
Enantioselective Synthesis and Pharmacological Evaluation of Aza-CGP37157–Lipoic Acid Hybrids for the Treatment of Alzheimer’s Disease


 and
and
Abstract
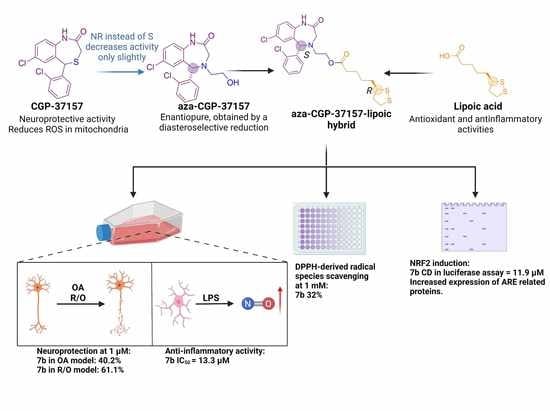
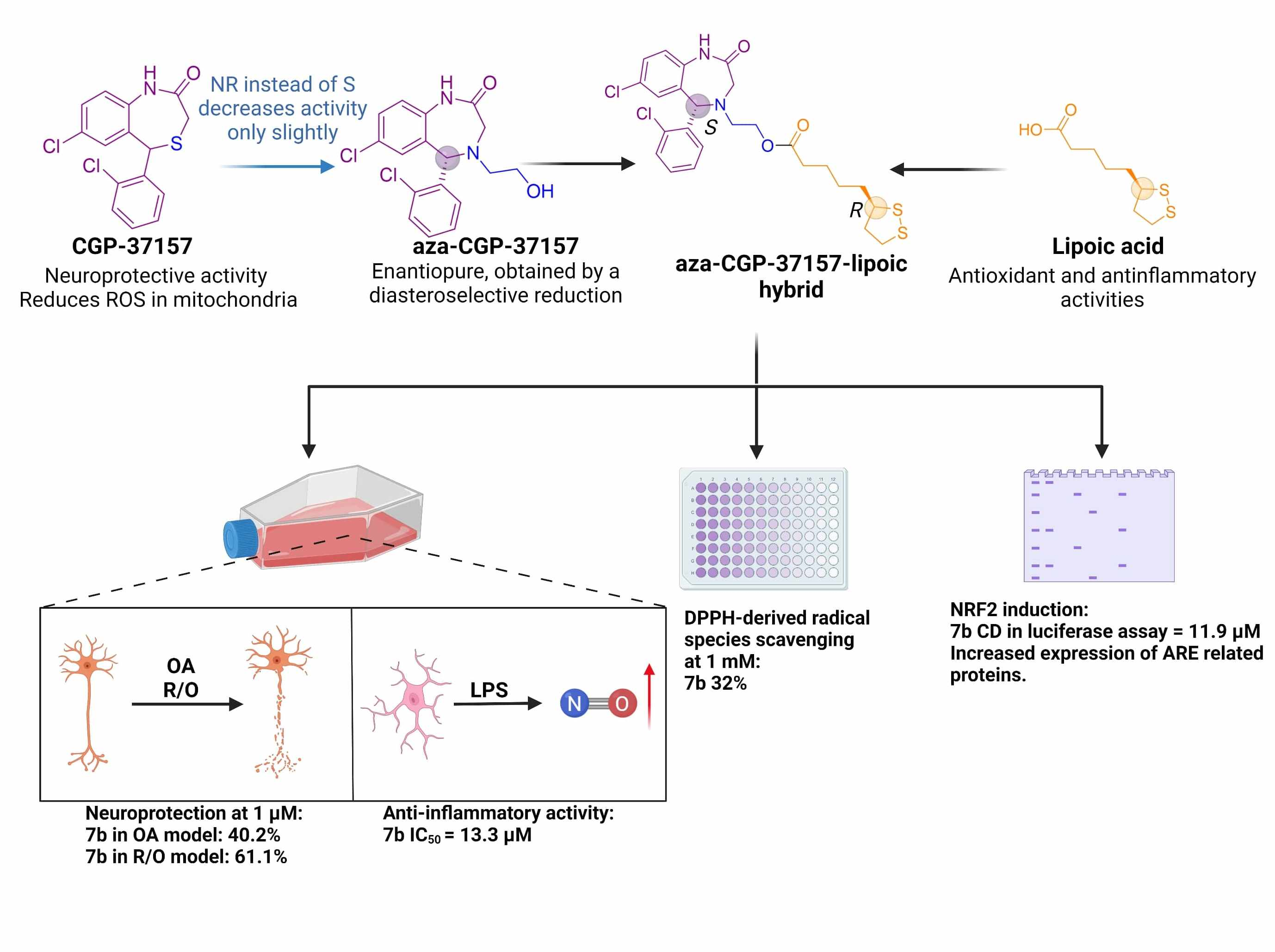
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Experimental Details
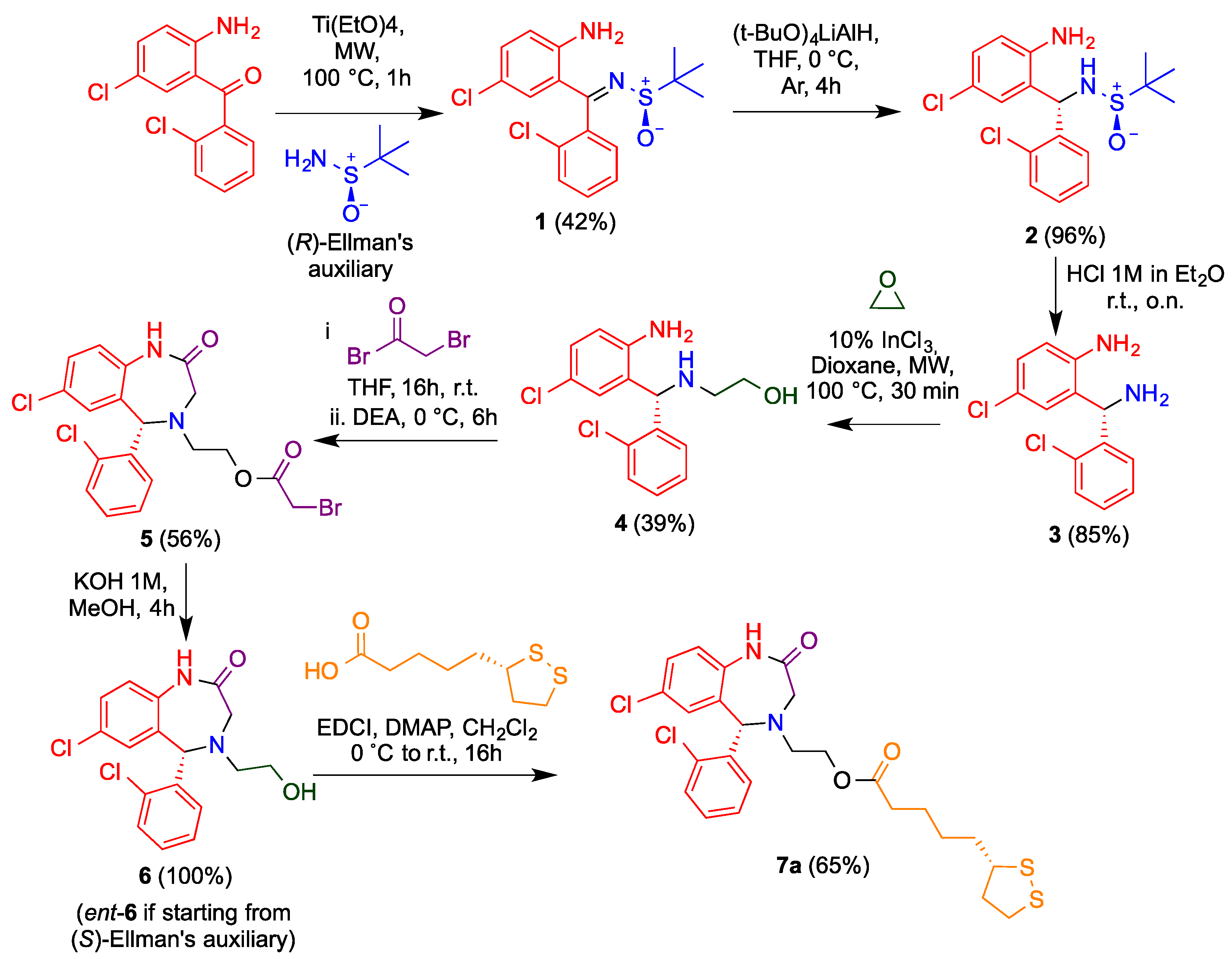
2.1.2. Synthesis of Sulfinilimine 1
(R,Z)-N-((2-Amino-5-chlorophenyl)(2-chlorophenyl)methylene)-2-methylpropane-2-sulfinamide (R-1)
(S,Z)-N-((2-Amino-5-chlorophenyl)(2-chlorophenyl)methylene)-2-methylpropane-2-sulfinamide (S-1)
2.1.3. Diastereoselective Reduction of Sulfinylimine 2
(R)-N-[(S)-(2-Amino-5-chlorophenyl)(2-chlorophenyl)methyl]-2-methylpropane-2-sulfinamide (R,S-2)
(S)-N-[(R)-(2-Amino-5-chlorophenyl)(2-chlorophenyl)methyl]-2-methylpropane-2-sulfinamide(S,R-2)
2.1.4. Cleavage of the N-Tert-Butylsulfinyl Chiral Auxiliary 3
(S)-2-[Amino(2-chlorophenyl)methyl]-4-chloroaniline (S-3)
(R)-2-(Amino(2-chlorophenyl)methyl)-4-chloroaniline (R-3)
2.1.5. Synthesis of Aminoethanol 4
(S)-2-(((2-Amino-5-chlorophenyl)(2-chlorophenyl)methyl)amino)ethan-1-ol (S-4)
(R)-2-(((2-amino-5-chlorophenyl)(2-chlorophenyl)methyl)amino)ethan-1-ol (R-4)
2.1.6. Synthesis of Benzodiazepine 5
(S)-2-(7-Chloro-5-(2-chlorophenyl)-2-oxo-1,2,3,5-tetrahydro-4H-benzo[e][1,4]diazepin-4-yl)ethyl 2-bromoacetate (S-5)
(R)-2-(7-Chloro-5-(2-chlorophenyl)-2-oxo-1,2,3,5-tetrahydro-4H-benzo[e][1,4]diazepin-4-yl)ethyl 2-bromoacetate (R-5)
2.1.7. Ester Hydrolysis 6
(5S)-7-Chloro-5-(2-chlorophenyl)-4-(2-hydroxyethyl)-4,5-dihydro-1H-benzo[e][1,4]diazepin-2(3H)-one (S-6)
(5R)-7-Chloro-5-(2-chlorophenyl)-4-(2-hydroxyethyl)-4,5-dihydro-1H-benzo[e][1,4]diazepin-2(3H)-one (R-6)
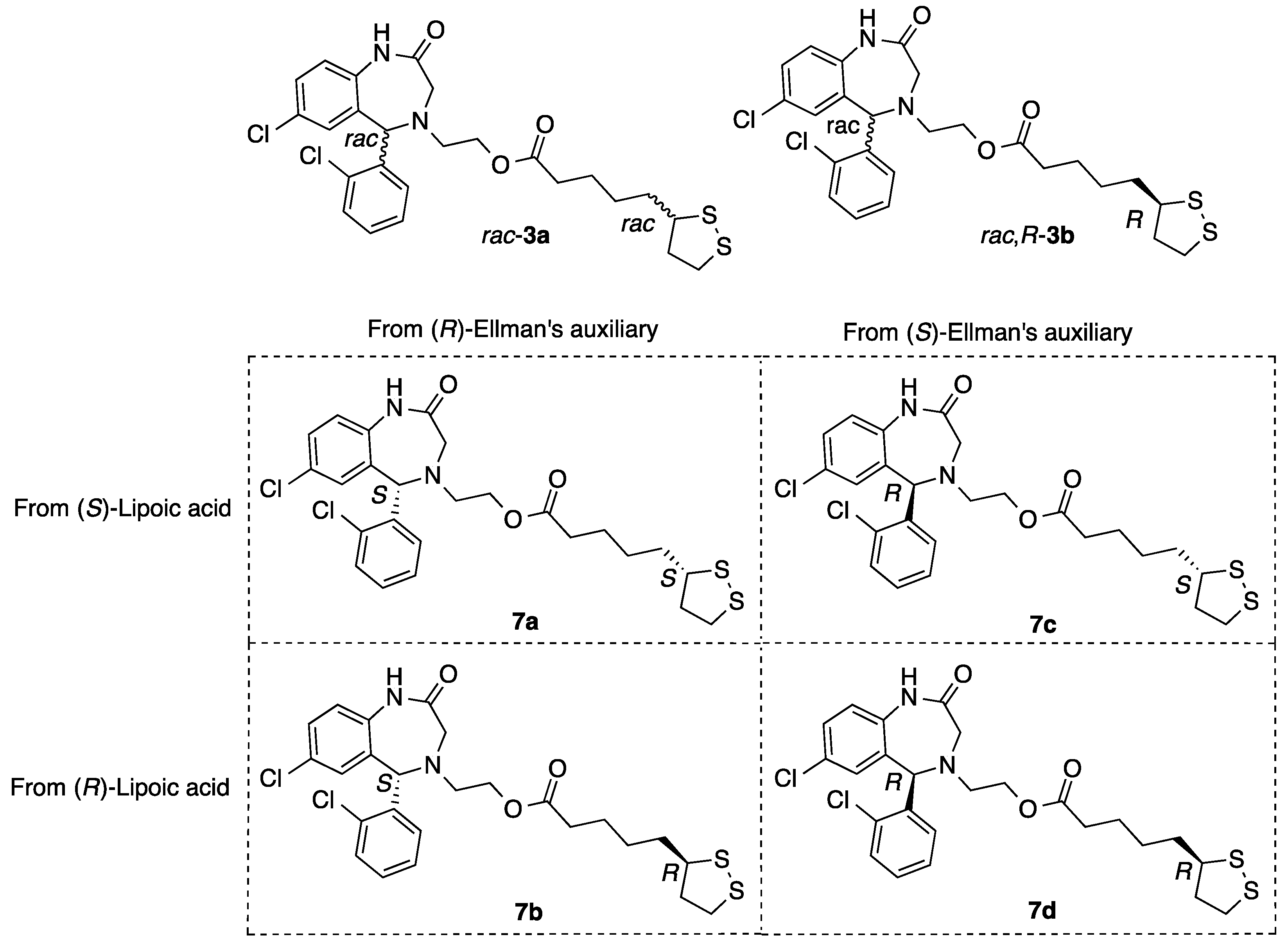
2.1.8. Synthesis of Aza-CGP37157–Lipoic Hybrids (7)
2-((S)-7-Chloro-5-(2-chlorophenyl)-2-oxo-1,2,3,5-tetrahydro-4H-benzo[e][1,4]diazepin-4-yl)ethyl 5-((S)-1,2-dithiolan-3-yl)pentanoate (7a)
