
Deciphering the Path of S-nitrosation of Human Thioredoxin: Evidence of an Internal NO Transfer and Implication for the Cellular Responses to NO

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
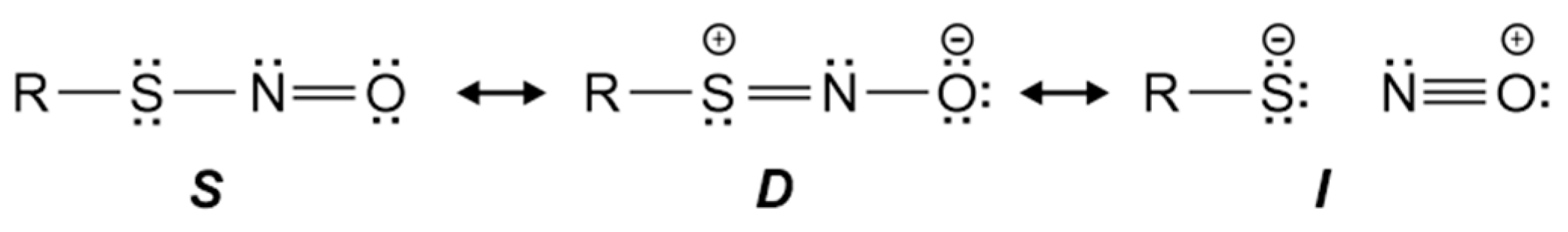{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. hTrs Constructs and Experimental Design
2.2. Expression and Purification of Human Thioredoxin and Mutants
2.3. GSNO Synthesis
2.4. Preparation of C62only(SNO), C69only(SNO), and C73only(SNO) Mutants
2.5. NMR Assignment Experiments
2.6. NMR Kinetic Experiments
2.7. Mass Spectrometry Analysis
3. Results
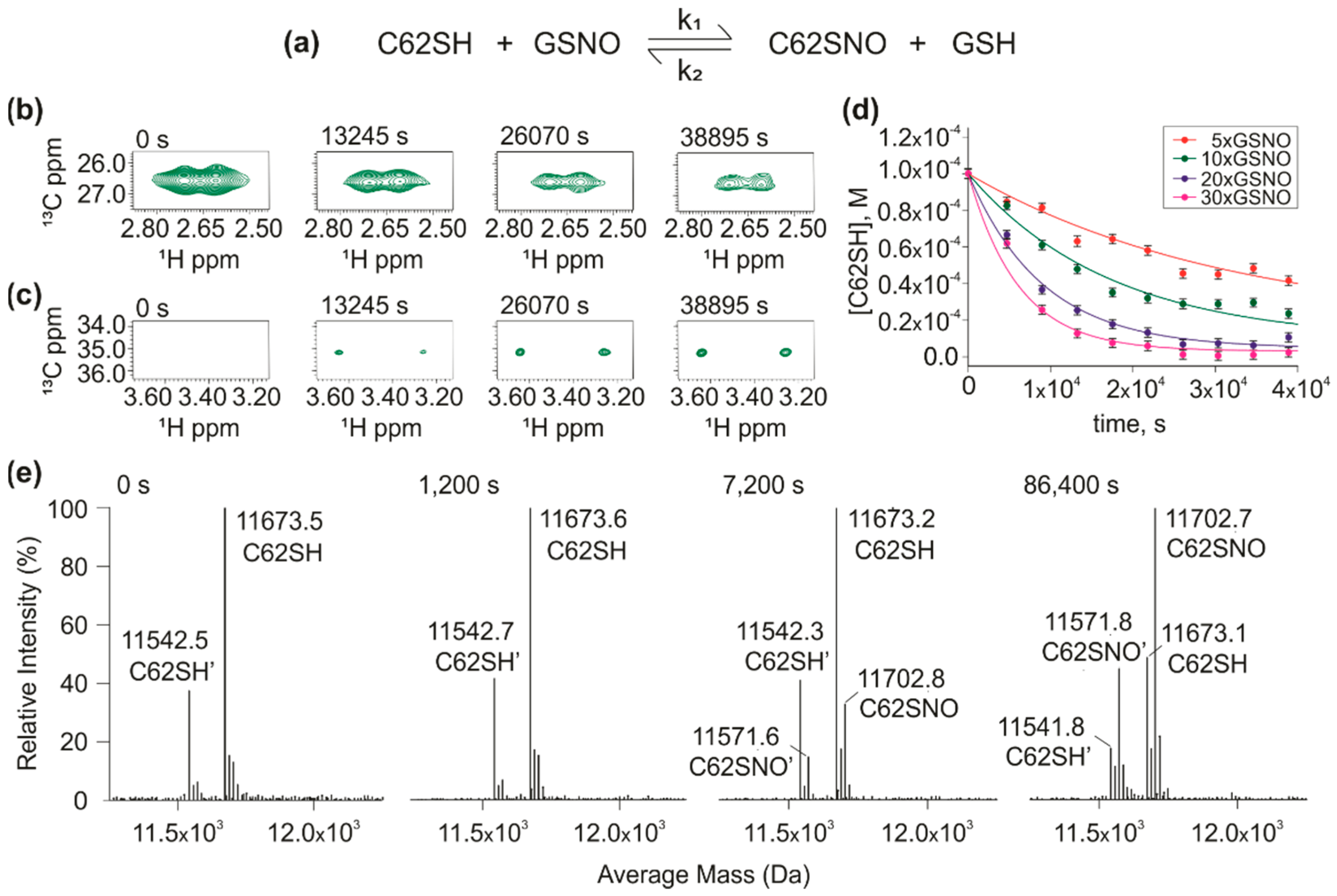
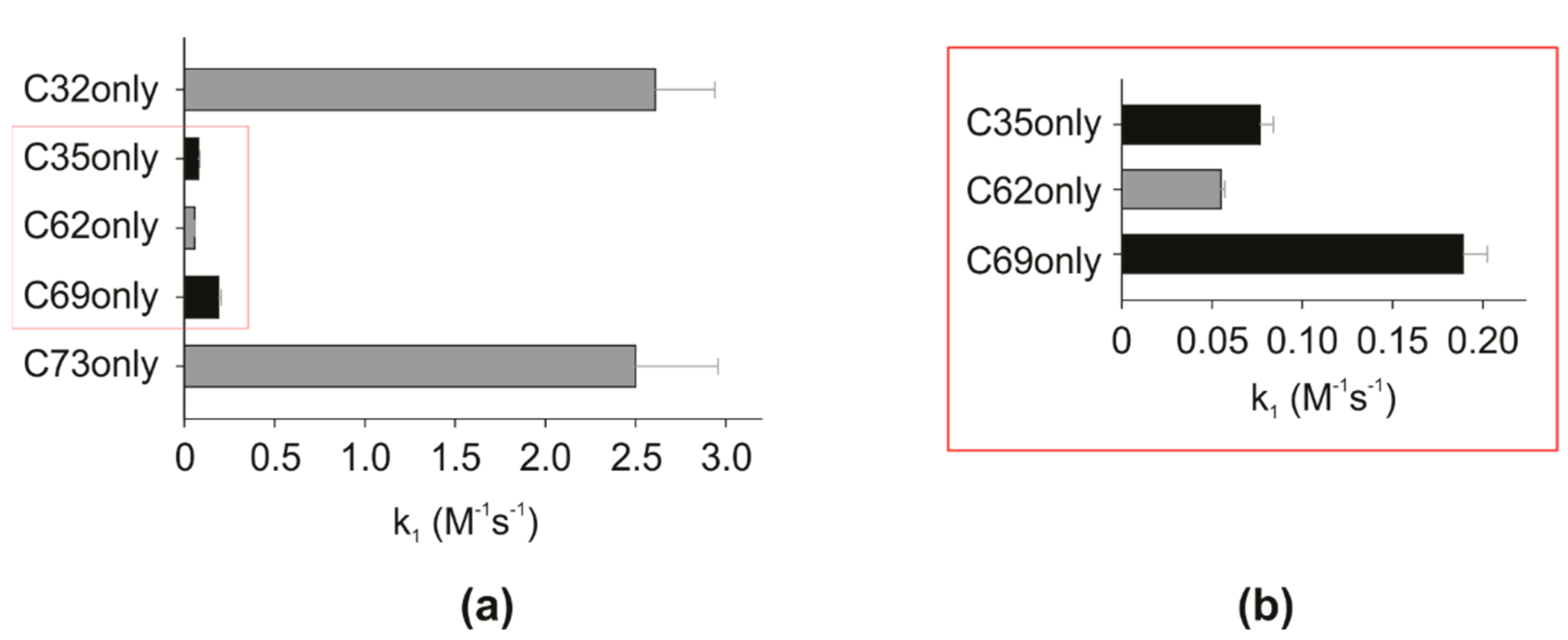
3.1. Reactivity of hTrx Cysteines
3.2. S-nitrosation Kinetics of Wild Type hTrx
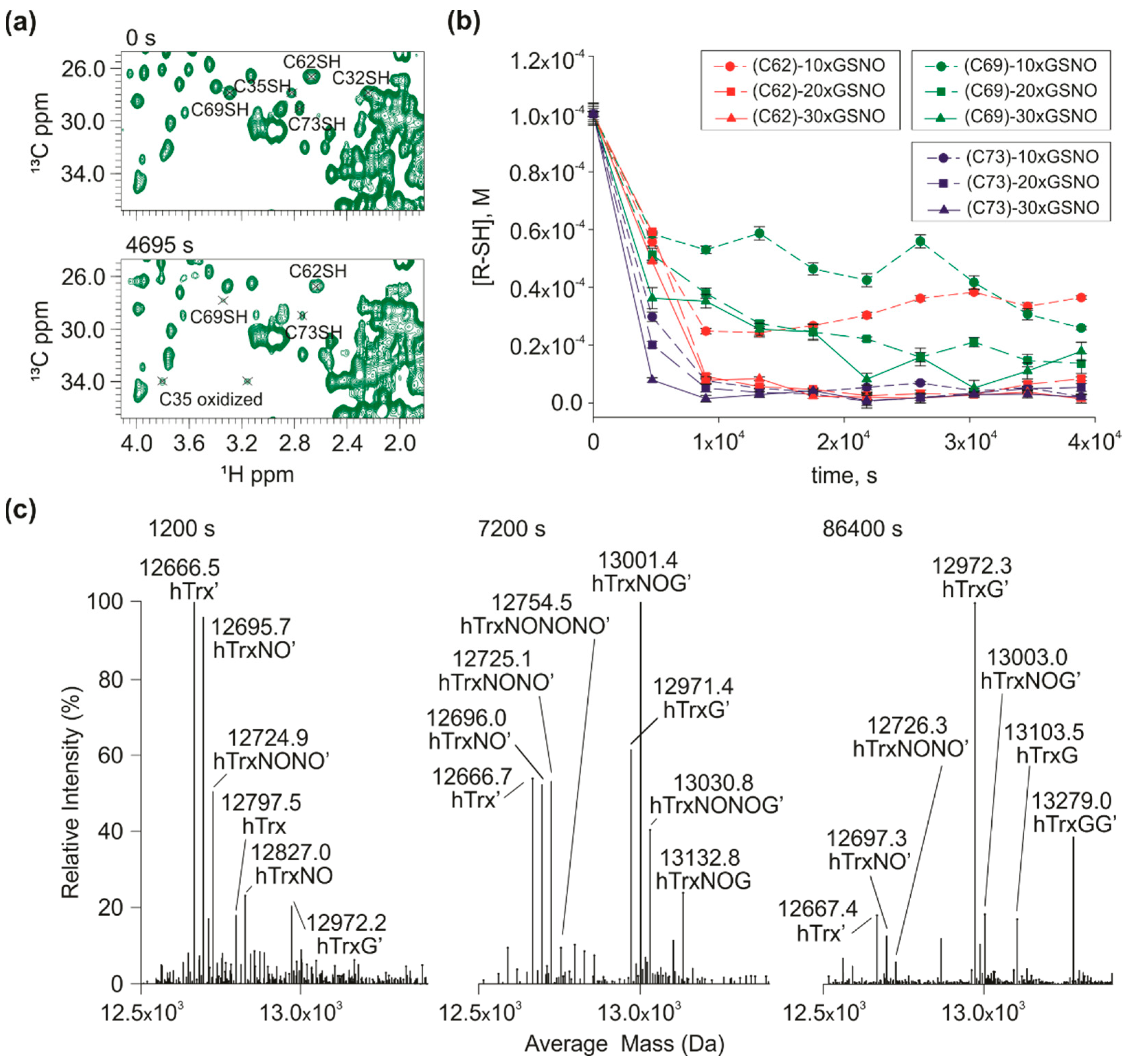
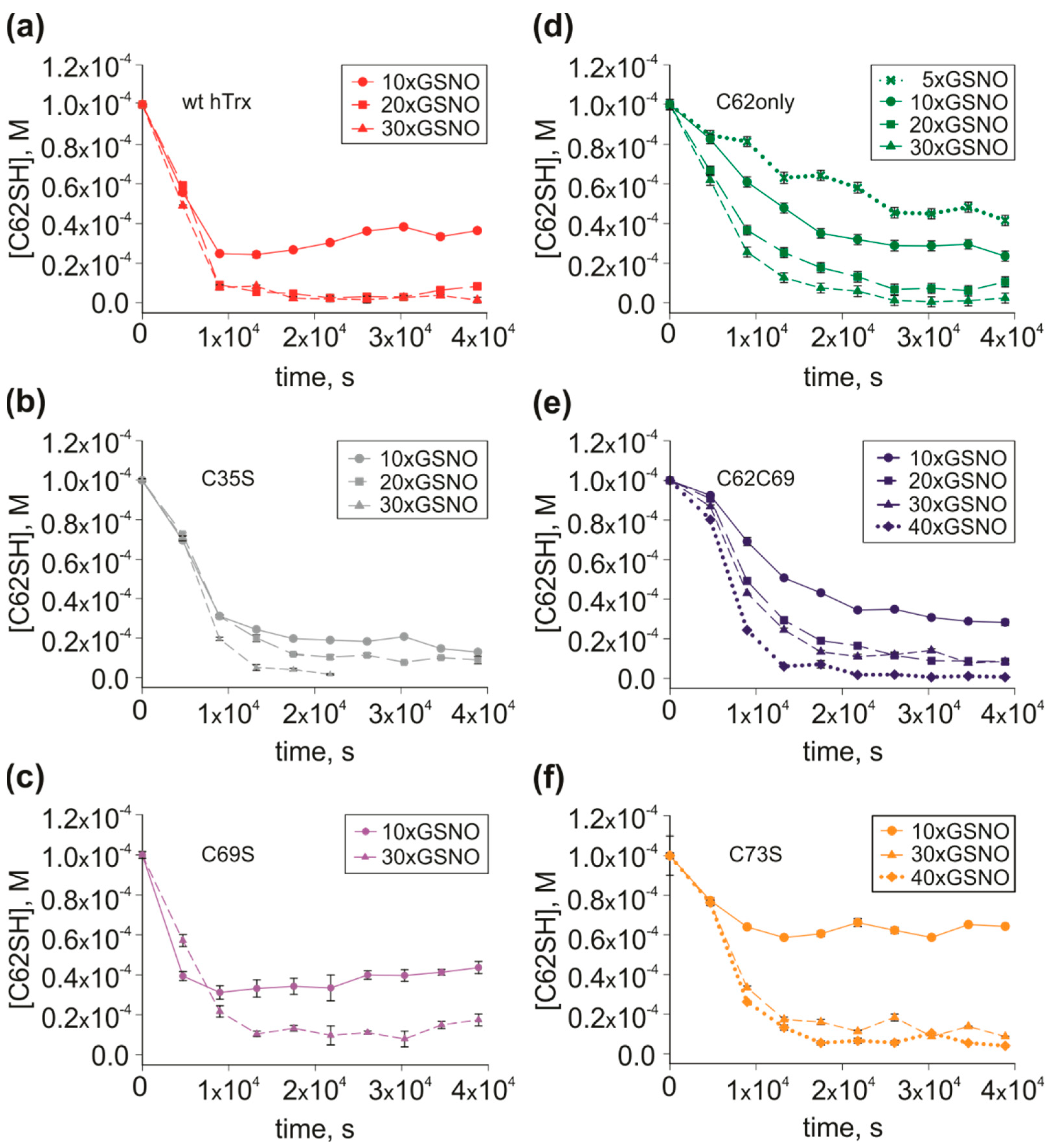
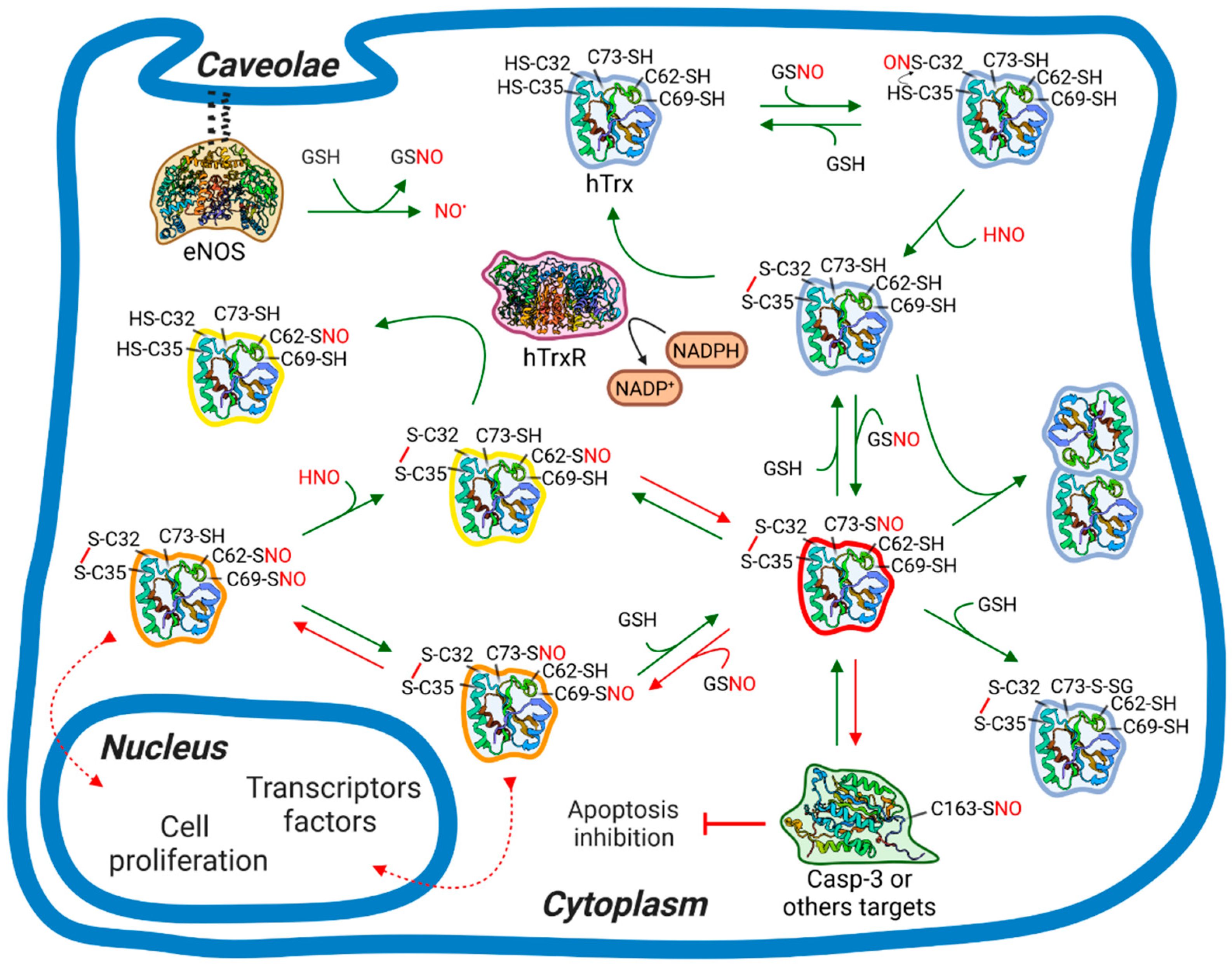
3.3. Mutants with Multiple S-nitrosation Sites to Define the Path of NO
3.4. Fine Details in the Changes in Protein Structure by Nitrosation
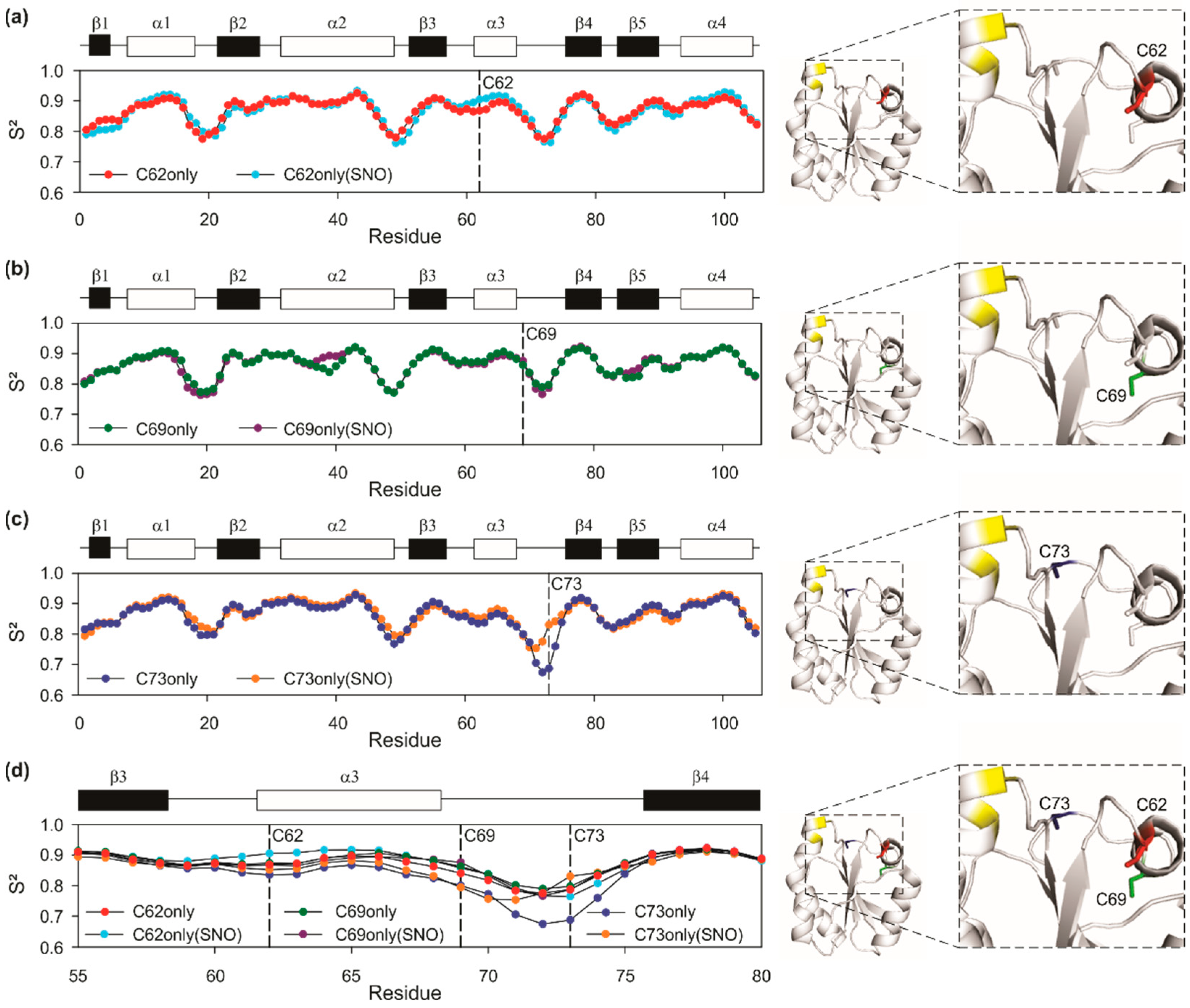
3.4.1. General Flexibility Changes after Nitrosation as Observed by S2 Measurements
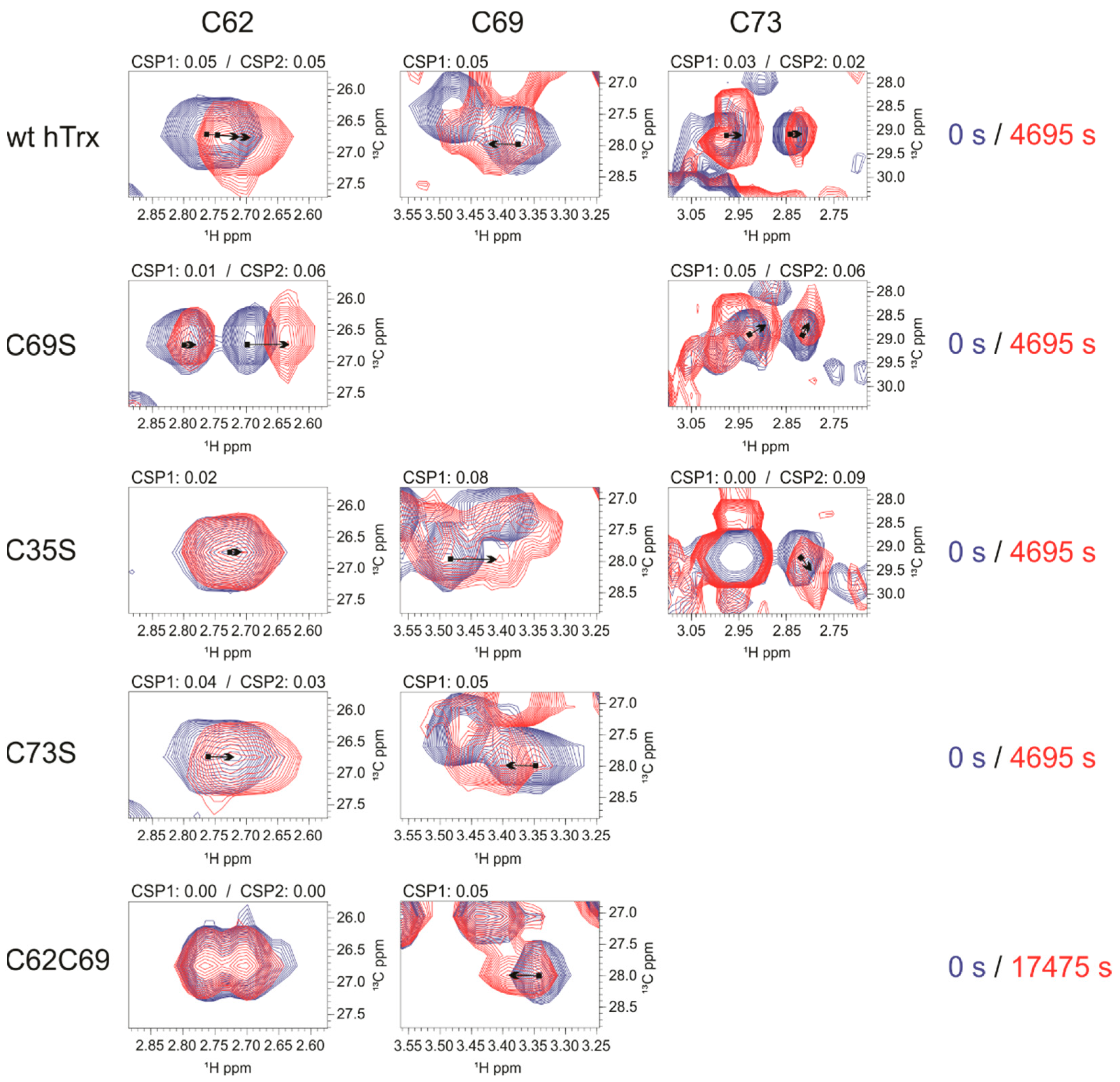
3.4.2. Changes around Nitrosated Sites as Analyzed by CSP
3.5. S-transnitrosation of hTrx-SNO to GSH and hTrx Mutants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weichsel, A.; Brailey, J.L.; Montfort, W.R. Buried S-Nitrosocysteine Revealed in Crystal Structures of Human Thioredoxin. Biochemistry 2007, 46, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M. Nitric Oxide and the Thioredoxin System: A Complex Interplay in Redox Regulation. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 2476–2484. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-Nitrosylation: Purview and Parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Steenbergen, C.; Murphy, E. S-Nitrosylation: NO-Related Redox Signaling to Protect against Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, I.; Lindermayr, C. Nitric Oxide-Based Protein Modification: Formation and Site-Specificity of Protein S-Nitrosylation. Front. Plant Sci. 2013, 4, 137. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. Protein S-Nitrosylation and Cancer. Cancer Lett. 2012, 320, 123–129. [Google Scholar] [CrossRef]
