
Changing Perspectives from Oxidative Stress to Redox Signaling—Extracellular Redox Control in Translational Medicine


Abstract
:1. Changing Perspectives—From Oxidative Stress to Redox Signaling
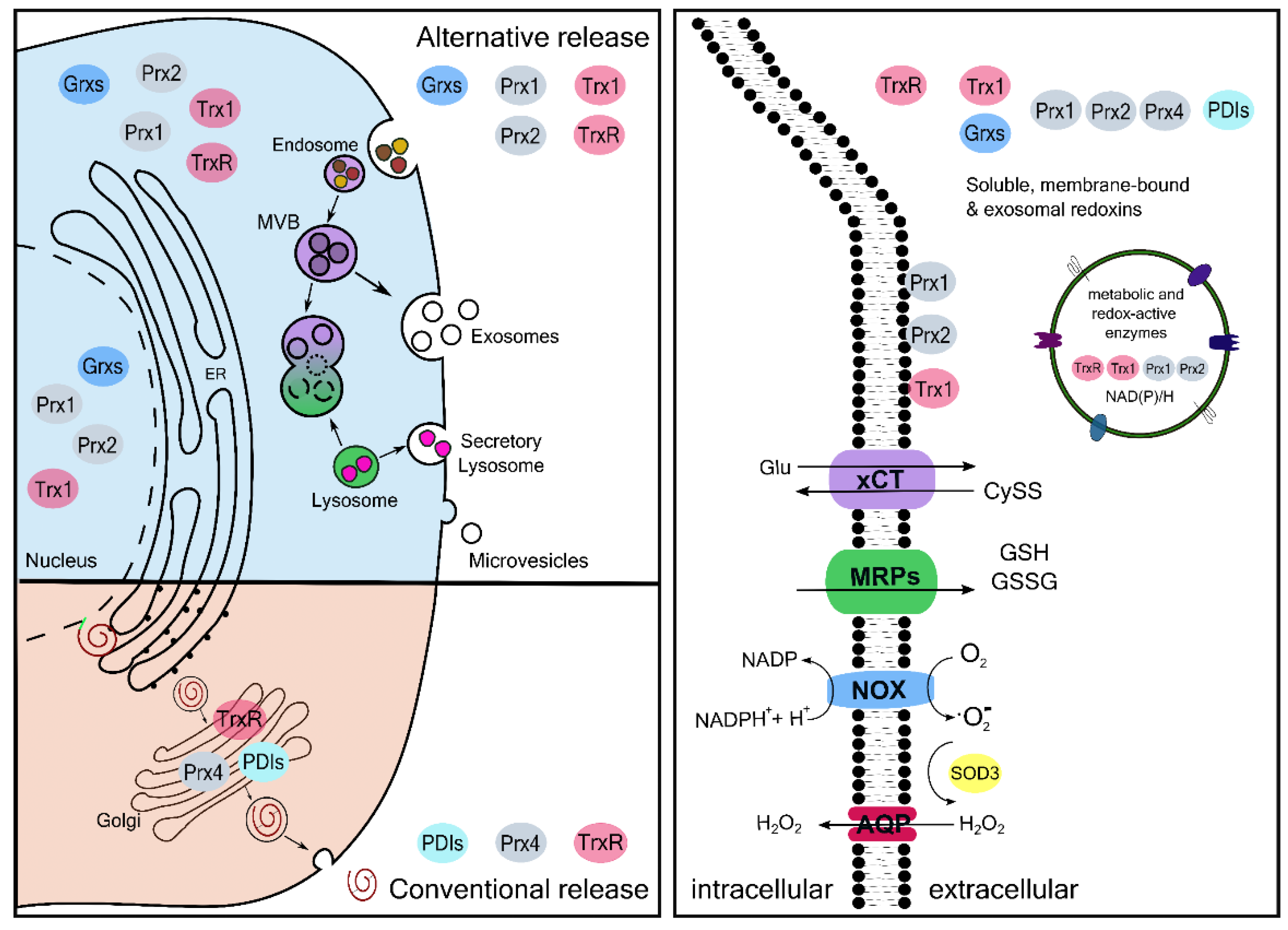
Extracellular Redoxins
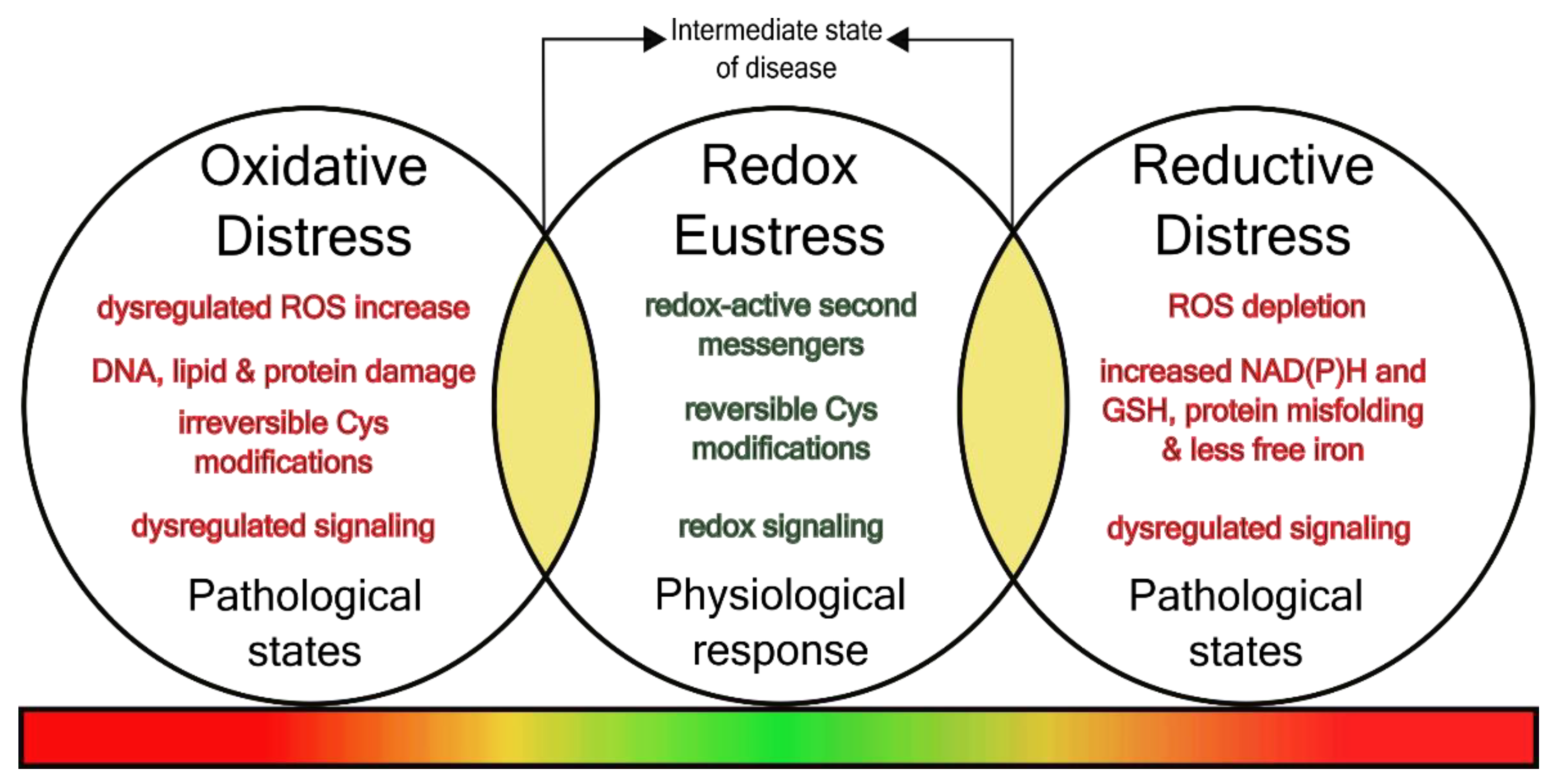
2. Redox Regulation in and between Health and Disease
2.1. Extracellular Redox Changes in Intermediate Pathophenotypes
2.1.1. Thrombosis
2.1.2. Fibrosis
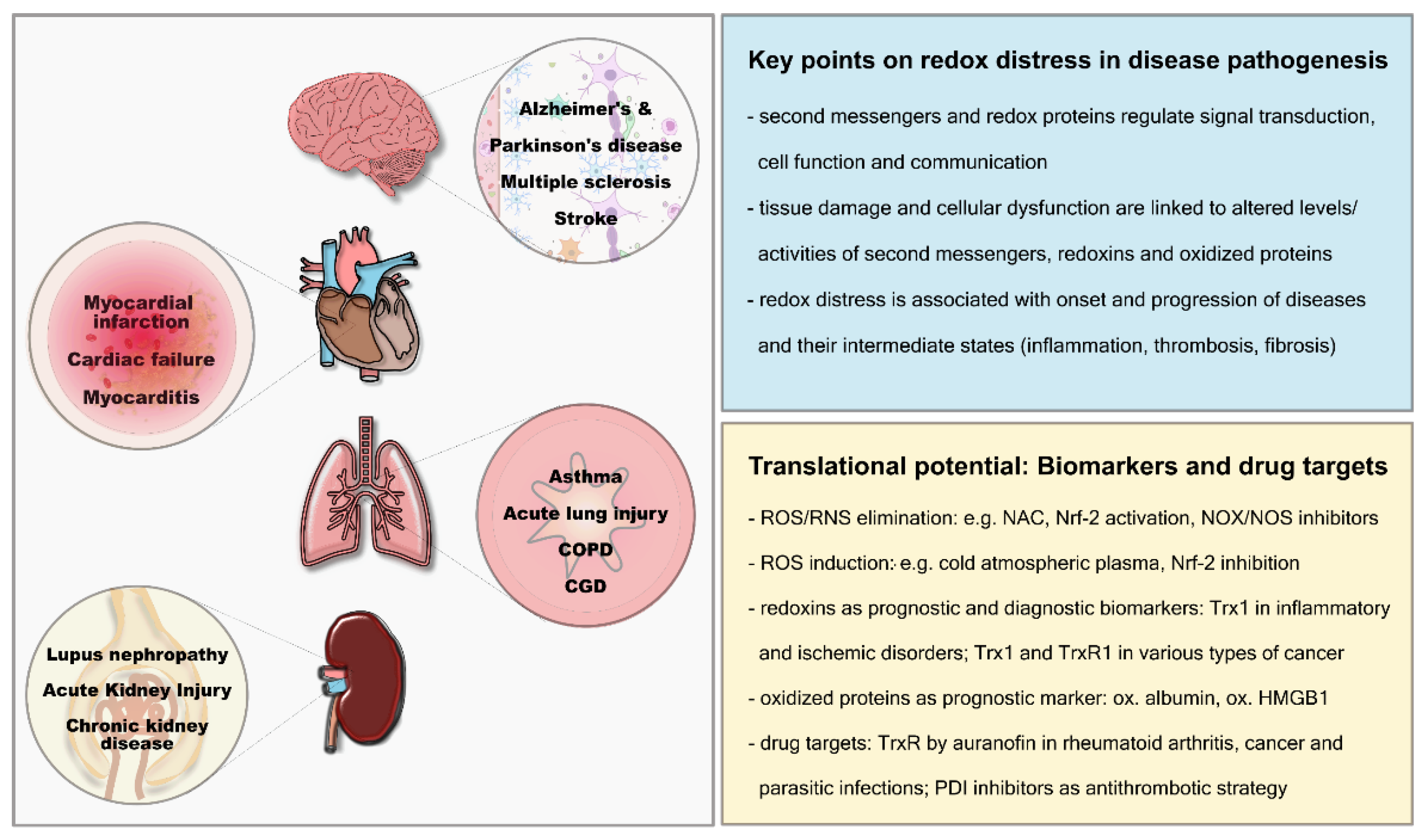
2.2. Extracellular Redox Changes in Pathologies
2.2.1. Cardiovascular System
2.2.2. Respiratory System
2.2.3. Neurological Disorders
2.2.4. Autoimmune Diseases
2.2.5. Cancer
3. Redoxins and Thiols as Biomarkers, Risk Factors and/or Drug Targets
3.1. Reactive Oxygen and Nitrogen Species
3.2. Oxidation Products
3.3. Antioxidants and Redox Enzymes
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, 14D–19D. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.M.; Holt, A.G. Why antioxidant therapies have failed in clinical trials. J. Theor. Biol. 2018, 457, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dangubic, V.; Tomic, I.; Sikimic, S.; Mijailovic, Z.; Videnovic, J.; Batakovic-Stupar, V.; Predragovic, R.; Karlicic, V. [Personal experience in the diagnosis and treatment of pulmonary embolism]. Vojn. Pregl. 1990, 47, 182–185. [Google Scholar]
- Narasimhan, M.; Rajasekaran, N.S. Reductive potential—A savior turns stressor in protein aggregation cardiomyopathy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. The redox proteome. J. Biol. Chem. 2013, 288, 26512–26520. [Google Scholar] [CrossRef] [Green Version]
- Zeke, A.; Dobson, L.; Szekeres, L.I.; Lango, T.; Tusnady, G.E. PolarProtDb: A Database of Transmembrane and Secreted Proteins showing Apical-Basal Polarity. J. Mol. Biol. 2021, 433, 166705. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Tanaka, L.Y.; Oliveira, P.V.S.; Laurindo, F.R.M. Peri/Epicellular Thiol Oxidoreductases as Mediators of Extracellular Redox Signaling. Antioxid. Redox Signal. 2020, 33, 280–307. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Eble, J.A.; Hanschmann, E.M. Thiol switches in membrane proteins—Extracellular redox regulation in cell biology. Biol. Chem. 2021, 402, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Carlson, J.L.; Mody, V.C.; Cai, J.; Lynn, M.J.; Sternberg, P. Redox state of glutathione in human plasma. Free Radic. Biol. Med. 2000, 28, 625–635. [Google Scholar] [CrossRef]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [CrossRef]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular superoxide dismutase and its role in cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef]
- Forman, H.J.; Bernardo, A.; Davies, K.J. What is the concentration of hydrogen peroxide in blood and plasma? Arch. Biochem. Biophys. 2016, 603, 48–53. [Google Scholar] [CrossRef]
- Bouayed, J.; Bohn, T. Exogenous antioxidants—Double-edged swords in cellular redox state: Health beneficial effects at physiologic doses versus deleterious effects at high doses. Oxidative Med. Cell. Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef]
- Cantin, A.M.; North, S.L.; Hubbard, R.C.; Crystal, R.G. Normal alveolar epithelial lining fluid contains high levels of glutathione. J. Appl. Physiol. 1987, 63, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Venglarik, C.J.; Giron-Calle, J.; Wigley, A.F.; Malle, E.; Watanabe, N.; Forman, H.J. Hypochlorous acid alters bronchial epithelial cell membrane properties and prevention by extracellular glutathione. J. Appl. Physiol. 2003, 95, 2444–2452. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar] [CrossRef]
- McGann, J.C.; Mandel, G. Neuronal activity induces glutathione metabolism gene expression in astrocytes. Glia 2018, 66, 2024–2039. [Google Scholar] [CrossRef] [PubMed]
- Gil-Bea, F.; Akterin, S.; Persson, T.; Mateos, L.; Sandebring, A.; Avila-Carino, J.; Gutierrez-Rodriguez, A.; Sundstrom, E.; Holmgren, A.; Winblad, B.; et al. Thioredoxin-80 is a product of alpha-secretase cleavage that inhibits amyloid-beta aggregation and is decreased in Alzheimer’s disease brain. EMBO Mol. Med. 2012, 4, 1097–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanschmann, E.M.; Petry, S.F.; Eitner, S.; Maresch, C.C.; Lingwal, N.; Lillig, C.H.; Linn, T. Paracrine regulation and improvement of beta-cell function by thioredoxin. Redox Biol. 2020, 34, 101570. [Google Scholar] [CrossRef] [PubMed]
- Yodoi, J.; Tian, H.; Masutani, H.; Nakamura, H. Thiol redox barrier; local and systemic surveillance against stress and inflammatory diseases. Arch. Biochem. Biophys. 2016, 595, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Pekkari, K.; Avila-Carino, J.; Bengtsson, A.; Gurunath, R.; Scheynius, A.; Holmgren, A. Truncated thioredoxin (Trx80) induces production of interleukin-12 and enhances CD14 expression in human monocytes. Blood 2001, 97, 3184–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.Z.; Sukumar, P.; Zeng, F.; Li, J.; Jairaman, A.; English, A.; Naylor, J.; Ciurtin, C.; Majeed, Y.; Milligan, C.J.; et al. TRPC channel activation by extracellular thioredoxin. Nature 2008, 451, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Bergerhausen, L.; Grosche, J.; Meissner, J.; Hecker, C.; Caliandro, M.F.; Westerhausen, C.; Kamenac, A.; Rezaei, M.; Morgelin, M.; Poschmann, G.; et al. Extracellular Redox Regulation of alpha7beta Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch. Antioxidants 2020, 9, 227. [Google Scholar] [CrossRef] [Green Version]
- Schwertassek, U.; Balmer, Y.; Gutscher, M.; Weingarten, L.; Preuss, M.; Engelhard, J.; Winkler, M.; Dick, T.P. Selective redox regulation of cytokine receptor signaling by extracellular thioredoxin-1. EMBO J. 2007, 26, 3086–3097. [Google Scholar] [CrossRef]
- Soderberg, A.; Sahaf, B.; Rosen, A. Thioredoxin reductase, a redox-active selenoprotein, is secreted by normal and neoplastic cells: Presence in human plasma. Cancer Res. 2000, 60, 2281–2289. [Google Scholar]