2-((R)-7-Chloro-5-(2-chlorophenyl)-2-oxo-1,2,3,5-tetrahydro-4H-benzo[e][1,4]diazepin-4-yl)ethyl 5-((S)-1,2-dithiolan-3-yl)pentanoate (7b)
2-((S)-7-Chloro-5-(2-chlorophenyl)-2-oxo-1,2,3,5-tetrahydro-4H-benzo[e][1,4]diazepin-4-yl)ethyl 5-((R)-1,2-dithiolan-3-yl)pentanoate (7c)
2-((R)-7-Chloro-5-(2-chlorophenyl)-2-oxo-1,2,3,5-tetrahydro-4H-benzo[e][1,4]diazepin-4-yl)ethyl 5-((R)-1,2-dithiolan-3-yl)pentanoate (7d)
2.2. Pharmacological Evaluation
2.2.1. Physicochemical and ADME Properties Calculation
2.2.2. Reduction Assay of 1,1-Diphenyl-2-picryl-hydrazyl (DPPH): Antioxidant Capacity
2.2.3. Determination of NRF2 Transcription Factor Induction
2.2.4. Immunocytochemistry
2.2.5. Western Blot Analysis
2.2.6. SH-SY5Y Neuroblastoma Cell Culture
2.2.7. Neuroprotection Assays in the SH-SY5Y Cell Line
2.2.8. Cytotoxicity Assay in the SH-SY5Y Cell Line (CC50)
2.2.9. Viability Assessment by MTT Reduction
2.2.10. BV2 Cell Line Culture
2.2.11. Nitrite Production Reduction Assay
2.2.12. Statistical Analysis
3. Results
3.1. Chemistry
3.2. Pharmacological Characterization
3.2.1. Computational Druggability Study of Compounds 7
3.2.2. Blood–Brain Barrier Permeability
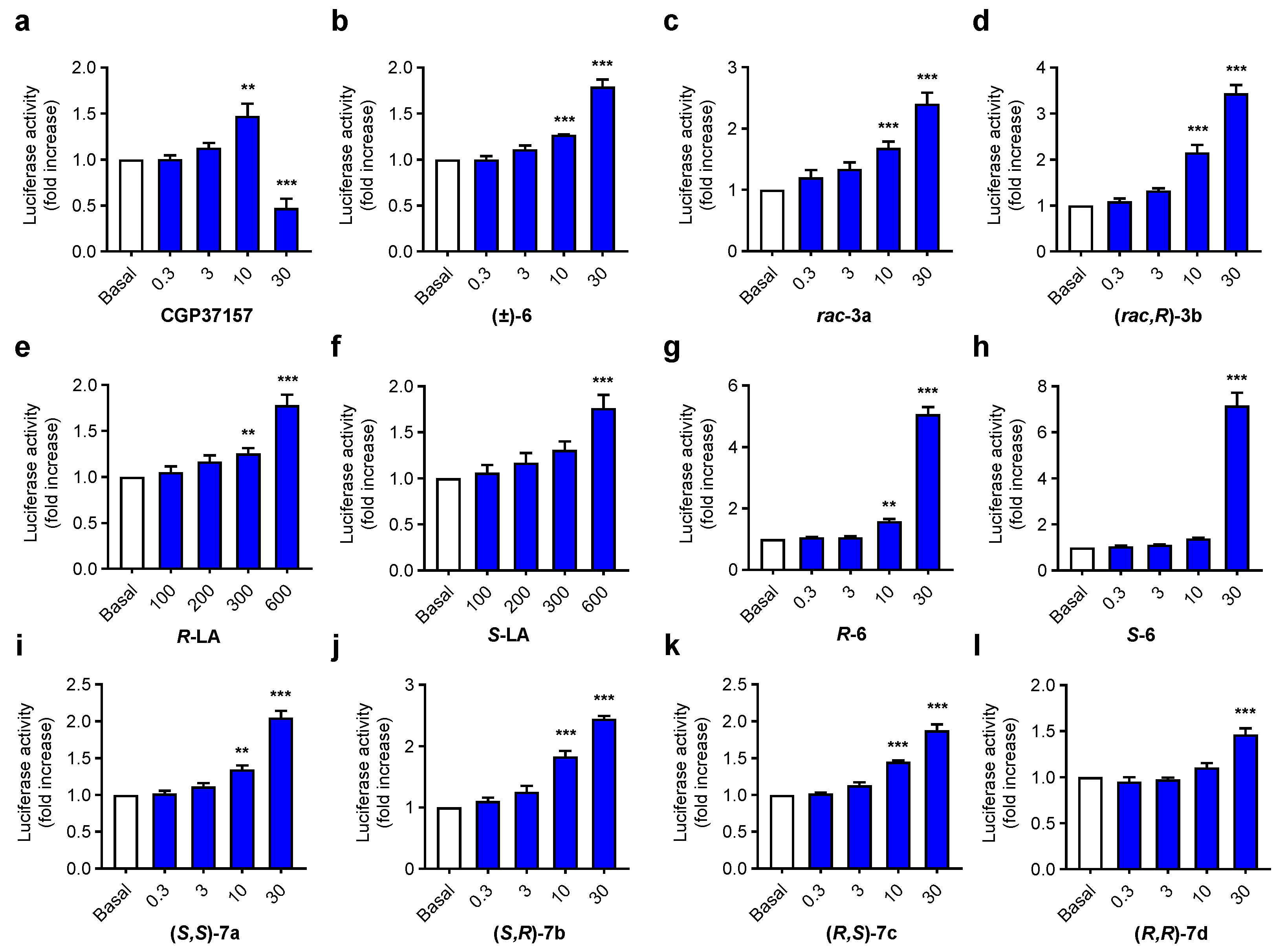
3.2.3. Antioxidant Capacity of Aza-CGP37157-LA 7a–d Derivatives
3.3. Biological Evaluation
3.3.1. Cytotoxicity Evaluation in the SH-SY5Y Cell Line
3.3.2. NRF2 Induction
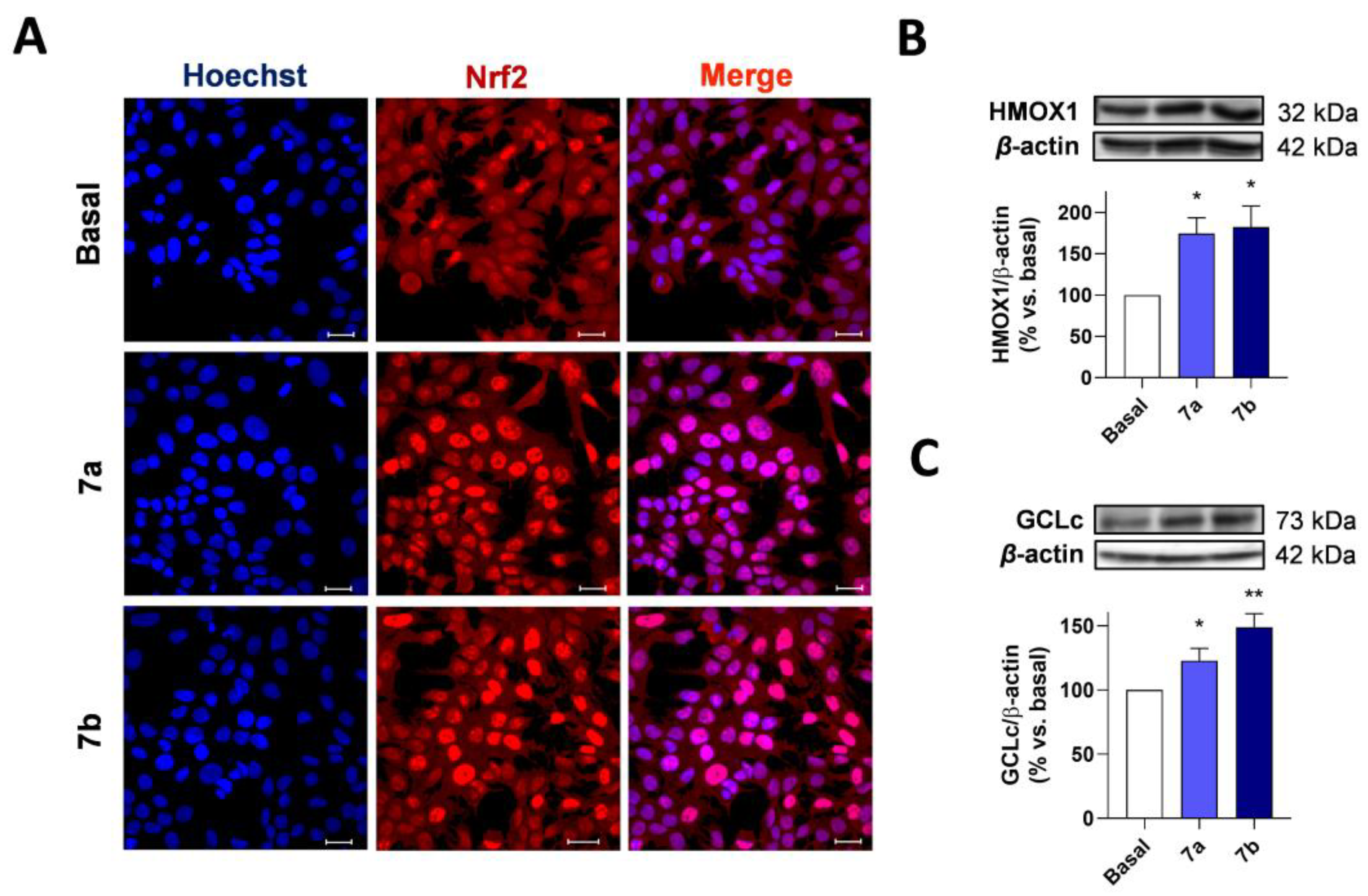
3.3.3. Compounds 7a and 7b Induce NRF2 Nuclear Translocation and Upregulate the Expression of NRF2 Dependent Genes
3.3.4. Anti-Inflammatory Properties of Compounds 7a–d
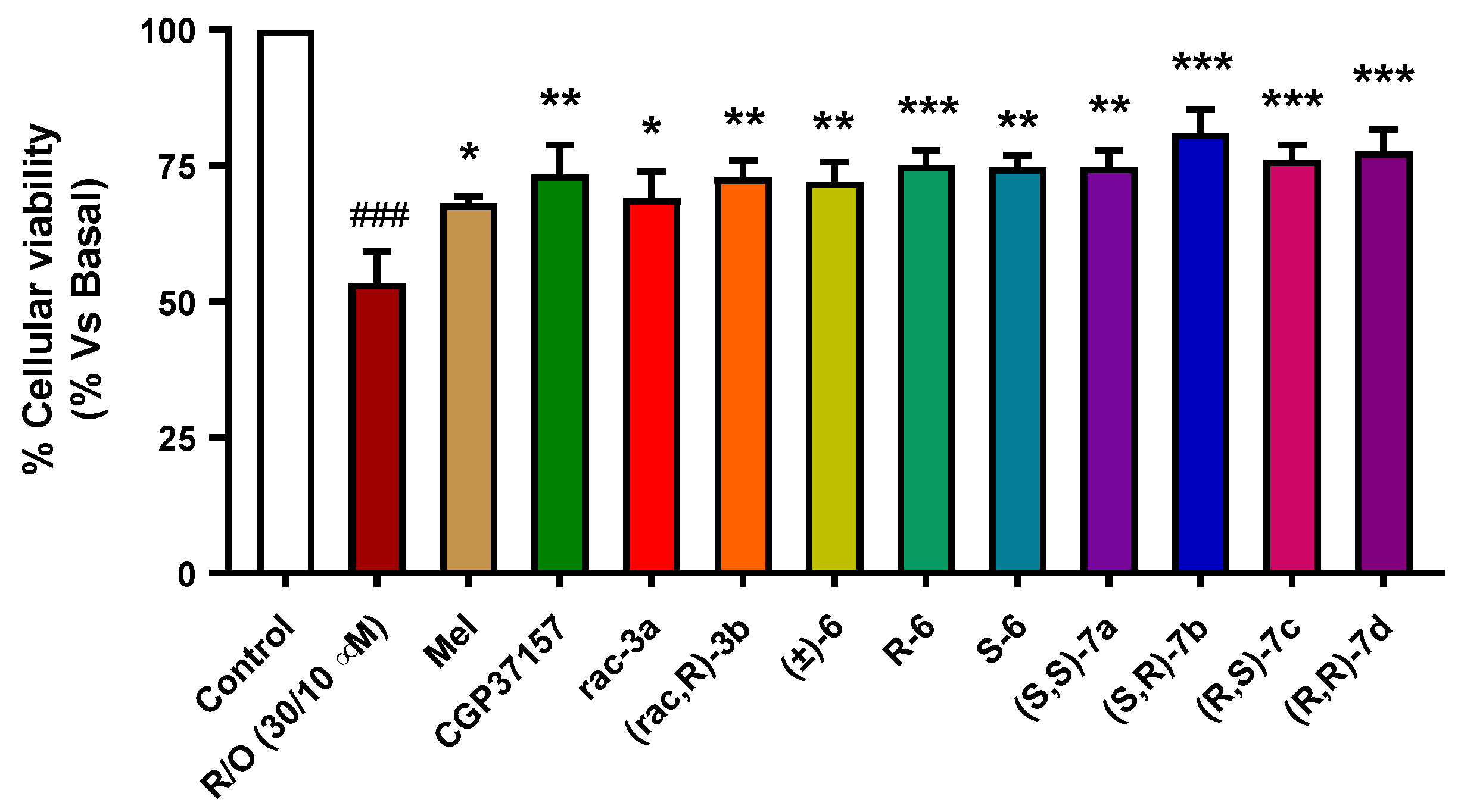
3.3.5. Neuroprotection in a Rotenone/Oligomycin A Oxidative Stress Model
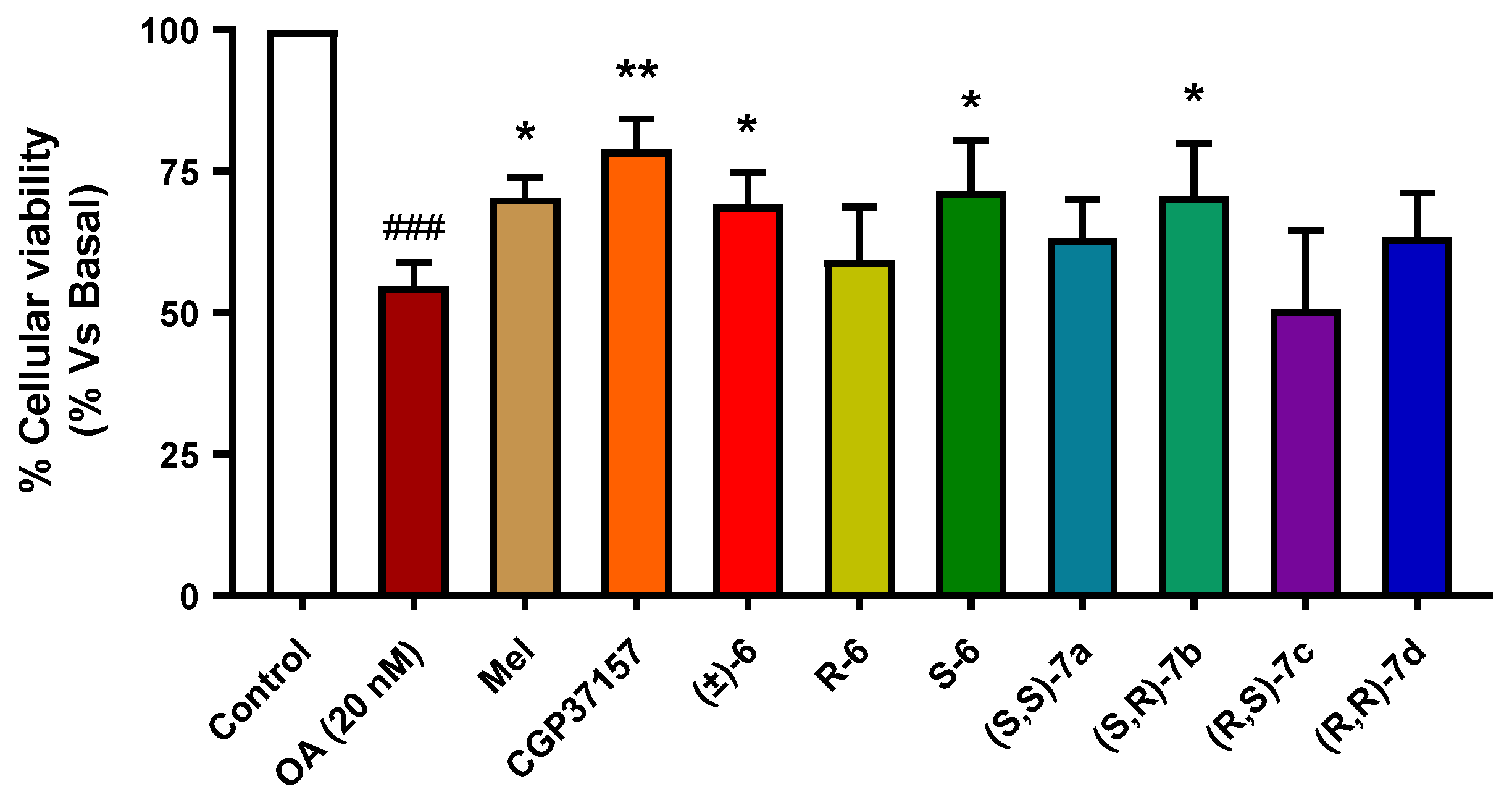
3.3.6. Neuroprotection against Tau Hyperphosphorylation Induced by Okadaic Acid
