
Glutathione Induces Keap1 S-Glutathionylation and Mitigates Oscillating Glucose-Induced β-Cell Dysfunction by Activating Nrf2
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vivo Studies
2.2. Cell Culture and Treatments
2.3. Pancreatic Islet Isolation and Primary Culture
2.4. Glucose-Stimulated Insulin Secretion (GSIS) Assay
2.5. RNA Isolation and Real-Time Quantitative PCR
2.6. Western Blotting
2.7. Co-Immunoprecipitation and Determination of S-Glutathionylation of Keap1
2.8. Construction of Keap1 Cysteine Mutants
2.9. Immunofluorescence Staining
2.10. GSH/GSSG Assay
2.11. Detection of Mitochondrial ROS Level
2.12. Apoptosis Assay
2.13. Statistical Analysis
3. Results
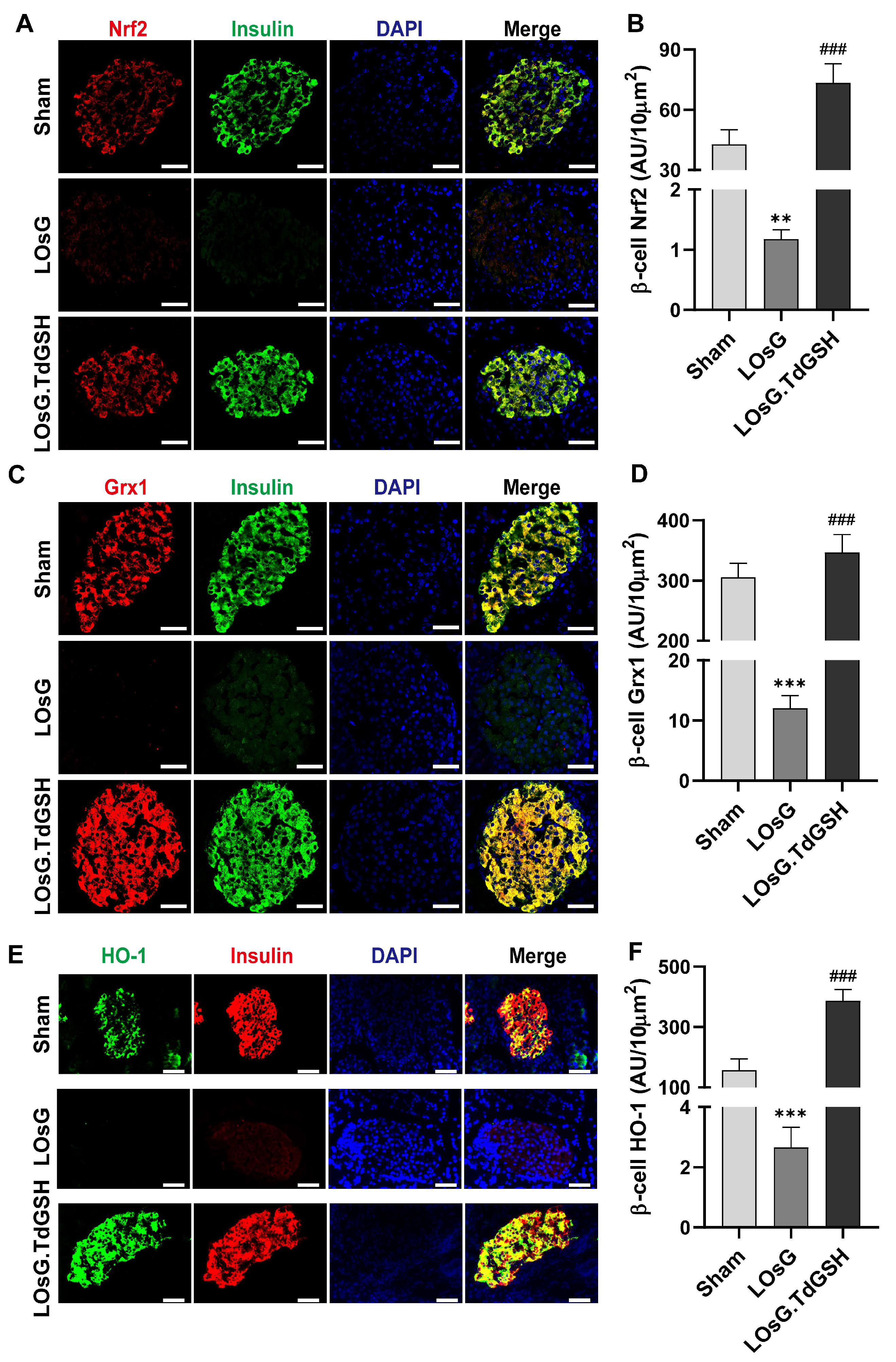
3.1. GSH Prevents the Inhibition of Nrf2, Grx1, and HO-1 Expressions in Islets of Rats Induced by LOsG
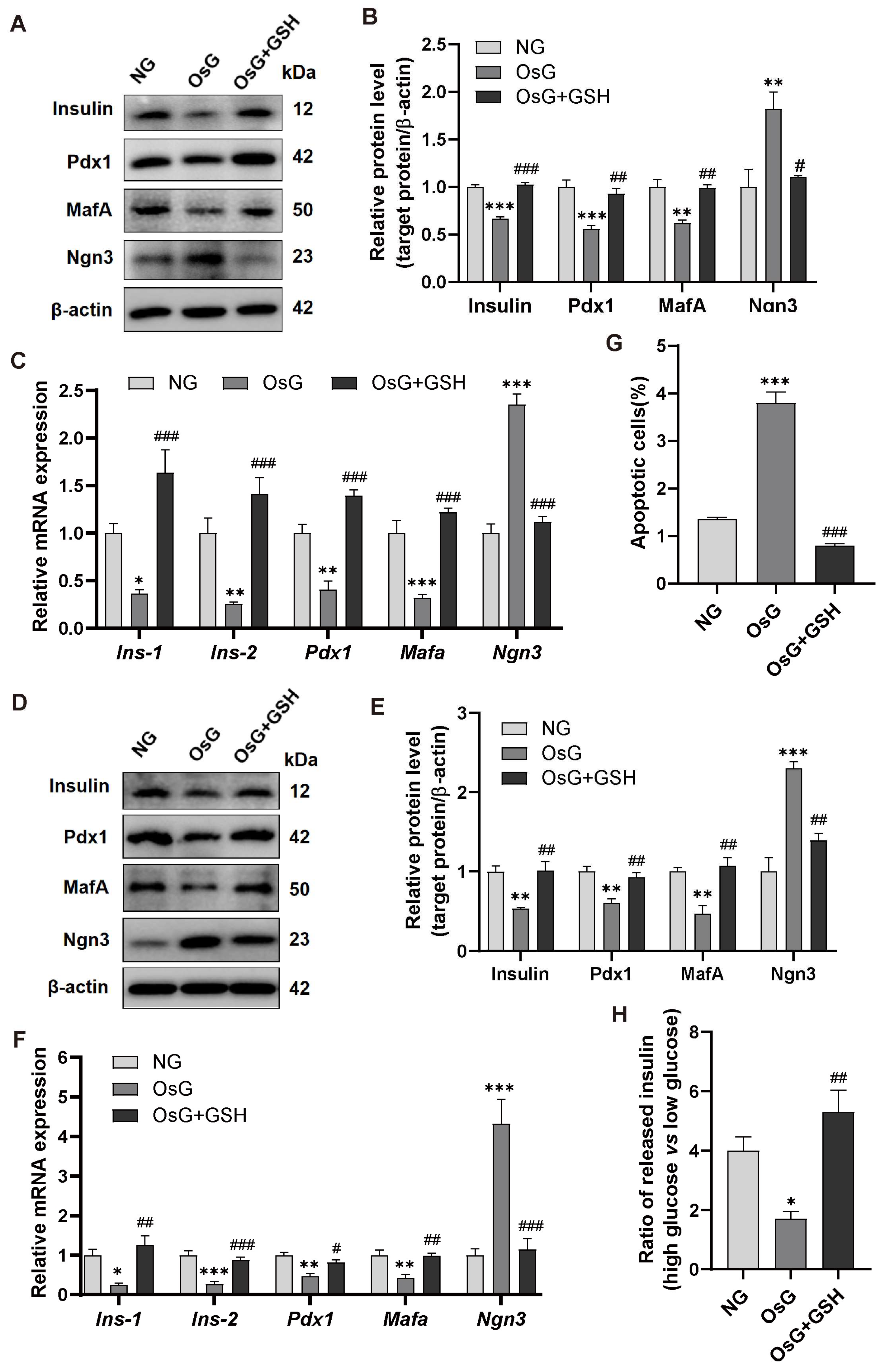
3.2. GSH Prevents OsG-Induced β-Cell Failure