- Bodega, G.; Alique, M.; Bohorquez, L.; Moran, M.; Magro, L.; Puebla, L.; Ciordia, S.; Mena, M.C.; Arza, E.; Ramirez, M.R. Young and Especially Senescent Endothelial Microvesicles Produce NADPH: The Fuel for Their Antioxidant Machinery. Oxidative Med. Cell. Longev. 2018, 2018, 3183794. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, J.E.; Martin, E.M.; Aksamitiene, E.; Mukherjee, P.; Alex, A.; Chaney, E.J.; Marjanovic, M.; Boppart, S.A. Label-free characterization of single extracellular vesicles using two-photon fluorescence lifetime imaging microscopy of NAD(P)H. Sci. Rep. 2021, 11, 3308. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, M.; Fernandes, A.P.; Kumar, S.; Holmgren, A. Cellular and plasma levels of human glutaredoxin 1 and 2 detected by sensitive ELISA systems. Biochem. Biophys. Res. Commun. 2004, 319, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.-T.; Van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, O.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; et al. EV-TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods 2017, 14, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Kubota, K.; Yamada, N.; Tagami, U.; Takehana, K.; Sonaka, I.; Suzuki, E.; Hirayama, K. Identification and characterization of oxidized human serum albumin. A slight structural change impairs its ligand-binding and antioxidant functions. FEBS J. 2006, 273, 3346–3357. [Google Scholar] [CrossRef]
- Schweigert, F.J.; Wirth, K.; Raila, J. Characterization of the microheterogeneity of transthyretin in plasma and urine using SELDI-TOF-MS immunoassay. Proteome Sci. 2004, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Checconi, P.; Salzano, S.; Bowler, L.; Mullen, L.; Mengozzi, M.; Hanschmann, E.M.; Lillig, C.H.; Sgarbanti, R.; Panella, S.; Nencioni, L.; et al. Redox proteomics of the inflammatory secretome identifies a common set of redoxins and other glutathionylated proteins released in inflammation, influenza virus infection and oxidative stress. PLoS ONE 2015, 10, e0127086. [Google Scholar] [CrossRef] [Green Version]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine Oxidation Targets Peroxiredoxins 1 and 2 for Exosomal Release through a Novel Mechanism of Redox-Dependent Secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef]
- Shau, H.; Butterfield, L.H.; Chiu, R.; Kim, A. Cloning and sequence analysis of candidate human natural killer-enhancing factor genes. Immunogenetics 1994, 40, 129–134. [Google Scholar] [CrossRef]
- Kramer, J.M.; Yi, L.; Shen, F.; Maitra, A.; Jiao, X.; Jin, T.; Gaffen, S.L. Evidence for ligand-independent multimerization of the IL-17 receptor. J. Immunol. 2006, 176, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.N.; Zhou, H.J.; Yang, X.J.; Zhao, L.X.; Kuang, X.; Chen, C.; Liu, D.L.; Du, J.R. Neuroprotective effect of a novel gastrodin derivative against ischemic brain injury: Involvement of peroxiredoxin and TLR4 signaling inhibition. Oncotarget 2017, 8, 90979–90995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossin, K.L. Nitric oxide (NO): A versatile second messenger in brain. Trends Biochem. Sci. 1991, 16, 81–82. [Google Scholar] [CrossRef]
- Garcia-Gimenez, J.L.; Roma-Mateo, C.; Perez-Machado, G.; Peiro-Chova, L.; Pallardo, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef]
- Ottaviano, F.G.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Regulation of the extracellular antioxidant selenoprotein plasma glutathione peroxidase (GPx-3) in mammalian cells. Mol. Cell. Biochem. 2009, 327, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000. [Google Scholar] [CrossRef]
- Haridas, V.; Ni, J.; Meager, A.; Su, J.; Yu, G.L.; Zhai, Y.; Kyaw, H.; Akama, K.T.; Hu, J.; Van Eldik, L.J.; et al. TRANK, a novel cytokine that activates NF-kappa B and c-Jun N-terminal kinase. J. Immunol. 1998, 161, 1–6. [Google Scholar]
- Yan, Z.; Spaulding, H.R. Extracellular superoxide dismutase, a molecular transducer of health benefits of exercise. Redox Biol. 2020, 32, 101508. [Google Scholar] [CrossRef]
- Manabe, Y.; Takagi, M.; Nakamura-Yamada, M.; Goto-Inoue, N.; Taoka, M.; Isobe, T.; Fujii, N.L. Redox proteins are constitutively secreted by skeletal muscle. J. Physiol. Sci. 2014, 64, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Couchie, D.; Vaisman, B.; Abderrazak, A.; Mahmood, D.F.D.; Hamza, M.M.; Canesi, F.; Diderot, V.; El Hadri, K.; Negre-Salvayre, A.; Le Page, A.; et al. Human Plasma Thioredoxin-80 Increases with Age and in ApoE(-/-) Mice Induces Inflammation, Angiogenesis, and Atherosclerosis. Circulation 2017, 136, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, L.; Mengozzi, M.; Hanschmann, E.M.; Alberts, B.; Ghezzi, P. How the redox state regulates immunity. Free Radic. Biol. Med. 2020, 157, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.M. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxidative Med. Cell. Longev. 2017, 2017, 8459402. [Google Scholar] [CrossRef] [Green Version]
- Knoops, B.; Argyropoulou, V.; Becker, S.; Ferte, L.; Kuznetsova, O. Multiple Roles of Peroxiredoxins in Inflammation. Mol. Cells 2016, 39, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Bayer, S.B.; Maghzal, G.; Stocker, R.; Hampton, M.B.; Winterbourn, C.C. Neutrophil-mediated oxidation of erythrocyte peroxiredoxin 2 as a potential marker of oxidative stress in inflammation. FASEB J. 2013, 27, 3315–3322. [Google Scholar] [CrossRef]
- Real, J.M.; Ferreira, L.R.P.; Esteves, G.H.; Koyama, F.C.; Dias, M.V.S.; Bezerra-Neto, J.E.; Cunha-Neto, E.; Machado, F.R.; Salomao, R.; Azevedo, L.C.P. Exosomes from patients with septic shock convey miRNAs related to inflammation and cell cycle regulation: New signaling pathways in sepsis? Crit. Care 2018, 22, 68. [Google Scholar] [CrossRef] [Green Version]