- Plenchette, S.; Romagny, S.; Laurens, V.; Bettaieb, A. S-Nitrosylation in TNF Superfamily Signaling Pathway: Implication in Cancer. Redox Biol. 2015, 6, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, H.; Costa, P.; Reis, A.; Stern, A. Nitric Oxide: Protein Tyrosine Phosphorylation and Protein S-Nitrosylation in Cancer. Biomed. J. 2015, 38, 380–388. [Google Scholar] [CrossRef]
- Yin, L.; Xie, Y.; Yin, S.; Lv, X.; Zhang, J.; Gu, Z.; Sun, H.; Liu, S. The S-Nitrosylation Status of PCNA Localized in Cytosol Impacts the Apoptotic Pathway in a Parkinson’s Disease Paradigm. PLoS ONE 2015, 10, e0117546. [Google Scholar] [CrossRef] [Green Version]
- Conway, M.E.; Harris, M. S-Nitrosylation of the Thioredoxin-like Domains of Protein Disulfide Isomerase and Its Role in Neurodegenerative Conditions. Front. Chem. 2015, 3, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhar, M.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. Regulated Protein Denitrosylation by Cytosolic and Mitochondrial Thioredoxins. Science 2008, 320, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wang, Z.-E.; Doulias, P.-T.; Wei, W.; Ischiropoulos, H.; Locksley, R.M.; Liu, L. Lymphocyte Development Requires S -Nitrosoglutathione Reductase. J. Immunol. 2010, 185, 6664–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, D.; Stamler, J.S. The SNO-Proteome: Causation and Classifications. Curr. Opin. Chem. Biol. 2012, 15, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Lu, C.T.; Su, M.G.; Huang, K.Y.; Ching, W.C.; Yang, H.H.; Liao, Y.C.; Chen, Y.J.; Lee, T.Y. DbSNO 2.0: A Resource for Exploring Structural Environment, Functional and Disease Association and Regulatory Network of Protein S-Nitrosylation. Nucleic Acids Res. 2015, 43, D503–D511. [Google Scholar] [CrossRef] [Green Version]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, R.; Holmgren, A. The Role of Thioredoxin in the Regulation of Cellular Processes by S-Nitrosylation. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 689–700. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Marletta, M.A. Thioredoxin Catalyzes the S-nitrosation of the Caspase-3 Active Site Cysteine. Nat. Chem. Biol. 2005, 1, 154–158. [Google Scholar] [CrossRef]