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Dementia: A Public Health Priority; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug. Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Cores, A.; Piquero, M.; Villacampa, M.; Leon, R.; Menendez, J.C. NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules 2020, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef]
- Moura, F.A.; de Andrade, K.Q.; dos Santos, J.C.; Goulart, M.O. Lipoic Acid: Its antioxidant and anti-inflammatory role and clinical applications. Curr. Top. Med. Chem. 2015, 15, 458–483. [Google Scholar] [CrossRef]
- Pilar Valdecantos, M.; Prieto-Hontoria, P.L.; Pardo, V.; Modol, T.; Santamaria, B.; Weber, M.; Herrero, L.; Serra, D.; Muntane, J.; Cuadrado, A.; et al. Essential role of Nrf2 in the protective effect of lipoic acid against lipoapoptosis in hepatocytes. Free Radic. Biol. Med. 2015, 84, 263–278. [Google Scholar] [CrossRef]
- Pei, Y.; Lilly, M.J.; Owen, D.J.; D’Souza, L.J.; Tang, X.Q.; Yu, J.; Nazarbaghi, R.; Hunter, A.; Anderson, C.M.; Glasco, S.; et al. Efficient syntheses of benzothiazepines as antagonists for the mitochondrial sodium-calcium exchanger: Potential therapeutics for type II diabetes. J. Org. Chem. 2003, 68, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanz, F.J.; Lajarin-Cuesta, R.; Moreno-Ortega, A.J.; Gonzalez-Lafuente, L.; Fernandez-Morales, J.C.; Lopez-Arribas, R.; Cano-Abad, M.F.; de los Rios, C. Benzothiazepine CGP37157 Analogues Exert Cytoprotection in Various in Vitro Models of Neurodegeneration. ACS Chem. Neurosci. 2015, 6, 1626–1636. [Google Scholar] [CrossRef]
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Gonzalez, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; Lopez, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalska, P.; Tenti, G.; Satriani, M.; Cores, A.; Ramos, M.T.; Garcia, A.G.; Menendez, J.C.; Leon, R. Aza-CGP37157-lipoic hybrids designed as novel Nrf2-inducers and antioxidants exert neuroprotection against oxidative stress and show neuroinflammation inhibitory properties. Drug Dev. Res. 2020, 81, 283–294. [Google Scholar] [CrossRef]