- Zandarashvili, L.; Sahu, D.; Lee, K.; Lee, Y.S.; Singh, P.; Rajarathnam, K.; Iwahara, J. Real-time kinetics of high-mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J. Biol. Chem. 2013, 288, 11621–11627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.S.; Kim, H.S.; Lkhamsuren, K.; Kim, Y.H.; Han, M.G.; Shin, J.M.; Park, I.H.; Rhee, W.J.; Lee, S.K.; Rhee, S.G.; et al. Peroxiredoxin-mediated disulfide bond formation is required for nucleocytoplasmic translocation and secretion of HMGB1 in response to inflammatory stimuli. Redox Biol. 2019, 24, 101203. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.; Chialli, G.; Ferreira, L.M.; Ruggieri, E.; Careccia, G.; Preti, A.; Piccirillo, R.; Bianchi, M.E.; Sitia, G.; Venereau, E. Oxidation of HMGB1 Is a Dynamically Regulated Process in Physiological and Pathological Conditions. Front. Immunol. 2020, 11, 1122. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High Mobility Group Box 1: Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases. Cells 2022, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ren, L.; Chen, Y.; Fang, J.; Ge, Z.; Li, X. Redox status of high-mobility group box 1 performs a dual role in angiogenesis of colorectal carcinoma. J. Cell. Mol. Med. 2015, 19, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.J.; Gong, H.; Wu, T.Y.; Su, W.J.; Zhang, Y.; Yang, Y.Y.; Peng, W.; Zhang, T.; Zhou, J.R.; Jiang, C.L.; et al. Ds-HMGB1 and fr-HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid-HMGB1. Brain. Behav. Immun. 2017, 59, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Abdulmahdi, W.; Patel, D.; Rabadi, M.M.; Azar, T.; Jules, E.; Lipphardt, M.; Hashemiyoon, R.; Ratliff, B.B. HMGB1 redox during sepsis. Redox Biol. 2017, 13, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, J.; Schneider, H.; Pagel, J.I.; Rehberg, M.; Singer, M.; Hellfritsch, J.; Chillo, O.; Schubert, K.M.; Qiu, J.; Pogoda, K.; et al. Endothelial Dysfunction, and A Prothrombotic, Proinflammatory Phenotype Is Caused by Loss of Mitochondrial Thioredoxin Reductase in Endothelium. Arter. Thromb. Vasc. Biol. 2016, 36, 1891–1899. [Google Scholar] [CrossRef] [Green Version]
- Okuda, M.; Inoue, N.; Azumi, H.; Seno, T.; Sumi, Y.; Hirata, K.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yodoi, J.; et al. Expression of glutaredoxin in human coronary arteries: Its potential role in antioxidant protection against atherosclerosis. Arter. Thromb. Vasc. Biol. 2001, 21, 1483–1487. [Google Scholar] [CrossRef] [Green Version]
- Stopa, J.D.; Neuberg, D.; Puligandla, M.; Furie, B.; Flaumenhaft, R.; Zwicker, J.I. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI Insight 2017, 2, e89373. [Google Scholar] [CrossRef] [Green Version]
- Giannakopoulos, B.; Gao, L.; Qi, M.; Wong, J.W.; Yu, D.M.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M.; Atsumi, T.; Koike, T.; Hogg, P.; et al. Factor XI is a substrate for oxidoreductases: Enhanced activation of reduced FXI and its role in antiphospholipid syndrome thrombosis. J. Autoimmun. 2012, 39, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, J.; Versteeg, H.H.; Kerver, M.; Chen, V.M.; Mueller, B.M.; Hogg, P.J.; Ruf, W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13932–13937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, F.; Ruf, W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb. Haemost. 2014, 111, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Chesterman, C.N.; Hogg, P.J. Reduction of von Willebrand factor by endothelial cells. Thromb. Haemost. 2000, 84, 506–513. [Google Scholar] [CrossRef]
- Passam, F.H.; Rahgozar, S.; Qi, M.; Raftery, M.J.; Wong, J.W.; Tanaka, K.; Ioannou, Y.; Zhang, J.Y.; Gemmell, R.; Qi, J.C.; et al. Redox control of beta2-glycoprotein I-von Willebrand factor interaction by thioredoxin-1. J. Thromb. Haemost. 2010, 8, 1754–1762. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, X.; Lv, X.; Jin, Q.; Shang, H.; Wang, C.C.; Wang, L. The extracellular Ero1alpha/PDI electron transport system regulates platelet function by increasing glutathione reduction potential. Redox Biol. 2022, 50, 102244. [Google Scholar] [CrossRef]
- Ulrich, M.M.; Verkerk, M.; Reijnen, L.; Vlig, M.; van den Bogaerdt, A.J.; Middelkoop, E. Expression profile of proteins involved in scar formation in the healing process of full-thickness excisional wounds in the porcine model. Wound Repair Regen. 2007, 15, 482–490. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, G.S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.Y.; Wu, L.C.; Dai, T.; Chen, S.Y.; Wang, A.Y.; Lin, K.; Lin, D.M.; Yang, J.Q.; Cheng, B.; Zhang, L.; et al. NADPH oxidase-2 is a key regulator of human dermal fibroblasts: A potential therapeutic strategy for the treatment of skin fibrosis. Exp. Dermatol. 2014, 23, 639–644. [Google Scholar] [CrossRef]
- Peltoniemi, M.; Kaarteenaho-Wiik, R.; Saily, M.; Sormunen, R.; Paakko, P.; Holmgren, A.; Soini, Y.; Kinnula, V.L. Expression of glutaredoxin is highly cell specific in human lung and is decreased by transforming growth factor-beta in vitro and in interstitial lung diseases in vivo. Hum. Pathol. 2004, 35, 1000–1007. [Google Scholar] [CrossRef]
- Islam, K.N.; Kayanoki, Y.; Kaneto, H.; Suzuki, K.; Asahi, M.; Fujii, J.; Taniguchi, N. TGF-beta1 triggers oxidative modifications and enhances apoptosis in HIT cells through accumulation of reactive oxygen species by suppression of catalase and glutathione peroxidase. Free Radic. Biol. Med. 1997, 22, 1007–1017. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal. 2014, 20, 2854–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarty-Craige, S.E.; Jones, D.P. Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 2004, 24, 481–509. [Google Scholar] [CrossRef]
- Banerjee, R. Redox outside the box: Linking extracellular redox remodeling with intracellular redox metabolism. J. Biol. Chem. 2012, 287, 4397–4402. [Google Scholar] [CrossRef] [Green Version]
- Soejima, H.; Suefuji, H.; Miyamoto, S.; Kajiwaram, I.; Kojima, S.; Hokamaki, J.; Sakamoto, T.; Yoshimura, M.; Nakamura, H.; Yodoi, J.; et al. Increased plasma thioredoxin in patients with acute myocardial infarction. Clin. Cardiol. 2003, 26, 583–587. [Google Scholar] [CrossRef]
- Mongardon, N.; Lemiale, V.; Borderie, D.; Burke-Gaffney, A.; Perbet, S.; Marin, N.; Charpentier, J.; Pene, F.; Chiche, J.D.; Mira, J.P.; et al. Plasma thioredoxin levels during post-cardiac arrest syndrome: Relationship with severity and outcome. Crit. Care 2013, 17, R18. [Google Scholar] [CrossRef] [Green Version]