- Atochina-Vasserman, E.N.; Gow, A.J.; Abramova, H.; Guo, C.-J.; Tomer, Y.; Preston, A.M.; Beck, J.M.; Beers, M.F. Immune Reconstitution during Pneumocystis Lung Infection: Disruption of Surfactant Component Expression and Function by S-Nitrosylation. J. Immunol. 2009, 182, 2277–2287. [Google Scholar] [CrossRef] [Green Version]
- Bentz, B.G.; Simmons, R.L.; Haines, G.K.; Radosevich, J.A. The Yin and Yang of Nitric Oxide: Reflections on the Physiology and Pathophysiology of NO. Head Neck 2000, 22, 71–83. [Google Scholar] [CrossRef]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation in Health and Disease: A Current Perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibiza, S.; Pérez-Rodríguez, A.; Ortega, Á.; Martínez-Ruiz, A.; Barreiro, O.; García-Domínguez, C.A.; Víctor, V.M.; Esplugues, J.V.; Rojas, J.M.; Sánchez-Madrid, F.; et al. Endothelial Nitric Oxide Synthase Regulates N-Ras Activation on the Golgi Complex of Antigen-Stimulated T Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10507–10512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.W.; Li, C.Y. Regulation of HIF-1α Stability through S-Nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manabe, S.I.; Gu, Z.; Lipton, S.A. Activation of Matrix Metalloproteinase-9 via Neuronal Nitric Oxide Synthase Contributes to NMDA-Induced Retinal Ganglion Cell Death. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4747–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannick, J.B. Regulation of Apoptosis by Protein S-Nitrosylation. Amino Acids 2007, 32, 523–526. [Google Scholar] [CrossRef]
- McCarthy, S.M.; Bove, P.F.; Matthews, D.E.; Akaike, T.; Van Der Vliet, A. Nitric Oxide Regulation of MMP-9 Activation and Its Relationship to Modifications of the Cysteine Switch. Biochemistry 2008, 47, 5832–5840. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Lipton, S.A. Emerging Roles of S-Nitrosylation in Protein Misfolding and Neurodegenerative Diseases. Antioxid. Redox Signal. 2008, 10, 87–101. [Google Scholar] [CrossRef]