- Cores, A. Natural Product-Related Multitarget-Directed Ligands for the Potential Treatment of Neurodegenerative Diseases; E-Prints Complutense; Universidad Complutense de Madrid: Madrid, Spain, 2019. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2021-4 LLC. QikProp, Schrödinger, 2021–2024; Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger Release 2021-4 LLC. LigPrep, Schrödinger; Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Dudonne, S.; Vitrac, X.; Coutiere, P.; Woillez, M.; Merillon, J.M. Comparative study of antioxidant properties and total phenolic content of 30 plant extracts of industrial interest using DPPH, ABTS, FRAP, SOD, and ORAC assays. J. Agric. Food Chem. 2009, 57, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colyer, J.T.; Andersen, N.G.; Tedrow, J.S.; Soukup, T.S.; Faul, M.M. Reversal of diastereofacial selectivity in hydride reductions of N-tert-butanesulfinyl imines. J. Org. Chem. 2006, 71, 6859–6862. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Luco, J.M. Prediction of the brain-blood distribution of a large set of drugs from structurally derived descriptors using partial least-squares (PLS) modeling. J. Chem. Inf. Comput. Sci. 1999, 39, 396–404. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar Molecular Surface as a Dominating Determinant for Oral Absorption and Brain Penetration of Drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Apak, R.; Ozyurek, M.; Guclu, K.; Capanoglu, E. Antioxidant Activity/Capacity Measurement. 2. Hydrogen Atom Transfer (HAT)-Based, Mixed-Mode (Electron Transfer (ET)/HAT), and Lipid Peroxidation Assays. J. Agric. Food Chem. 2016, 64, 1028–1045. [Google Scholar] [CrossRef]