- Shim, Y.K.; Kim, J.T.; Seong, M.H.; Kim, Y.J.; Shim, T.J.; Kim, S.M.; Lee, S.Y.; Bae, J.W.; Kim, K.S.; Hwang, K.K.; et al. Serum thioredoxin 1 level has close relation with myocardial damage amount in acute myocardial infarction patients. J. Korean Med. Sci. 2012, 27, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Jekell, A.; Hossain, A.; Alehagen, U.; Dahlstrom, U.; Rosen, A. Elevated circulating levels of thioredoxin and stress in chronic heart failure. Eur. J. Heart Fail. 2004, 6, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Wahlgren, C.M.; Pekkari, K. Elevated thioredoxin after angioplasty in peripheral arterial disease. Eur. J. Vasc. Endovasc. Surg. 2005, 29, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Godoy, J.R.; Pittrich, S.; Slavic, S.; Lillig, C.H.; Hanschmann, E.M.; Erben, R.G. Thioredoxin 1 is upregulated in the bone and bone marrow following experimental myocardial infarction: Evidence for a remote organ response. Histochem. Cell. Biol. 2021, 155, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Bertini, R.; Howard, O.M.; Dong, H.F.; Oppenheim, J.J.; Bizzarri, C.; Sergi, R.; Caselli, G.; Pagliei, S.; Romines, B.; Wilshire, J.A.; et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J. Exp. Med. 1999, 189, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.R.; Dias, I.H.; Willetts, R.S.; Devitt, A. Redox regulation of protein damage in plasma. Redox Biol. 2014, 2, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornstedt, M.; Xue, J.; Huang, W.; Akesson, B.; Holmgren, A. The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J. Biol. Chem. 1994, 269, 29382–29384. [Google Scholar] [CrossRef]
- Furukawa, M.; Tanaka, R.; Chuang, V.T.; Ishima, Y.; Taguchi, K.; Watanabe, H.; Maruyama, T.; Otagiri, M. Human serum albumin-thioredoxin fusion protein with long blood retention property is effective in suppressing lung injury. J. Control. Release 2011, 154, 189–195. [Google Scholar] [CrossRef]
- Kramer, P.A.; Chacko, B.K.; Ravi, S.; Johnson, M.S.; Mitchell, T.; Barnes, S.; Arabshahi, A.; Dell’Italia, L.J.; George, D.J.; Steele, C.; et al. Hemoglobin-associated oxidative stress in the pericardial compartment of postoperative cardiac surgery patients. Lab. Investig. 2015, 95, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Checa, J.; Aran, J.M. Airway Redox Homeostasis and Inflammation Gone Awry: From Molecular Pathogenesis to Emerging Therapeutics in Respiratory Pathology. Int. J. Mol. Sci. 2020, 21, 9317. [Google Scholar] [CrossRef]
- Tanaka, K.I.; Kubota, M.; Shimoda, M.; Hayase, T.; Miyaguchi, M.; Kobayashi, N.; Ikeda, M.; Ishima, Y.; Kawahara, M. Thioredoxin-albumin fusion protein prevents urban aerosol-induced lung injury via suppressing oxidative stress-related neutrophil extracellular trap formation. Environ. Pollut. 2021, 268, 115787. [Google Scholar] [CrossRef]
- Yamada, Y.; Nakamura, H.; Adachi, T.; Sannohe, S.; Oyamada, H.; Kayaba, H.; Yodoi, J.; Chihara, J. Elevated serum levels of thioredoxin in patients with acute exacerbation of asthma. Immunol. Lett. 2003, 86, 199–205. [Google Scholar] [CrossRef]
- Moriarty, S.E.; Shah, J.H.; Lynn, M.; Jiang, S.; Openo, K.; Jones, D.P.; Sternberg, P. Oxidation of glutathione and cysteine in human plasma associated with smoking. Free Radic. Biol. Med. 2003, 35, 1582–1588. [Google Scholar] [CrossRef]
- Peltoniemi, M.J.; Rytila, P.H.; Harju, T.H.; Soini, Y.M.; Salmenkivi, K.M.; Ruddock, L.W.; Kinnula, V.L. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir. Res. 2006, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, S.B.; Nolin, J.D.; Aboushousha, R.; Erikson, C.; Irvin, C.G.; Poynter, M.E.; van der Velden, J.; Taatjes, D.J.; van der Vliet, A.; Anathy, V.; et al. Glutaredoxin deficiency promotes activation of the transforming growth factor beta pathway in airway epithelial cells, in association with fibrotic airway remodeling. Redox Biol. 2020, 37, 101720. [Google Scholar] [CrossRef] [PubMed]
- Rostila, A.M.; Anttila, S.L.; Lalowski, M.M.; Vuopala, K.S.; Toljamo, T.I.; Lindstrom, I.; Baumann, M.H.; Puustinen, A.M. Reactive oxygen species-regulating proteins peroxiredoxin 2 and thioredoxin, and glyceraldehyde-3-phosphate dehydrogenase are differentially abundant in induced sputum from smokers with lung cancer or asbestos exposure. Eur. J. Cancer Prev. 2020, 29, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, I.; Louis, R.; Manise, M.; Dentener, M.A.; Irvin, C.G.; Janssen-Heininger, Y.M.; Brightling, C.E.; Wouters, E.F.; Reynaert, N.L. Increased glutaredoxin-1 and decreased protein S-glutathionylation in sputum of asthmatics. Eur. Respir. J. 2013, 41, 469–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, K.; Nakamura, H.; Nakamura, T.; Hoshino, Y.; Ueda, S.; Ichikawa, M.; Tabata, C.; Fujita, S.; Masago, K.; Yodoi, J.; et al. Elevation of serum thioredoxin in patients with gefitinib-induced interstitial lung disease. Intern. Med. 2007, 46, 1905–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anathy, V.; Lahue, K.G.; Chapman, D.G.; Chia, S.B.; Casey, D.T.; Aboushousha, R.; van der Velden, J.L.J.; Elko, E.; Hoffman, S.M.; McMillan, D.H.; et al. Reducing protein oxidation reverses lung fibrosis. Nat. Med. 2018, 24, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.M.; Torres, S.; Siedlak, S.L.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. Antimicrobial peptide beta-defensin-1 expression is upregulated in Alzheimer’s brain. J. Neuroinflamm. 2013, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Rocha-Ferreira, E.; Hristova, M. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front. Immunol. 2015, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Arodin, L.; Lamparter, H.; Karlsson, H.; Nennesmo, I.; Bjornstedt, M.; Schroder, J.; Fernandes, A.P. Alteration of thioredoxin and glutaredoxin in the progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 787–797. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Tavassolifar, M.J.; Vodjgani, M.; Salehi, Z.; Izad, M. The Influence of Reactive Oxygen Species in the Immune System and Pathogenesis of Multiple Sclerosis. Autoimmune Dis. 2020, 2020, 5793817. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.Z.; Guan, W.P.; Maeda, T.; Guoqing, X.; GuangZhi, W.; Makino, N. Patients with multiple sclerosis show increased oxidative stress markers and somatic telomere length shortening. Mol. Cell. Biochem. 2015, 400, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Karlik, M.; Valkovic, P.; Hancinova, V.; Krizova, L.; Tothova, L.; Celec, P. Markers of oxidative stress in plasma and saliva in patients with multiple sclerosis. Clin. Biochem. 2015, 48, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, E.; Khalilnezhad, A.; Gharagozli, K.; Amani, D. Thioredoxin-1, redox factor-1 and thioredoxin-interacting protein, mRNAs are differentially expressed in Multiple Sclerosis patients exposed and non-exposed to interferon and immunosuppressive treatments. Gene 2017, 634, 29–36. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Adamczyk-Sowa, M.; Gajewska, A.; Bartosz, G. Oxidative modification of blood serum proteins in multiple sclerosis after interferon or mitoxantrone treatment. J. Neuroimmunol. 2014, 266, 67–74. [Google Scholar] [CrossRef]