- Hashemy, S.I.; Holmgren, A. Regulation of the Catalytic Activity and Structure of Human Thioredoxin 1 via Oxidation and S-Nitrosylation of Cysteine Residues. J. Biol. Chem. 2008, 283, 21890–21898. [Google Scholar] [CrossRef] [Green Version]
- Barglow, K.T.; Knutson, C.G.; Wishnok, J.S.; Tannenbaum, S.R.; Marletta, M.A. Site-Specific and Redox-Controlled S-nitrosation of Thioredoxin. Proc. Natl. Acad. Sci. USA 2011, 108, E600–E606. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Morton, S.U.; Fernhoff, N.B.; Marletta, M.A. Thioredoxin Is Required for S-nitrosation of Procaspase-3 and the Inhibition of Apoptosis in Jurkat Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 11609–11614. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, T.; Wu, C.; Li, H. A Strategy for Direct Identification of Protein S-Nitrosylation Sites by Quadrupole Time-of-Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Liu, T.; Chen, W.; Oka, S.-I.; Fu, C.; Jain, R.; Parrott, A.M.; Tarik Baykal, A.; Sadoshima, J.; Li, H. Redox Regulatory Mechanism of Transnitrosylation by Thioredoxin. Mol. Cell. Proteom. 2010, 9, 2262–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haendeler, J.; Hoffmann, J.; Tischler, V.; Berk, B.C.; Zeiher, A.M.; Dimmeler, S. Redox Regulatory and Anti-Apoptotic Functions of Thioredoxin Depend on S-Nitrosylation at Cysteine 69. Nat. Cell Biol. 2002, 4, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Weichsel, A.; Gasdaska, J.R.; Powis, G.; Montfort, W.R. Crystal Structures of Reduced, Oxidized, and Mutated Human Thioredoxins: Evidence for a Regulatory Homodimer. Structure 1996, 4, 735–751. [Google Scholar] [CrossRef] [Green Version]