- Yamada, T.; Hashida, K.; Takarada-Iemata, M.; Matsugo, S.; Hori, O. alpha-Lipoic acid (LA) enantiomers protect SH-SY5Y cells against glutathione depletion. Neurochem. Int. 2011, 59, 1003–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buendia, I.; Tenti, G.; Michalska, P.; Mendez-Lopez, I.; Luengo, E.; Satriani, M.; Padin-Nogueira, F.; Lopez, M.G.; Ramos, M.T.; Garcia, A.G.; et al. ITH14001, a CGP37157-Nimodipine Hybrid Designed to Regulate Calcium Homeostasis and Oxidative Stress, Exerts Neuroprotection in Cerebral Ischemia. ACS Chem. Neurosci. 2017, 8, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Shiosaka, M.; Ogino, T.; Okimura, Y.; Utsumi, T.; Sato, E.F.; Akagi, R.; Inoue, M.; Utsumi, K.; Sasaki, J. Alpha-lipoic acid suppresses 6-hydroxydopamine-induced ROS generation and apoptosis through the stimulation of glutathione synthesis but not by the expression of heme oxygenase-1. Brain Res. 2008, 1206, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lii, C.K.; Liu, K.L.; Cheng, Y.P.; Lin, A.H.; Chen, H.W.; Tsai, C.W. Sulforaphane and alpha-lipoic acid upregulate the expression of the pi class of glutathione S-transferase through c-jun and Nrf2 activation. J. Nutr. 2010, 140, 885–892. [Google Scholar] [CrossRef] [Green Version]
- Kilic, F.; Handelman, G.J.; Traber, K.; Tsang, K.; Packer, L.; Trevithick, J.R. Modelling cortical cataractogenesis XX. In vitro effect of alpha-lipoic acid on glutathione concentrations in lens in model diabetic cataractogenesis. Biochem. Mol. Biol. Int. 1998, 46, 585–595. [Google Scholar] [PubMed]
- Wolz, P.; Krieglstein, J. Neuroprotective effects of alpha-lipoic acid and its enantiomers demonstrated in rodent models of focal cerebral ischemia. Neuropharmacology 1996, 35, 369–375. [Google Scholar] [CrossRef]
