driven by Biology to Chemistry. Med. Chem. 2015, 11, 317–328. [Google Scholar] [CrossRef]
- Ehrenkranz, J.R.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A review. Diabetes/Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, D.; Laparra-Llopis, J.M.; Zielinski, H.; Szawara-Nowak, D.; Giménez-Bastida, J.A. Role of Apple Phytochemicals, Phloretin and Phloridzin, in Modulating Processes Related to Intestinal Inflammation. Nutrients 2019, 11, 1173. [Google Scholar]
- Prabhakar, P.; Ahmed, A.B.; Chidambaram, S. The Role of Phloridzin and its Possible Potential Therapeutic Effect on Parkinson’s Disease. Int. J. Nutr. Pharmacol. Neurol. Dis. 2020, 10, 69–74. [Google Scholar] [CrossRef]
- Osorio, H.; Coronel, I.; Arellano, A.; Pacheco, U.; Bautista, R.; Franco, M.; Escalante, B. Sodium-glucose cotransporter inhibition prevents oxidative stress in the kidney of diabetic rats. Oxidative Med. Cell. Longev. 2012, 2012, 542042. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnölzer, M.; Lasitschka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef]
- Oshima, H.; Miki, T.; Kuno, A.; Mizuno, M.; Sato, T.; Tanno, M.; Yano, T.; Nakata, K.; Kimura, Y.; Abe, K.; et al. Empagliflozin, an SGLT2 Inhibitor, Reduced the Mortality Rate after Acute Myocardial Infarction with Modification of Cardiac Metabolomes and Antioxidants in Diabetic Rats. J. Pharmacol. Exp. Ther. 2019, 368, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Han, F.; Lu, Q.; Li, X.; Ren, D.; Zhang, J.; Han, Y.; Xiang, Y.K.; Li, J. Empagliflozin Ameliorates Obesity-Related Cardiac Dysfunction by Regulating Sestrin2-Mediated AMPK-mTOR Signaling and Redox Homeostasis in High-Fat Diet-Induced Obese Mice. Diabetes 2020, 69, 1292–1305. [Google Scholar] [CrossRef]
- Andreadou, I.; Efentakis, P.; Balafas, E.; Togliatto, G.; Davos, C.H.; Varela, A.; Dimitriou, C.A.; Nikolaou, P.-E.; Maratou, E.; Lambadiari, V.; et al. Empagliflozin Limits Myocardial Infarction in Vivo and Cell Death in Vitro: Role of STAT3, Mitochondria, and Redox Aspects. Front. Physiol. 2017, 8, 1077. [Google Scholar] [CrossRef] [Green Version]
- Iannantuoni, F.; de Marañon, M.A.; Diaz-Morales, N.; Falcon, R.; Bañuls, C.; Abad-Jimenez, Z.; Victor, V.M.; Hernandez-Mijares, A.; Rovira-Llopis, S. The SGLT2 Inhibitor Empagliflozin Ameliorates the Inflammatory Profile in Type 2 Diabetic Patients and Promotes an Antioxidant Response in Leukocytes. J. Clin. Med. 2019, 8, 1814. [Google Scholar] [CrossRef] [Green Version]
- Takagi, S.; Li, J.; Takagaki, Y.; Kitada, M.; Nitta, K.; Takasu, T.; Kanasaki, K.; Koya, D. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J. Diabetes Investig. 2018, 9, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of sodium-glucose cotransporter 2 selective inhibitor ipragliflozin on hyperglycaemia, oxidative stress, inflammation and liver injury in streptozotocin-induced type 1 diabetic rats. J. Pharm. Pharmacol. 2014, 66, 975–987. [Google Scholar] [CrossRef]
- Lin, T.K.; Chen, S.D.; Lin, K.J.; Chuang, Y.C. Seizure-Induced Oxidative Stress in Status Epilepticus: Is Antioxidant Beneficial? Antioxidants 2020, 9, 1029. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Q.; Li, Y.; Tang, Q.; Wu, T.; Chen, L.; Pu, S.; Zhao, Y.; Zhang, G.; Huang, C.; et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1α signalling pathway. Adipocyte 2020, 9, 484–494. [Google Scholar] [CrossRef]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef] [Green Version]