- Weichsel, A.; Kem, M.; Montfort, W.R. Crystal Structure of Human Thioredoxin Revealing an Unraveled Helix and Exposed S-nitrosation Site. Protein Sci. 2010, 19, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, P.; Popp, R.; Wiegand, B.; Altschmied, J.; Haendeler, J. Nuclear Redox-Signaling Is Essential for Apoptosis Inhibition in Endothelial Cells—Important Role for Nuclear Thioredoxin-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2325–2331. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Broniowska, K.A.; Diers, A.R.; Hogg, N. S-Nitrosoglutathione. Biochim. Biophys. Acta 2013, 1830, 3173–3181. [Google Scholar] [CrossRef] [Green Version]
- Almeida, V.S.; Iqbal, A.; Almeida, F.C.L. 1H, 15N and 13C Backbone and Side-chain Assignments of Reduced and S-Nitrosated C62only Mutant of Human Thioredoxin. Biomol. NMR Assign. 2021, 15, 261–265. [Google Scholar] [CrossRef]
- Palmer, A.G.; Cavanagh, J.; Wright, P.E.; Rance, M. Sensitivity Improvement in Proton-Detected Two-Dimensional Heteronuclear Correlation NMR Spectroscopy. J. Magn. Reson. 1991, 93, 151–170. [Google Scholar] [CrossRef]
- Kay, L.E.; Keifer, P.; Saarinen, T. Pure Absorption Gradient Enhanced Heteronuclear Single Quantum Correlation Spectroscopy with Improved Sensitivity. J. Am. Chem. Soc. 1992, 114, 10663–10665. [Google Scholar] [CrossRef]
- Grzesiek, S.; Bax, A. Improved 3D Triple-Resonance NMR Techniques Applied to a 31 KDa Protein. J. Magn. Reson. 1992, 96, 432–440. [Google Scholar] [CrossRef]
- Schleucher, J.; Schwendinger, M.; Sattler, M.; Schmidt, P.; Schedletzky, O.; Glaser, S.J.; Sørensen, O.W.; Griesinger, C. A General Enhancement Scheme in Heteronuclear Multidimensional NMR Employing Pulsed Field Gradients. J. Biomol. NMR 1994, 4, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Muhandiram, D.R.; Kay, L.E. Gradient-Enhanced Triple-Resonance Three-Dimensional NMR Experiments with Improved Sensitivity. J. Magn. Reson. Ser. B 1994, 103, 203–216. [Google Scholar] [CrossRef]
- Wittekind, M.; Mueller, L. HNCACB, a High-Sensitivity 3D NMR Experiment to Correlate Amide-Proton and Nitrogen Resonances with the Alpha- and Beta-Carbon Resonances in Proteins. J. Magn. Reson. Ser. B 1993, 101, 201–205. [Google Scholar] [CrossRef]