- Buonora, J.E.; Mousseau, M.; Jacobowitz, D.M.; Lazarus, R.C.; Yarnell, A.M.; Olsen, C.H.; Pollard, H.B.; Diaz-Arrastia, R.; Latour, L.; Mueller, G.P. Autoimmune Profiling Reveals Peroxiredoxin 6 as a Candidate Traumatic Brain Injury Biomarker. J. Neurotrauma 2015, 32, 1805–1814. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, X.S.; Zhang, Z.H.; Zhou, X.M.; Gao, Y.Y.; Liu, G.J.; Wang, H.; Wu, L.Y.; Li, W.; Hang, C.H. Peroxiredoxin 2 activates microglia by interacting with Toll-like receptor 4 after subarachnoid hemorrhage. J. Neuroinflamm. 2018, 15, 87. [Google Scholar] [CrossRef]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef]
- Hayem, G.; Nicaise-Roland, P.; Palazzo, E.; de Bandt, M.; Tubach, F.; Weber, M.; Meyer, O. Anti-oxidized low-density-lipoprotein (OxLDL) antibodies in systemic lupus erythematosus with and without antiphospholipid syndrome. Lupus 2001, 10, 346–351. [Google Scholar] [CrossRef]
- Mannering, S.I.; Harrison, L.C.; Williamson, N.A.; Morris, J.S.; Thearle, D.J.; Jensen, K.P.; Kay, T.W.; Rossjohn, J.; Falk, B.A.; Nepom, G.T.; et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J. Exp. Med. 2005, 202, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Khojah, H.M.; Ahmed, S.; Abdel-Rahman, M.S.; Hamza, A.B. Reactive oxygen and nitrogen species in patients with rheumatoid arthritis as potential biomarkers for disease activity and the role of antioxidants. Free Radic. Biol. Med. 2016, 97, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Maurice, M.M.; Nakamura, H.; Gringhuis, S.; Okamoto, T.; Yoshida, S.; Kullmann, F.; Lechner, S.; van der Voort, E.A.; Leow, A.; Versendaal, J.; et al. Expression of the thioredoxin-thioredoxin reductase system in the inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum 1999, 42, 2430–2439. [Google Scholar] [CrossRef]
- Jikimoto, T.; Nishikubo, Y.; Koshiba, M.; Kanagawa, S.; Morinobu, S.; Morinobu, A.; Saura, R.; Mizuno, K.; Kondo, S.; Toyokuni, S.; et al. Thioredoxin as a biomarker for oxidative stress in patients with rheumatoid arthritis. Mol. Immunol. 2002, 38, 765–772. [Google Scholar] [CrossRef]
- Pavlakis, E.; Neumann, M.; Stiewe, T. Extracellular Vesicles: Messengers of p53 in Tumor-Stroma Communication and Cancer Metastasis. Int. J. Mol. Sci. 2020, 21, 9648. [Google Scholar] [CrossRef]
- Abbaszade Dibavar, M.; Pourbagheri-Sigaroodi, A.; Asemani, Y.; Salari, S.; Bashash, D. Extracellular vesicles (EVs): What we know of the mesmerizing roles of these tiny vesicles in hematological malignancies? Life Sci. 2021, 271, 119177. [Google Scholar] [CrossRef]
- Li, J.; Cheng, Z.J.; Liu, Y.; Yan, Z.L.; Wang, K.; Wu, D.; Wan, X.Y.; Xia, Y.; Lau, W.Y.; Wu, M.C.; et al. Serum thioredoxin is a diagnostic marker for hepatocellular carcinoma. Oncotarget 2015, 6, 9551–9563. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Bai, J.; Nishinaka, Y.; Ueda, S.; Sasada, T.; Ohshio, G.; Imamura, M.; Takabayashi, A.; Yamaoka, Y.; Yodoi, J. Expression of thioredoxin and glutaredoxin, redox-regulating proteins, in pancreatic cancer. Cancer Detect. Prev. 2000, 24, 53–60. [Google Scholar]
- Peng, W.; Zhou, Z.; Zhong, Y.; Sun, Y.; Wang, Y.; Zhu, Z.; Jiao, W.; Bai, M.; Sun, J.; Lu, J.; et al. Plasma activity of Thioredoxin Reductase as a Novel Biomarker in Gastric Cancer. Sci. Rep. 2019, 9, 19084. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Chen, X.; Yao, Y.; Li, Y.; Sun, R.; Zeng, H.; Shu, Y.; Yin, H. Thioredoxin Reductase as a Novel and Efficient Plasma Biomarker for the Detection of Non-Small Cell Lung Cancer: A Large-scale, Multicenter study. Sci. Rep. 2019, 9, 2652. [Google Scholar] [CrossRef]
- Hughes, N.P.; Xu, L.; Nielsen, C.H.; Chang, E.; Hori, S.S.; Natarajan, A.; Lee, S.; Kjaer, A.; Kani, K.; Wang, S.X.; et al. A blood biomarker for monitoring response to anti-EGFR therapy. Cancer Biomark 2018, 22, 333–344. [Google Scholar] [CrossRef]
- Chaiswing, L.; Zhong, W.; Cullen, J.J.; Oberley, L.W.; Oberley, T.D. Extracellular redox state regulates features associated with prostate cancer cell invasion. Cancer Res. 2008, 68, 5820–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.P.; Yu, G.; Tseng, G.; Cieply, K.; Nelson, J.; Defrances, M.; Zarnegar, R.; Michalopoulos, G.; Luo, J.H. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007, 67, 8043–8050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Weiss, H.L.; Jayswal, R.D.; Hensley, P.J.; Downes, L.M.; St Clair, D.K.; Chaiswing, L. Extracellular redox state shift: A novel approach to target prostate cancer invasion. Free Radic. Biol. Med. 2018, 117, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef]
- Lukaszewicz, A.C.; Faivre, V.; Bout, H.; Gayat, E.; Lagergren, T.; Damoisel, C.; Bresson, D.; Paugam, C.; Mantz, J.; Payen, D. Multicenter testing of the rapid quantification of radical oxygen species in cerebrospinal fluid to diagnose bacterial meningitis. PLoS ONE 2015, 10, e0128286. [Google Scholar] [CrossRef] [Green Version]
- Savastano, M.; Brescia, G.; Marioni, G. Antioxidant therapy in idiopathic tinnitus: Preliminary outcomes. Arch. Med. Res. 2007, 38, 456–459. [Google Scholar] [CrossRef]
- Tavassolifar, M.J.; Moghadasi, A.N.; Esmaeili, B.; Sadatpour, O.; Vodjgani, M.; Izad, M. Redox Imbalance in CD4+ T Cells of Relapsing-Remitting Multiple Sclerosis Patients. Oxidative Med. Cell. Longev. 2020, 2020, 8860813. [Google Scholar] [CrossRef]
- De Riccardis, L.; Rizzello, A.; Ferramosca, A.; Urso, E.; De Robertis, F.; Danieli, A.; Giudetti, A.M.; Trianni, G.; Zara, V.; Maffia, M. Bioenergetics profile of CD4(+) T cells in relapsing remitting multiple sclerosis subjects. J. Biotechnol. 2015, 202, 31–39. [Google Scholar] [CrossRef]
- Sokol, G.M.; Konduri, G.G.; Van Meurs, K.P. Inhaled nitric oxide therapy for pulmonary disorders of the term and preterm infant. Semin. Perinatol. 2016, 40, 356–369. [Google Scholar] [CrossRef] [Green Version]
- Mathru, M.; Huda, R.; Solanki, D.R.; Hays, S.; Lang, J.D. Inhaled nitric oxide attenuates reperfusion inflammatory responses in humans. Anesthesiology 2007, 106, 275–282. [Google Scholar] [CrossRef]