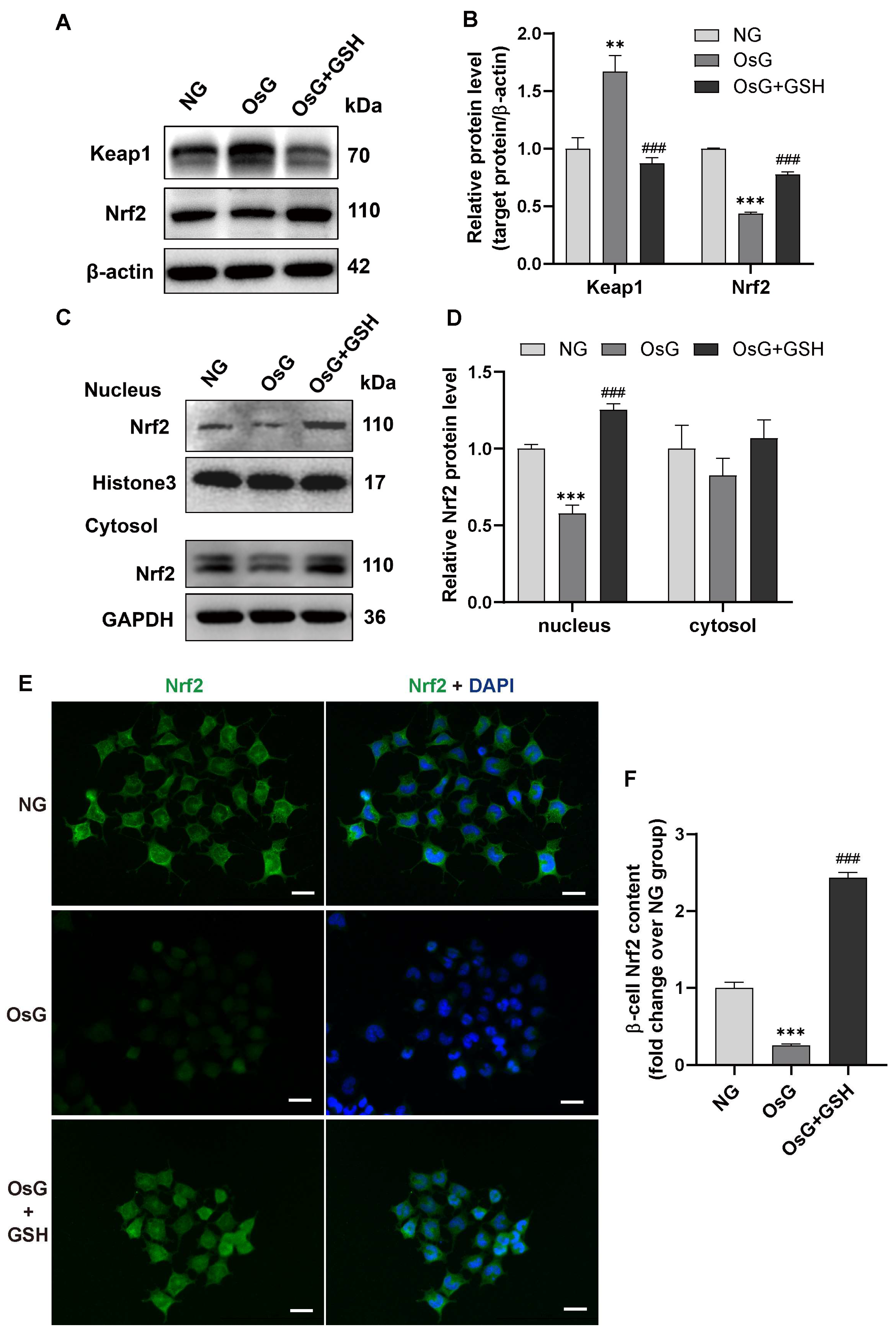
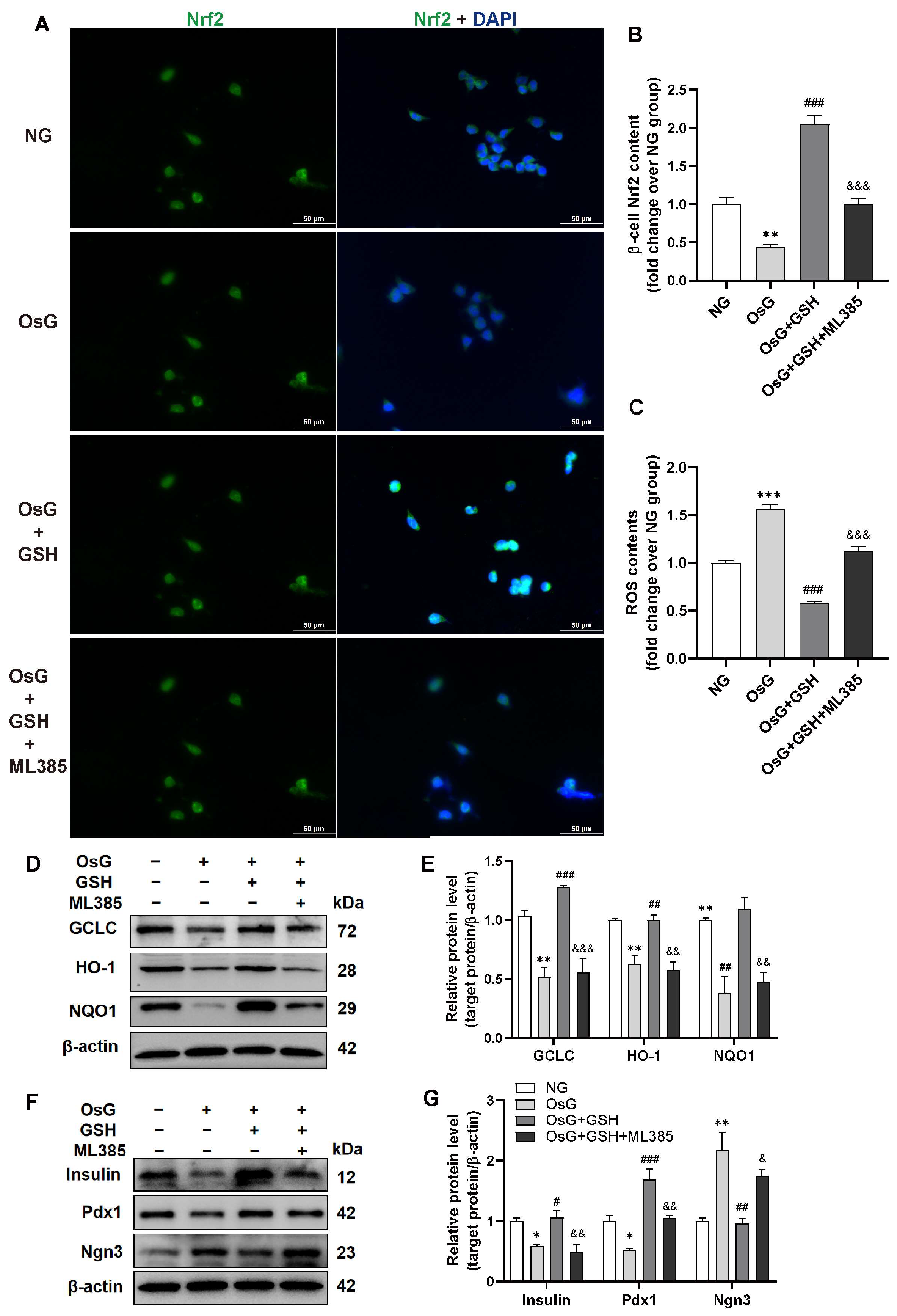
3.3. GSH Activates Nrf2 Pathway in OsG-Treated β-Cells
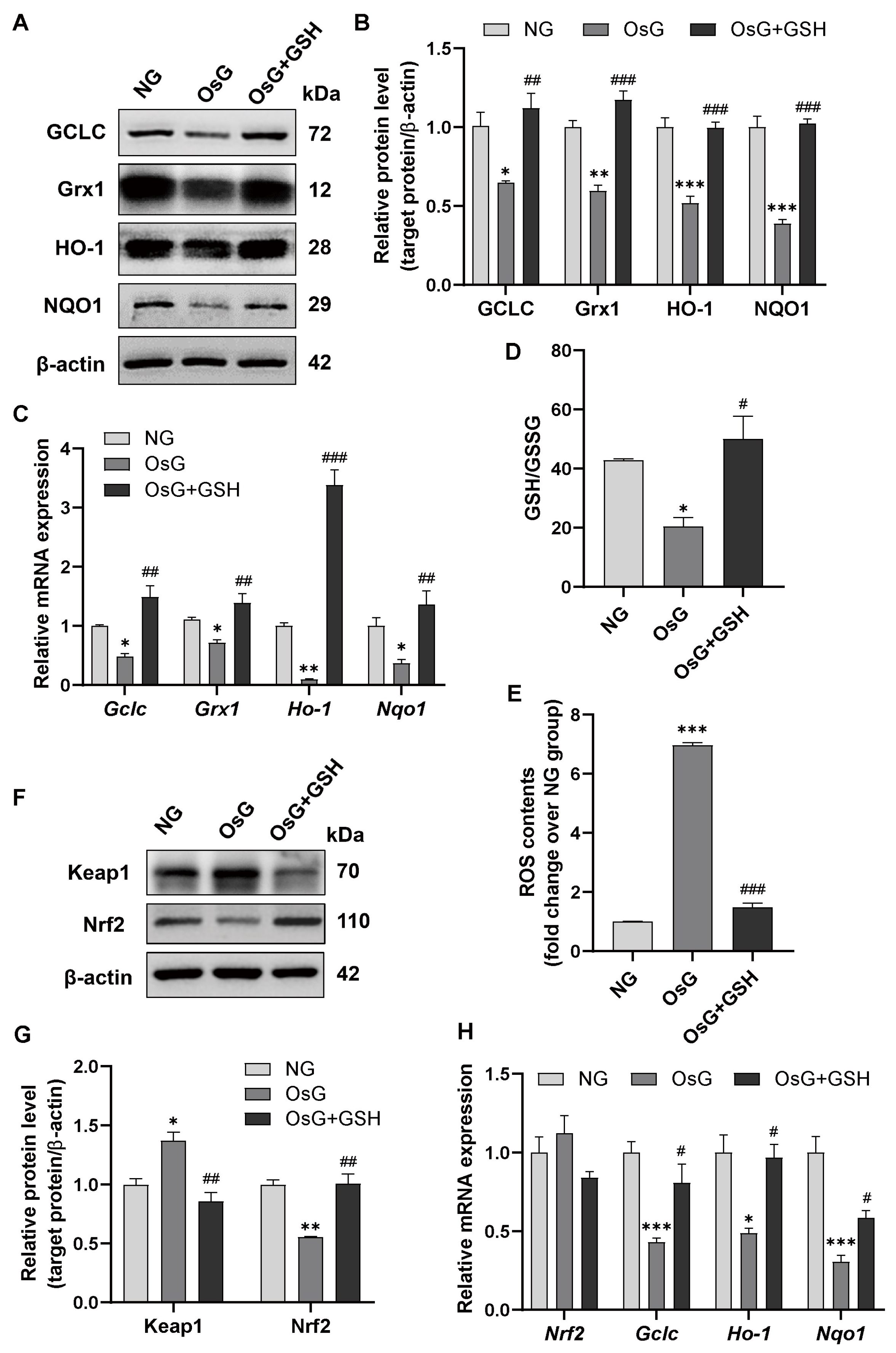
3.4. GSH Promotes Nrf2-Related Antioxidant Enzyme Expression to Attenuate Oxidative Stress in OsG-Treated β-Cells and Primary Islets

3.5. Inhibition of Nrf2 Abrogates the Cytoprotective Effects of GSH in OsG-Treated β-Cells
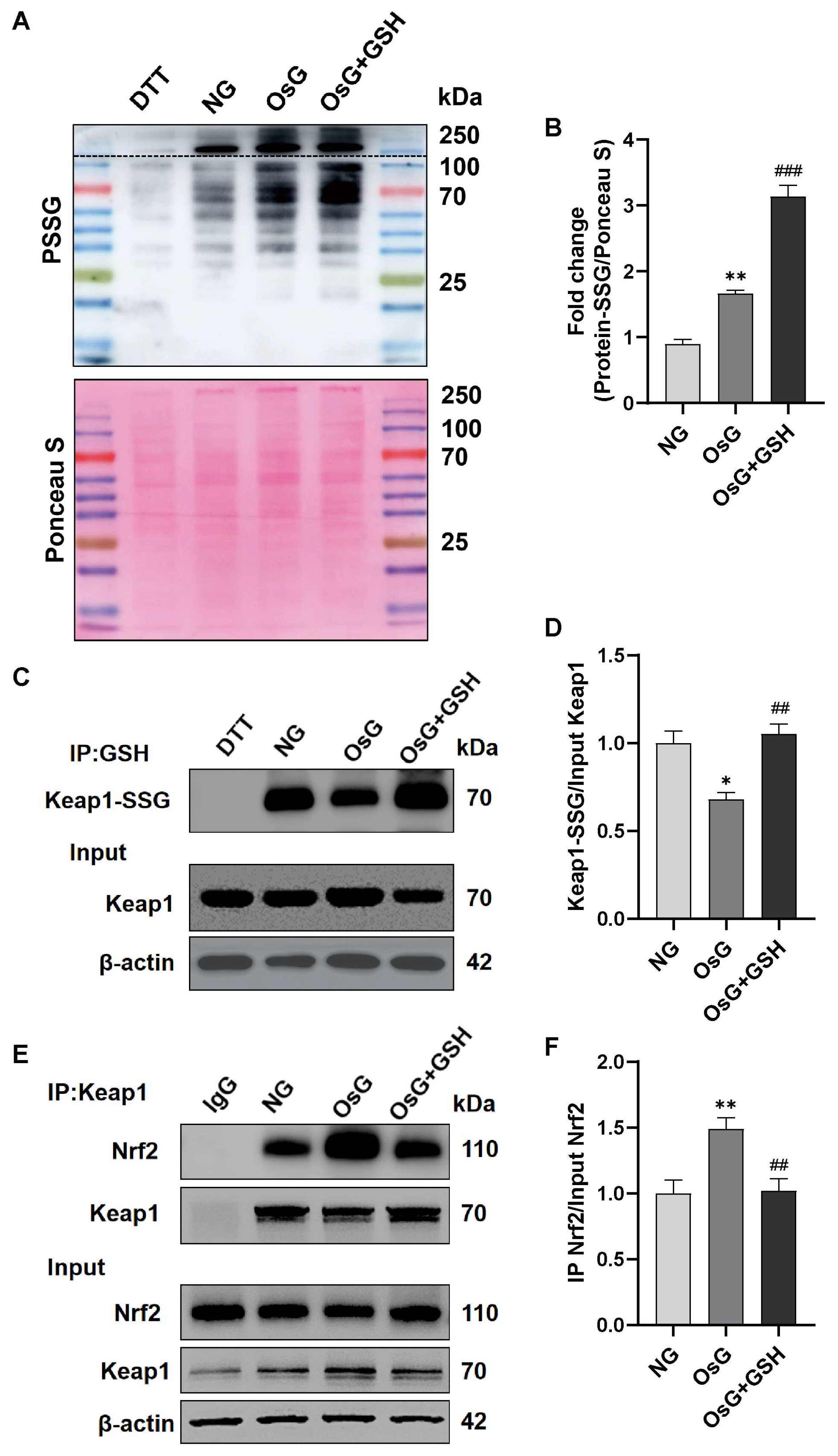
3.6. GSH Increases the S-Glutathionylation of Keap1 to Activate the Nrf2 Pathway in OsG-Treated β-Cells
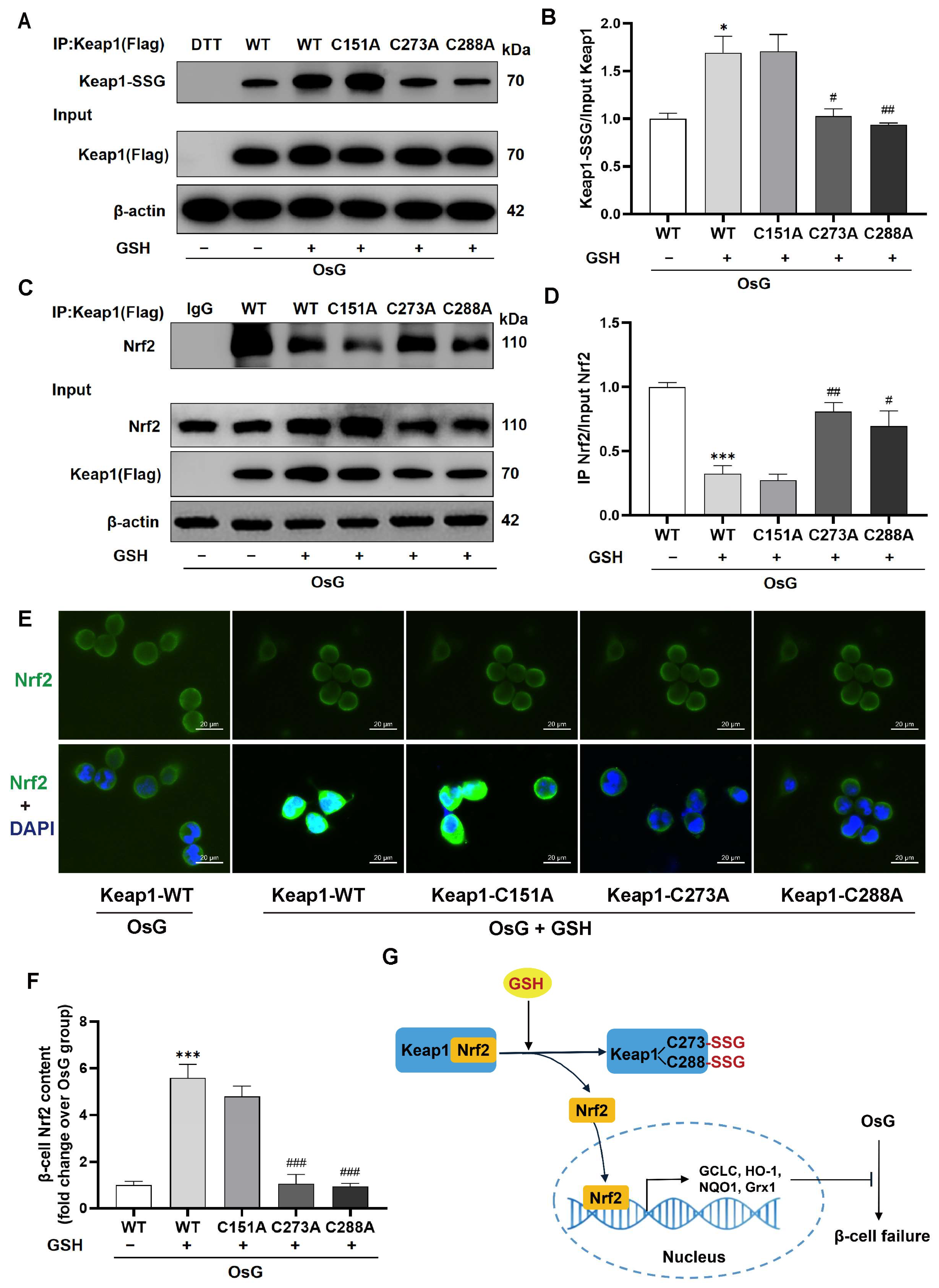
3.7. The S-Glutathionylation of Keap1 at Cys273 and Cys288 Caused the Activation of the Keap1/Nrf2 Pathway in GSH-Treated β-Cells under OsG Condition
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, J.; Jin, L.; Chen, M.; Xu, K.; Huang, Q.; He, B. Application of natural compounds in the treatment and prevention of prediabetes. Front. Nutr. 2023, 10, 1301129. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Selvin, E. Prediabetes and What It Means: The Epidemiological Evidence. Annu. Rev. Public Health 2021, 42, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Tabak, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimaki, M. Prediabetes: A high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bai, J.; Zhou, Q.; Hu, Y.; Wang, Q.; Yang, L.; Chen, H.; An, H.; Zhou, C.; Wang, Y.; et al. Glutathione prevents high glucose-induced pancreatic fibrosis by suppressing pancreatic stellate cell activation via the ROS/TGFbeta/SMAD pathway. Cell Death Dis. 2022, 13, 440. [Google Scholar] [CrossRef]
- Zhang, J.; An, H.; Ni, K.; Chen, B.; Li, H.; Li, Y.; Sheng, G.; Zhou, C.; Xie, M.; Chen, S.; et al. Glutathione prevents chronic oscillating glucose intake-induced beta-cell dedifferentiation and failure. Cell Death Dis. 2019, 10, 321. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Leso, V.; Trovato-Salinaro, A.; Ventimiglia, B.; Cavallaro, M.; Scuto, M.; Rizza, S.; Zanoli, L.; Neri, S.; et al. Oxidative stress, glutathione status, sirtuin and cellular stress response in type 2 diabetes. Biochim. Biophys. Acta 2012, 1822, 729–736. [Google Scholar] [CrossRef]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011, 34, 162–167. [Google Scholar] [CrossRef]
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.E.; Collins, S.; Andersen, M.E. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharmacol. 2010, 244, 77–83. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S.H. Mechanism of Generation of Oxidative Stress and Pathophysiology of Type 2 Diabetes Mellitus: How Are They Interlinked? J. Cell. Biochem. 2017, 118, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Brief Review and Perspective: Antioxidants for early treatment of Type 2 Diabetes in Rodents and Humans: Lost in Translation? Diabetes 2024, 73, db230901. [Google Scholar] [CrossRef] [PubMed]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Butler, P.C. Relationship between beta-cell mass and diabetes onset. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 23–31. [Google Scholar] [CrossRef] [PubMed]
- Cassier-Chauvat, C.; Marceau, F.; Farci, S.; Ouchane, S.; Chauvat, F. The Glutathione System: A Journey from Cyanobacteria to Higher Eukaryotes. Antioxidants 2023, 12, 1199. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef]
- Hill, B.G.; Bhatnagar, A. Protein S-glutathiolation: Redox-sensitive regulation of protein function. J. Mol. Cell. Cardiol. 2012, 52, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef]
- Carvalho, A.N.; Marques, C.; Guedes, R.C.; Castro-Caldas, M.; Rodrigues, E.; van Horssen, J.; Gama, M.J. S-Glutathionylation of Keap1: A new role for glutathione S-transferase pi in neuronal protection. FEBS Lett. 2016, 590, 1455–1466. [Google Scholar] [CrossRef]
- Gambhir, L.; Checker, R.; Thoh, M.; Patwardhan, R.S.; Sharma, D.; Kumar, M.; Sandur, S.K. 1,4-Naphthoquinone, a pro-oxidant, suppresses immune responses via KEAP-1 glutathionylation. Biochem. Pharmacol. 2014, 88, 95–105. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Song, M.Y.; Kim, E.K.; Moon, W.S.; Park, J.W.; Kim, H.J.; So, H.S.; Park, R.; Kwon, K.B.; Park, B.H. Sulforaphane protects against cytokine- and streptozotocin-induced beta-cell damage by suppressing the NF-kappaB pathway. Toxicol. Appl. Pharmacol. 2009, 235, 57–67. [Google Scholar] [CrossRef]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 protects pancreatic beta-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef]
- Liu, R.; Liu, C.; He, X.; Sun, P.; Zhang, B.; Yang, H.; Shi, W.; Ruan, Q. MicroRNA-21 promotes pancreatic beta cell function through modulating glucose uptake. Nat. Commun. 