- Hyberts, S.G.; Milbradt, A.G.; Wagner, A.B.; Arthanari, H.; Wagner, G. Application of Iterative Soft Thresholding for Fast Reconstruction of NMR Data Non-Uniformly Sampled with Multidimensional Poisson Gap Scheduling. J. Biomol. NMR 2012, 52, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Hyberts, S.G.; Frueh, D.P.; Arthanari, H.; Wagner, G. FM Reconstruction of Non-Uniformly Sampled Protein NMR Data at Higher Dimensions and Optimization by Distillation. J. Biomol. NMR 2009, 45, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Hyberts, S.G.; Takeuchi, K.; Wagner, G. Poisson-Gap Sampling and Forward Maximum Entropy Reconstruction for Enhancing the Resolution and Sensitivity of Protein NMR Data. J. Am. Chem. Soc. 2010, 132, 2145–2147. [Google Scholar] [CrossRef] [Green Version]
- Hyberts, S.G.; Arthanari, H.; Robson, S.A.; Wagner, G. Perspectives in Magnetic Resonance: NMR in the Post-FFT Era. J. Magn. Reson. 2014, 241, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Hyberts, S.G.; Robson, S.A.; Wagner, G. Exploring Signal-to-Noise Ratio and Sensitivity in Non-Uniformly Sampled Multi-Dimensional NMR Spectra. J. Biomol. NMR 2013, 55, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN Data Model for NMR Spectroscopy: Development of a Software Pipeline. Proteins Struct. Funct. Genet. 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, M.W.; Schuyler, A.D.; Gryk, M.R.; Moraru, I.I.; Romero, P.R.; Ulrich, E.L.; Eghbalnia, H.R.; Livny, M.; Delaglio, F.; Hoch, J.C. NMRbox: A Resource for Biomolecular NMR Computation. Biophys. J. 2017, 112, 1529–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berjanskii, M.V.; Wishart, D.S. A Simple Method to Predict Protein Flexibility Using Secondary Chemical Shifts. J. Am. Chem. Soc. 2005, 127, 14970–14971. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Simpson, Z.B.; Blom, T. Global Kinetic Explorer: A New Computer Program for Dynamic Simulation and Fitting of Kinetic Data. Anal. Biochem. 2009, 387, 20–29. [Google Scholar] [CrossRef]
- Johnson, K.A.; Simpson, Z.B.; Blom, T. FitSpace Explorer: An Algorithm to Evaluate Multidimensional Parameter Space in Fitting Kinetic Data. Anal. Biochem. 2009, 387, 30–41. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein Backbone and Sidechain Torsion Angles Predicted from NMR Chemical Shifts Using Artificial Neural Networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Nguyen, L.T.; Jeon, Y.H.; Lee, C.Y.; Kim, M.H. Crystal Structure of Fully Oxidized Human Thioredoxin. Biochem. Biophys. Res. Commun. 2015, 467, 218–222. [Google Scholar] [CrossRef]