- Lang, J.D., Jr.; Teng, X.; Chumley, P.; Crawford, J.H.; Isbell, T.S.; Chacko, B.K.; Liu, Y.; Jhala, N.; Crowe, D.R.; Smith, A.B.; et al. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. J. Clin. Investig. 2007, 117, 2583–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streeter, E.; Ng, H.H.; Hart, J.L. Hydrogen sulfide as a vasculoprotective factor. Med. Gas. Res. 2013, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Privat-Maldonado, A.; Schmidt, A.; Lin, A.; Weltmann, K.D.; Wende, K.; Bogaerts, A.; Bekeschus, S. ROS from Physical Plasmas: Redox Chemistry for Biomedical Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 9062098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ding, P.; Xie, C.; Ye, C.; Ye, M.; Pan, C.; Cao, X.; Zhang, S.; Zheng, S. Potential application of the oxidative nucleic acid damage biomarkers in detection of diseases. Oncotarget 2017, 8, 75767–75777. [Google Scholar] [CrossRef] [Green Version]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxidative Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Hohn, A. Protein oxidation—Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef]
- Pascovici, D.; Wu, J.X.; McKay, M.J.; Joseph, C.; Noor, Z.; Kamath, K.; Wu, Y.; Ranganathan, S.; Gupta, V.; Mirzaei, M. Clinically Relevant Post-Translational Modification Analyses-Maturing Workflows and Bioinformatics Tools. Int. J. Mol. Sci. 2018, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Sinha, M.K.; Gaze, D.C.; Tippins, J.R.; Collinson, P.O.; Kaski, J.C. Ischemia modified albumin is a sensitive marker of myocardial ischemia after percutaneous coronary intervention. Circulation 2003, 107, 2403–2405. [Google Scholar] [CrossRef] [Green Version]
- Jiao, D.; Guo, F.; Yue, M.; Tian, Z. Ischemia-Modified Albumin Is Associated with Arterial Stiffness in Hemodialysis Patients. Int. Heart J. 2020, 61, 332–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, E.; Yasuda, K.; Takeda, N.; Sakata, S.; Era, S.; Kuwata, K.; Sogami, M.; Miura, K. Increased oxidized form of human serum albumin in patients with diabetes mellitus. Diabetes Res. Clin. Pract. 1992, 18, 153–158. [Google Scholar] [CrossRef]
- Watanabe, A.; Matsuzaki, S.; Moriwaki, H.; Suzuki, K.; Nishiguchi, S. Problems in serum albumin measurement and clinical significance of albumin microheterogeneity in cirrhotics. Nutrition 2004, 20, 351–357. [Google Scholar] [CrossRef]
- Nagumo, K.; Tanaka, M.; Chuang, V.T.; Setoyama, H.; Watanabe, H.; Yamada, N.; Kubota, K.; Tanaka, M.; Matsushita, K.; Yoshida, A.; et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS ONE 2014, 9, e85216. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Imafuku, T.; Suzuki, Y.; Nishida, K.; Matsusaka, K.; Shin, T.; Sato, Y.; Ishima, Y.; Watanabe, H.; Mimata, H.; et al. Changes in redox state of albumin before and after kidney transplantation in patients with end-stage renal disease. Clin. Biochem. 2020, 81, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Gericke, B.; Raila, J.; Deja, M.; Rohn, S.; Donaubauer, B.; Nagl, B.; Haebel, S.; Schweigert, F.J.; Kaisers, U. Alteration of transthyretin microheterogeneity in serum of multiple trauma patients. Biomark. Insights 2007, 2, 299–306. [Google Scholar] [CrossRef]
- Zhang, Q.; Kelly, J.W. Cys10 mixed disulfides make transthyretin more amyloidogenic under mildly acidic conditions. Biochemistry 2003, 42, 8756–8761. [Google Scholar] [CrossRef]
- Kaushik, V.; Brunnert, D.; Hanschmann, E.M.; Sharma, P.K.; Anand, B.G.; Kar, K.; Kateriya, S.; Goyal, P. The intrinsic amyloidogenic propensity of cofilin-1 is aggravated by Cys-80 oxidation: A possible link with neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2021, 569, 187–192. [Google Scholar] [CrossRef]
- Fiorini, A.; Koudriavtseva, T.; Bucaj, E.; Coccia, R.; Foppoli, C.; Giorgi, A.; Schinina, M.E.; Di Domenico, F.; De Marco, F.; Perluigi, M. Involvement of oxidative stress in occurrence of relapses in multiple sclerosis: The spectrum of oxidatively modified serum proteins detected by proteomics and redox proteomics analysis. PLoS ONE 2013, 8, e65184. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.W.; Tian, M.; Song, F.; Venereau, E.; Preti, A.; Park, S.W.; Hamilton, K.; Swapna, G.V.; Manohar, M.; Moreau, M.; et al. Aspirin’s Active Metabolite Salicylic Acid Targets High Mobility Group Box 1 to Modulate Inflammatory Responses. Mol. Med. 2015, 21, 526–535. [Google Scholar] [CrossRef]
- Cuello, F.; Shankar-Hari, M.; Mayr, U.; Yin, X.; Marshall, M.; Suna, G.; Willeit, P.; Langley, S.R.; Jayawardhana, T.; Zeller, T.; et al. Redox state of pentraxin 3 as a novel biomarker for resolution of inflammation and survival in sepsis. Mol. Cell. Proteom. 2014, 13, 2545–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Kodera, Y.; Saito, T.; Fujimoto, K.; Momozono, A.; Hayashi, A.; Kamata, Y.; Shichiri, M. Methionine sulfoxides in serum proteins as potential clinical biomarkers of oxidative stress. Sci. Rep. 2016, 6, 38299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momozono, A.; Kodera, Y.; Sasaki, S.; Nakagawa, Y.; Konno, R.; Shichiri, M. Oxidised Met(147) of human serum albumin is a biomarker of oxidative stress, reflecting glycaemic fluctuations and hypoglycaemia in diabetes. Sci. Rep. 2020, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Yang, T.C.; Hu, S.X.; Candy Chen, H.J. Multiple oxidative and advanced oxidative modifications of hemoglobin in gastric cancer patients measured by nanoflow LC-MS/MS. Clin. Chim. Acta 2022, 531, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Shet, A.S.; Pinto, S.M.; Mitra, G.; Mandal, A.K. Glutathionyl hemoglobin is elevated in iron deficiency anemia. Acta Haematol. 2012, 127, 26–30. [Google Scholar] [CrossRef]
- Verhoye, E.; Langlois, M.R.; Asklepios, I. Circulating oxidized low-density lipoprotein: A biomarker of atherosclerosis and cardiovascular risk? Clin. Chem. Lab. Med. 2009, 47, 128–137. [Google Scholar] [CrossRef]
- Winklhofer-Roob, B.M.; Faustmann, G.; Roob, J.M. Low-density lipoprotein oxidation biomarkers in human health and disease and effects of bioactive compounds. Free Radic. Biol. Med. 2017, 111, 38–86. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hubbard, R.C.; Crystal, R.G. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 1989, 139, 370–372. [Google Scholar] [CrossRef]