- Hagen, T.M.; Vinarsky, V.; Wehr, C.M.; Ames, B.N. (R)-alpha-lipoic acid reverses the age-associated increase in susceptibility of hepatocytes to tert-butylhydroperoxide both in vitro and in vivo. Antioxid. Redox Signal. 2000, 2, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Biewenga, G.P.; Dorstijn, M.A.; Verhagen, J.V.; Haenen, G.R.; Bast, A. Reduction of lipoic acid by lipoamide dehydrogenase. Biochem. Pharmacol. 1996, 51, 233–238. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Gyengesi, E.; Munch, G. In search of an anti-inflammatory drug for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev. Immunol 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Coneski, P.N.; Schoenfisch, M.H. Nitric oxide release: Part III. Measurement and reporting. Chem. Soc. Rev. 2012, 41, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Tenti, G.; Parada, E.; Leon, R.; Egea, J.; Martinez-Revelles, S.; Briones, A.M.; Sridharan, V.; Lopez, M.G.; Ramos, M.T.; Menendez, J.C. New 5-unsubstituted dihydropyridines with improved CaV1.3 selectivity as potential neuroprotective agents against ischemic injury. J. Med. Chem. 2014, 57, 4313–4323. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Perry, G.; Zhu, X.; Moreira, P.I.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 940603. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xiong, S.; Xie, C.; Davies, P.; Markesbery, W.R. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimer’s Dis. 2004, 6, 659–671; discussion 673–681. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badia, M.C.; Giraldo, E.; Ermak, G.; Alonso, M.D.; Pallardo, F.V.; Davies, K.J.; Vina, J. Amyloid-beta toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 701–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid. Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, J.; Gupta, N.; Agrawal, M.; Bala Bhaskar, A.S.; Lakshmana Rao, P.V. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 2011, 16, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Cores, A.; Abril, S.; Michalska, P.; Duarte, P.; Olives, A.I.; Martin, M.A.; Villacampa, M.; Leon, R.; Menendez, J.C. Bisavenathramide Analogues as Nrf2 Inductors and Neuroprotectors in In Vitro Models of Oxidative Stress and Hyperphosphorylation. Antioxidants 2021, 10, 941. [Google Scholar] [CrossRef]