- Talipov, M.R.; Timerghazin, Q.K. Protein Control of S-Nitrosothiol Reactivity: Interplay of Antagonistic Resonance Structures. J. Phys. Chem. B 2013, 117, 1827–1837. [Google Scholar] [CrossRef]
- Moran, E.E.; Timerghazin, Q.K.; Kwong, E.; English, A.M. Kinetics and Mechanism of S-Nitrosothiol Acid-Catalyzed Hydrolysis: Sulfur Activation Promotes Facile NO+ Release. J. Phys. Chem. B 2011, 115, 3112–3126. [Google Scholar] [CrossRef]
- Smith, B.C.; Marletta, M.A. Mechanisms of S-Nitrosothiol Formation and Selectivity in Nitric Oxide Signaling. Curr. Opin. Chem. Biol. 2012, 16, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Forman-Kay, J.D.; Marius Clore, G.; Wingfield, P.T.; Gronenborn, A.M. High-Resolution Three-Dimensional Structure of Reduced Recombinant Human Thioredoxin in Solution. Biochemistry 1991, 30, 2685–2698. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Wolf, K.; Urig, S.; Becker, K. The Structure of Human Thioredoxin Reductase 1 Provides Insights into C-Terminal Rearrangements During Catalysis. J. Mol. Biol. 2007, 370, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Long, S.A.; Adams, J.L.; Chan, G.; Vaidya, K.S.; Francis, T.A.; Kikly, K.; Winkler, J.D.; Sung, C.M.; Debouck, C.; et al. Potent and Selective Nonpeptide Inhibitors of Caspases 3 and 7 Inhibit Apoptosis and Maintain Cell Functionality. J. Biol. Chem. 2000, 275, 16007–16014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischmann, T.O.; Hruza, A.; Niu, X.D.; Fossetta, J.D.; Lunn, C.A.; Dolphin, E.; Prongay, A.J.; Reichert, P.; Lundell, D.J.; Narula, S.K.; et al. Structural Characterization of Nitric Oxide Synthase Isoforms Reveals Striking Active-Site Conservation. Nat. Struct. Biol. 1999, 6, 233–242. [Google Scholar] [CrossRef]