- Borok, Z.; Buhl, R.; Grimes, G.J.; Bokser, A.D.; Hubbard, R.C.; Holroyd, K.J.; Roum, J.H.; Czerski, D.B.; Cantin, A.M.; Crystal, R.G. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet 1991, 338, 215–216. [Google Scholar] [CrossRef]
- Meyer, A.; Buhl, R.; Kampf, S.; Magnussen, H. Intravenous N-acetylcysteine and lung glutathione of patients with pulmonary fibrosis and normals. Am. J. Respir. Crit. Care Med. 1995, 152, 1055–1060. [Google Scholar] [CrossRef]
- Meyer, A.; Buhl, R.; Magnussen, H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur. Respir. J. 1994, 7, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Pacht, E.R.; Diaz, P.; Clanton, T.; Hart, J.; Gadek, J.E. Alveolar fluid glutathione decreases in asymptomatic HIV-seropositive subjects over time. Chest 1997, 112, 785–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhl, R.; Jaffe, H.A.; Holroyd, K.J.; Wells, F.B.; Mastrangeli, A.; Saltini, C.; Cantin, A.M.; Crystal, R.G. Systemic glutathione deficiency in symptom-free HIV-seropositive individuals. Lancet 1989, 2, 1294–1298. [Google Scholar] [CrossRef]
- Pacht, E.R.; Diaz, P.; Clanton, T.; Hart, J.; Gadek, J.E. Alveolar fluid glutathione is not reduced in asymptomatic HIV-seropositive subjects. Am. J. Respir. Crit. Care Med. 1997, 155, 374–377. [Google Scholar] [CrossRef]
- Zhang, Q.; Ju, Y.; Ma, Y.; Wang, T. N-acetylcysteine improves oxidative stress and inflammatory response in patients with community acquired pneumonia: A randomized controlled trial. Medicine 2018, 97, e13087. [Google Scholar] [CrossRef]
- Moslehi, A.; Taghizadeh-Ghehi, M.; Gholami, K.; Hadjibabaie, M.; Jahangard-Rafsanjani, Z.; Sarayani, A.; Javadi, M.; Esfandbod, M.; Ghavamzadeh, A. N-acetyl cysteine for prevention of oral mucositis in hematopoietic SCT: A double-blind, randomized, placebo-controlled trial. Bone Marrow Transplant. 2014, 49, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Fowdar, K.; Chen, H.; He, Z.; Zhang, J.; Zhong, X.; Zhang, J.; Li, M.; Bai, J. The effect of N-acetylcysteine on exacerbations of chronic obstructive pulmonary disease: A meta-analysis and systematic review. Heart Lung 2017, 46, 120–128. [Google Scholar] [CrossRef]
- De Flora, S.; Grassi, C.; Carati, L. Attenuation of influenza-like symptomatology and improvement of cell-mediated immunity with long-term N-acetylcysteine treatment. Eur. Respir. J. 1997, 10, 1535–1541. [Google Scholar] [CrossRef]
- Poe, F.L.; Corn, J. N-Acetylcysteine: A potential therapeutic agent for SARS-CoV-2. Med. Hypotheses 2020, 143, 109862. [Google Scholar] [CrossRef]
- Zhou, N.; Yang, X.; Huang, A.; Chen, Z. The Potential Mechanism of N-acetylcysteine in Treating COVID-19. Curr. Pharm. Biotechnol. 2021, 22, 1584–1590. [Google Scholar] [CrossRef]
- Guerini, M.; Condro, G.; Friuli, V.; Maggi, L.; Perugini, P. N-acetylcysteine (NAC) and Its Role in Clinical Practice Management of Cystic Fibrosis (CF): A Review. Pharmaceuticals 2022, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Javaherforooshzadeh, F.; Shaker, Z.; Rashidi, M.; Akhondzadeh, R.; Hayati, F. The effect of N-acetyl cysteine injection on renal function after coronary artery bypass graft surgery: A randomized double blind clinical trial. J. Cardiothorac. Surg. 2021, 16, 161. [Google Scholar] [CrossRef] [PubMed]
- Koca, K.; Yurttas, Y.; Cayci, T.; Bilgic, S.; Kaldirim, U.; Durusu, M.; Cekli, Y.; Ozkan, H.; Hanci, V.; Purtuloglu, T.; et al. The role of preconditioning and N-acetylcysteine on oxidative stress resulting from tourniquet-induced ischemia-reperfusion in arthroscopic knee surgery. J. Trauma Acute Care Surg. 2011, 70, 717–723. [Google Scholar] [CrossRef]
- Momeni, M.; De Kock, M.; Devuyst, O.; Liistro, G. Effect of N-acetyl-cysteine and hyperoxia on erythropoietin production. Eur. J. Appl. Physiol. 2011, 111, 2681–2686. [Google Scholar] [CrossRef]
- Wang, J.; Wang, P.; Dong, C.; Zhao, Y.; Zhou, J.; Yuan, C.; Zou, L. Mechanisms of ebselen as a therapeutic and its pharmacology applications. Future Med. Chem. 2020, 12, 2141–2160. [Google Scholar] [CrossRef]
- Dao, V.T.; Casas, A.I.; Maghzal, G.J.; Seredenina, T.; Kaludercic, N.; Robledinos-Anton, N.; Di Lisa, F.; Stocker, R.; Ghezzi, P.; Jaquet, V.; et al. Pharmacology and Clinical Drug Candidates in Redox Medicine. Antioxid. Redox Signal. 2015, 23, 1113–1129. [Google Scholar] [CrossRef] [Green Version]
- Staurengo-Ferrari, L.; Badaro-Garcia, S.; Hohmann, M.S.N.; Manchope, M.F.; Zaninelli, T.H.; Casagrande, R.; Verri, W.A., Jr. Contribution of Nrf2 Modulation to the Mechanism of Action of Analgesic and Anti-inflammatory Drugs in Pre-clinical and Clinical Stages. Front. Pharmacol. 2018, 9, 1536. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult. Scler. J. 2017, 23, 1875–1883. [Google Scholar] [CrossRef]
- Xu, Z.M.; Li, M.J.; Tao, C. Serum and urinary thioredoxin concentrations are associated with severity of children hydronephrosis. Clin. Chim. Acta 2017, 466, 127–132. [Google Scholar] [CrossRef]
- Ahsan, M.K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox regulation of cell survival by the thioredoxin superfamily: An implication of redox gene therapy in the heart. Antioxid. Redox Signal. 2009, 11, 2741–2758. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shen, H.; Zhou, T.; Cao, X.; Chen, Y.; Liang, Y.; Lu, T.; He, J.; Dou, Z.; Liu, C.; et al. Early Elevation of Thioredoxin-1 Serum Levels Predicts 28-Day Mortality in Patients with Sepsis. J. Inflamm. Res. 2021, 14, 3837–3848. [Google Scholar] [CrossRef]
- Leaver, S.K.; MacCallum, N.S.; Pingle, V.; Hacking, M.B.; Quinlan, G.J.; Evans, T.W.; Burke-Gaffney, A. Increased plasma thioredoxin levels in patients with sepsis: Positive association with macrophage migration inhibitory factor. Intensive Care Med. 2010, 36, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.; Gidlof, A.; Eriksson, M.; Larsson, E.; Brattstrom, O.; Oldner, A. Thioredoxin a novel biomarker of post-injury sepsis. Free Radic. Biol. Med. 2017, 104, 138–143. [Google Scholar] [CrossRef]
- Wu, M.H.; Song, F.Y.; Wei, L.P.; Meng, Z.Y.; Zhang, Z.Q.; Qi, Q.D. Serum Levels of Thioredoxin Are Associated with Stroke Risk, Severity, and Lesion Volumes. Mol. Neurobiol. 2016, 53, 677–685. [Google Scholar] [CrossRef]