- Hierro-Bujalance, C.; Infante-Garcia, C.; del Marco, A.; Herrera, M.; Carranza-Naval, M.J.; Suarez, J.; Alves-Martinez, P.; Lubian-Lopez, S.; Garcia-Alloza, M. Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimer’s Res. Ther. 2020, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Amin, E.F.; Rifaai, R.A.; Abdel-Latif, R.G. Empagliflozin attenuates transient cerebral ischemia/reperfusion injury in hyperglycemic rats via repressing oxidative-inflammatory-apoptotic pathway. Fundam. Clin. Pharmacol. 2020, 34, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Arafa, N.M.S.; Ali, E.H.A.; Hassan, M.K. Canagliflozin prevents scopolamine-induced memory impairment in rats: Comparison with galantamine hydrobromide action. Chem.-Biol. Interact. 2017, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sa-nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef]
- Arab, H.H.; Safar, M.M.; Shahin, N.N. Targeting ROS-Dependent AKT/GSK-3β/NF-κB and DJ-1/Nrf2 Pathways by Dapagliflozin Attenuates Neuronal Injury and Motor Dysfunction in Rotenone-Induced Parkinson’s Disease Rat Model. ACS Chem. Neurosci. 2021, 12, 689–703. [Google Scholar] [CrossRef]
- Erdogan, M.A.; Yusuf, D.; Christy, J.; Solmaz, V.; Erdogan, A.; Taskiran, E.; Erbas, O. Highly selective SGLT2 inhibitor dapagliflozin reduces seizure activity in pentylenetetrazol-induced murine model of epilepsy. BMC Neurol. 2018, 18, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.J.; Tyagi, P.; Lin, T.K.; Huang, C.C.; Lee, W.C.; Chancellor, M.B.; Chuang, Y.C. Low Energy Shock Wave Therapy Attenuates Mitochondrial Dysfunction and Improves Bladder Function in HCl induced Cystitis in Rats. Biomed. J. 2021. [Google Scholar] [CrossRef]
- Barreca, D.; Currò, M.; Bellocco, E.; Ficarra, S.; Laganà, G.; Tellone, E.; Laura Giunta, M.; Visalli, G.; Caccamo, D.; Galtieri, A.; et al. Neuroprotective effects of phloretin and its glycosylated derivative on rotenone-induced toxicity in human SH-SY5Y neuronal-like cells. BioFactors 2017, 43, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci. 2019, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-latif, R.G.; Rifaai, R.A.; Amin, E.F. Empagliflozin alleviates neuronal apoptosis induced by cerebral ischemia/reperfusion injury through HIF-1α/VEGF signaling pathway. Arch. Pharmacal Res. 2020, 43, 514–525. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, K.-J.; Wang, T.-J.; Chen, S.-D.; Lin, K.-L.; Liou, C.-W.; Lan, M.-Y.; Chuang, Y.-C.; Chuang, J.-H.; Wang, P.-W.; Lee, J.-J.; et al. Two Birds One Stone: The Neuroprotective Effect of Antidiabetic Agents on Parkinson Disease—Focus on Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors. Antioxidants 2021, 10, 1935. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10121935
Lin K-J, Wang T-J, Chen S-D, Lin K-L, Liou C-W, Lan M-Y, Chuang Y-C, Chuang J-H, Wang P-W, Lee J-J, et al. Two Birds One Stone: The Neuroprotective Effect of Antidiabetic Agents on Parkinson Disease—Focus on Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors. Antioxidants. 2021; 10(12):1935. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10121935
Chicago/Turabian StyleLin, Kai-Jung, Tzu-Jou Wang, Shang-Der Chen, Kai-Lieh Lin, Chia-Wei Liou, Min-Yu Lan, Yao-Chung Chuang, Jiin-Haur Chuang, Pei-Wen Wang, Jong-Jer Lee, and et al. 2021. "Two Birds One Stone: The Neuroprotective Effect of Antidiabetic Agents on Parkinson Disease—Focus on Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors" Antioxidants 10, no. 12: 1935. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10121935