- Medina, M.; Avila, J.; Villanueva, N. Use of okadaic acid to identify relevant phosphoepitopes in pathology: A focus on neurodegeneration. Mar. Drugs 2013, 11, 1656–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagent | Yield (%) | dr 1 |
|---|---|---|---|
| 1 | NaBH4 | quant. | 71:29 |
| 2 | L-selectride | 0 | -- |
| 3 | DIBAL | quant. | 100:0 |
| 4 | (t-BuO)3LiAlH | quant. | 100:0 |
| Entry | Compound | MW (g/mol) a | TPSA (Å2) a | HBD a | HBA a | cLog P a | Oral Absorption b |
|---|---|---|---|---|---|---|---|
| 1 | CGP37157 | 324.22 | 54.40 | 1 | 1 | 3.9 | 100 |
| 2 | R-6 | 351.23 | 52.57 | 2 | 3 | 2.8 | 83 |
| 3 | S-6 | 351.23 | 52.57 | 2 | 3 | 2.8 | 85 |
| 4 | (S,S)-7a | 539.53 | 109.24 | 1 | 4 | 5.1 | 75 |
| 5 | (S,R)-7b | 539.53 | 109.24 | 1 | 4 | 5.2 | 75 |
| 6 | (R,S)-7c | 539.53 | 109.24 | 1 | 4 | 5.1 | 76 |
| 7 | (R,R)-7d | 539.53 | 109.24 | 1 | 4 | 5.2 | 81 |
| Entry | Compound | PPMDCK (nm/s) a | CNS MPO.v2 | CNS Prediction b |
|---|---|---|---|---|
| 1 | CGP37157 | 7461 | 5.2 | CNS + |
| 2 | R-6 | 465 | 5 | CNS + |
| 3 | S-6 | 668 | 5 | CNS + |
| 4 | (S,S)-7a | 1187 | 3 | CNS + |
| 5 | (S,R)-7b | 1203 | 3 | CNS + |
| 6 | (R,S)-7c | 1280 | 3 | CNS + |
| 7 | (R,R)-7d | 2613 | 3 | CNS + |
| Entry | Compound | DPPH | ||
|---|---|---|---|---|
| Scavenging at 100 µM, % | Scavenging at 1mM, % | IC50, μM | ||
| 1 | Trolox | 93.4 ± 0.5 | 92.2 ± 0.5 | 11.4 ± 1.0 |
| 2 | Ascorbic acid | - | - | 16.2 ± 0.7 |
| 3 | CGP37157 | 9.6 ± 1.8 | 24.3 ± 1.4 | - |
| 4 | rac-3a [19] | 15.1 ± 3.4 [19] | 37.4 ± 3.8 [19] | - |
| 5 | (rac,R)-3b [19] | 8.7 ± 5.1 [19] | 30.7 ± 3.9 [19] | - |
| 6 | R-6 | 10.30 ± 1.10 | 26.7 ± 1.3 | - |
| 7 | S-6 | 8.47 ± 2.42 | 25.3 ± 1.5 | - |