- Qi, A.Q.; Li, Y.; Liu, Q.; Si, J.Z.; Tang, X.M.; Zhang, Z.Q.; Qi, Q.D.; Chen, W.B. Thioredoxin is a novel diagnostic and prognostic marker in patients with ischemic stroke. Free Radic. Biol. Med. 2015, 80, 129–135. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, W.; Lin, Y.; Li, Q.; Xue, J.; Cai, Z.; Cheng, Y.; Shao, B. Prognostic value of serum thioredoxin levels in ischemic stroke. Neurol. Res. 2017, 39, 988–995. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, Y.; Choi, B.B.; Kim, J.R.; Ko, H.M.; Suh, K.H.; Lee, J.S. The blood level of thioredoxin 1 as a supporting biomarker in the detection of breast cancer. BMC Cancer 2022, 22, 12. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Q.; Lu, Y.; Zhang, J.; Yin, H.; Yi, Y. Clinical application of thioredoxin reductase as a novel biomarker in liver cancer. Sci. Rep. 2021, 11, 6069. [Google Scholar] [CrossRef]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arner, E.S.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Bombardier, C.; Ware, J.; Russell, I.J.; Larson, M.; Chalmers, A.; Read, J.L. Auranofin therapy and quality of life in patients with rheumatoid arthritis. Results of a multicenter trial. Am. J. Med. 1986, 81, 565–578. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; Furie, B.; Zwicker, J.I. Therapeutic implications of protein disulfide isomerase inhibition in thrombotic disease. Arter. Thromb. Vasc. Biol. 2015, 35, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerrits, E.G.; Alkhalaf, A.; Landman, G.W.; van Hateren, K.J.; Groenier, K.H.; Struck, J.; Schulte, J.; Gans, R.O.; Bakker, S.J.; Kleefstra, N.; et al. Serum peroxiredoxin 4: A marker of oxidative stress associated with mortality in type 2 diabetes (ZODIAC-28). PLoS ONE 2014, 9, e89719. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Lapierre, V.; Girerd, N.; Bonnerot, M.; Burkhard, P.R.; Lagerstedt, L.; Bracard, S.; Debouverie, M.; Turck, N.; Sanchez, J.C. Diagnostic performance of peroxiredoxin 1 to determine time-of-onset of acute cerebral infarction. Sci. Rep. 2016, 6, 38300. [Google Scholar] [CrossRef] [PubMed]
- Nazri, H.M.; Imran, M.; Fischer, R.; Heilig, R.; Manek, S.; Dragovic, R.A.; Kessler, B.M.; Zondervan, K.T.; Tapmeier, T.T.; Becker, C.M. Characterization of exosomes in peritoneal fluid of endometriosis patients. Fertil. Steril. 2020, 113, 364.e2–373.e2. [Google Scholar] [CrossRef] [Green Version]
- Rinalducci, S.; D’Amici, G.M.; Blasi, B.; Vaglio, S.; Grazzini, G.; Zolla, L. Peroxiredoxin-2 as a candidate biomarker to test oxidative stress levels of stored red blood cells under blood bank conditions. Transfusion 2011, 51, 1439–1449. [Google Scholar] [CrossRef]
- Krata, N.; Foroncewicz, B.; Zagozdzon, R.; Moszczuk, B.; Zielenkiewicz, M.; Paczek, L.; Mucha, K. Peroxiredoxins as Markers of Oxidative Stress in IgA Nephropathy, Membranous Nephropathy and Lupus Nephritis. Arch. Immunol. Ther. Exp. 2021, 70, 3. [Google Scholar] [CrossRef]
- Karasawa, R.; Ozaki, S.; Nishioka, K.; Kato, T. Autoantibodies to peroxiredoxin I and IV in patients with systemic autoimmune diseases. Microbiol. Immunol. 2005, 49, 57–65. [Google Scholar] [CrossRef]
- Gharesi-Fard, B.; Jafarzadeh, L.; Ghaderi-shabankareh, F.; Zolghadri, J.; Kamali-Sarvestani, E. Presence of autoantibody against two placental proteins, peroxiredoxin 3 and peroxiredoxin 4, in sera of recurrent pregnancy loss patients. Am. J. Reprod. Immunol. 2013, 69, 248–255. [Google Scholar] [CrossRef]
- Iizuka, N.; Okamoto, K.; Matsushita, R.; Kimura, M.; Nagai, K.; Arito, M.; Kurokawa, M.S.; Masuko, K.; Suematsu, N.; Hirohata, S.; et al. Identification of autoantigens specific for systemic lupus erythematosus with central nervous system involvement. Lupus 2010, 19, 717–726. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Redox Mediator | Release Mechanism | Function | Reference/(Vesiclepedia ID) |
|---|---|---|---|
| Second messengers | |||
| H2O2 | It crosses plasma membrane directly or via aquaporins | Signal transduction | [43] |
| H2S | It crosses plasma membrane directly | Signal transduction | [44] |
| NO | It crosses plasma membrane directly | Signal transduction, vasodilation | [45] |
| Low molecular weight redox couples | |||
| Cysteine/cystine | Carrier-mediated: CPx | [46] | |
| GSH/GSSG | Carrier-mediated: MRPs | ||
| Redox enzymes | |||
| Gpx3 | Alternative release in extracellular vesicles | It catalyzes the reduction of H2O2 and lipid hydroperoxides | [47] [33] (VP_2878) |
| Gpx7, 8 | Alternative release in extracellular vesicles | unknown | [33] (VP_493869, VP_2882) |
| Grx1, 2, 3, and 5 | Alternative release in extracellular vesicles | unknown | [48] [33] (VP_2745, VP_51022, VP_10539, VP_51218) |
| PDI A1, A3, and A6 | Classical release | Disulfide isomerisation | [12] |
| Prx1, 2, 3, 5, and 6 | Alternative release in extracellular vesicles and exosomes | TLR-mediated signaling | [48] [33] (VP_5052, VP_7001, VP_10935, VP_9588, VP_9588 |
| Prx4 | Classical release | TLR4-mediated signaling | [49] |
| SOD3 | Classical release | Catalysis of the dismutation of superoxide anion to H2O2 | [50] |
| Trx1 | Alternative release in extracellular vesicles and exosomes | Regulation of extracellular thiol switches, i.e., disulfide reduction | [48] [33] (VP_7295) [51] |
| Trx80 | Alternative secretion, processed and released via ADAM17 | Chemokine-like functions, no redox activity | [52] |
| TrxR1 | Classical release and alternative release in extracellular vesicles | Unknown. Potential reduction of Trx1 | [29] [33] (VP_7296) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loreto Palacio, P.; Godoy, J.R.; Aktas, O.; Hanschmann, E.-M. Changing Perspectives from Oxidative Stress to Redox Signaling—Extracellular Redox Control in Translational Medicine. Antioxidants 2022, 11, 1181. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061181
Loreto Palacio P, Godoy JR, Aktas O, Hanschmann E-M. Changing Perspectives from Oxidative Stress to Redox Signaling—Extracellular Redox Control in Translational Medicine. Antioxidants. 2022; 11(6):1181. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061181
Chicago/Turabian StyleLoreto Palacio, Paola, José R. Godoy, Orhan Aktas, and Eva-Maria Hanschmann. 2022. "Changing Perspectives from Oxidative Stress to Redox Signaling—Extracellular Redox Control in Translational Medicine" Antioxidants 11, no. 6: 1181. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061181