- Ferret, P.J.; Soum, E.; Negre, O.; Wollman, E.E.; Fradelizi, D. Protective Effect of Thioredoxin upon NO-Mediated Cell Injury in THP1 Monocytic Human Cells. Biochem. J. 2000, 346, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.J.; Ogata, F.T.; Batista, W.L.; Masutani, H.; Yodoi, J.; Debbas, V.; Augusto, O.; Stern, A.; Monteiro, H.P. Thioredoxin-1 Promotes Survival in Cells Exposed to S-Nitrosoglutathione: Correlation with Reduction of Intracellular Levels of Nitrosothiols and up-Regulation of the ERK1/2 MAP Kinases. Toxicol. Appl. Pharmacol. 2008, 233, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, H.P.; Ogata, F.T.; Stern, A. Thioredoxin Promotes Survival Signaling Events under Nitrosative/Oxidative Stress Associated with Cancer Development. Biomed. J. 2017, 40, 189–199. [Google Scholar] [CrossRef]
- Wu, C.; Parrott, A.M.; Fu, C.; Liu, T.; Marino, S.M.; Gladyshev, V.N.; Jain, M.R.; Baykal, A.T.; Li, Q.; Oka, S.; et al. Thioredoxin 1-Mediated Post-Translational Modifications: Reduction, Transnitrosylation, Denitrosylation, and Related Proteomics Methodologies. Antioxid. Redox Signal. 2011, 15, 2565–2604. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, V.S.; Miller, L.L.; Delia, J.P.G.; Magalhães, A.V.; Caruso, I.P.; Iqbal, A.; Almeida, F.C.L. Deciphering the Path of S-nitrosation of Human Thioredoxin: Evidence of an Internal NO Transfer and Implication for the Cellular Responses to NO. Antioxidants 2022, 11, 1236. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11071236
Almeida VS, Miller LL, Delia JPG, Magalhães AV, Caruso IP, Iqbal A, Almeida FCL. Deciphering the Path of S-nitrosation of Human Thioredoxin: Evidence of an Internal NO Transfer and Implication for the Cellular Responses to NO. Antioxidants. 2022; 11(7):1236. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11071236
Chicago/Turabian StyleAlmeida, Vitor S., Lara L. Miller, João P. G. Delia, Augusto V. Magalhães, Icaro P. Caruso, Anwar Iqbal, and Fabio C. L. Almeida. 2022. "Deciphering the Path of S-nitrosation of Human Thioredoxin: Evidence of an Internal NO Transfer and Implication for the Cellular Responses to NO" Antioxidants 11, no. 7: 1236. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11071236