| 8 | (S,S)-7a | 10.9 ± 3.5 | 30.3 ± 1.6 | - |
| 9 | (S,R)-7b | 8.46 ± 3.8 | 32.0 ± 1.7 | - |
| 10 | (R,S)-7c | 13.6 ± 3.8 | 35.9 ± 1.9 | - |
| 11 | (R,R)-7d | 8.66 ± 3.0 | 23.3 ± 3.5 | - |
| Entry | Compound | CC50 (μM) |
|---|---|---|
| 1 | CGP37157 | 57.3 ± 2.3 |
| 2 | rac-3a [19] | 97.1 ± 2.3 |
| 3 | (rac,R)-3b [19] | 76.2 ± 4.8 |
| 4 | R-6 | >100 |
| 5 | S-6 | >100 |
| 6 | (S,S)-7a | >100 |
| 7 | (S,R)-7b | >100 |
| 8 | (R,S)-7c | >100 |
| 9 | (R,R)-7d | >100 |
| Entry | Compound | IC50 (μM) BV2 |
|---|---|---|
| 1 | CGP37157 | 21.9 ± 5.01 |
| 2 | (S)-Lipoic acid | >30 |
| 3 | (R)-Lipoic acid | >30 |
| 4 | rac-3a | 13.3 ± 3.65 |
| 5 | (rac,R)-3b | 27.4 ± 1.60 |
| 6 | R-6 | 28.5 ± 1.21 |
| 7 | S-6 | 19.9 ± 5.66 |
| 8 | (S,S)-7a | >30 |
| 9 | (S,R)-7b | 13.3 ± 3.68 |
| 10 | (R,S)-7c | >30 |
| 11 | (R,R)-7d | >30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cores, Á.; Michalska, P.; Pérez, J.M.; Crisman, E.; Gómez, C.; Villacampa, M.; Menéndez, J.C.; León, R. Enantioselective Synthesis and Pharmacological Evaluation of Aza-CGP37157–Lipoic Acid Hybrids for the Treatment of Alzheimer’s Disease. Antioxidants 2022, 11, 112. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11010112
Cores Á, Michalska P, Pérez JM, Crisman E, Gómez C, Villacampa M, Menéndez JC, León R. Enantioselective Synthesis and Pharmacological Evaluation of Aza-CGP37157–Lipoic Acid Hybrids for the Treatment of Alzheimer’s Disease. Antioxidants. 2022; 11(1):112. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11010112
Chicago/Turabian StyleCores, Ángel, Patrycja Michalska, José Miguel Pérez, Enrique Crisman, Clara Gómez, Mercedes Villacampa, José Carlos Menéndez, and Rafael León. 2022. "Enantioselective Synthesis and Pharmacological Evaluation of Aza-CGP37157–Lipoic Acid Hybrids for the Treatment of Alzheimer’s Disease" Antioxidants 11, no. 1: 112. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11010112