2022, 13, 3545. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.D.; Lee, K.; Yang, D.; Amin, S.; Verma, N.; Li, Q.V.; Zhu, Z.; Soh, C.L.; Kumar, R.; Evans, T.; et al. Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction with GATA4 in Human Pancreatic Development. Cell Stem Cell 2017, 20, 675–688.e676. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.; Chen, Y.; Chen, X.; Fan, J.; Zhu, K.; Hu, Z.; Li, S.; Zhu, J.; Feng, J.; Wu, Z.; et al. FGF18 alleviates hepatic ischemia-reperfusion injury via the USP16-mediated KEAP1/Nrf2 signaling pathway in male mice. Nat. Commun. 2023, 14, 6107. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G.; Menini, S. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10, 727. [Google Scholar] [CrossRef] [PubMed]
- Hakki Kalkan, I.; Suher, M. The relationship between the level of glutathione, impairment of glucose metabolism and complications of diabetes mellitus. Pak. J. Med. Sci. 2013, 29, 938–942. [Google Scholar] [CrossRef]
- De Mattia, G.; Bravi, M.C.; Laurenti, O.; Cassone-Faldetta, M.; Armiento, A.; Ferri, C.; Balsano, F. Influence of reduced glutathione infusion on glucose metabolism in patients with non-insulin-dependent diabetes mellitus. Metabolism 1998, 47, 993–997. [Google Scholar] [CrossRef]
- Barrett, W.C.; DeGnore, J.P.; Konig, S.; Fales, H.M.; Keng, Y.F.; Zhang, Z.Y.; Yim, M.B.; Chock, P.B. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 1999, 38, 6699–6705. [Google Scholar] [CrossRef]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef]
- Liu, X.; Jann, J.; Xavier, C.; Wu, H. Glutaredoxin 1 (Grx1) Protects Human Retinal Pigment Epithelial Cells From Oxidative Damage by Preventing AKT Glutathionylation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, R.K.; Bhaskar, A.; Vijayalingam, S.; Viswanathan, M.; Mohan, R.; Shanmugasundaram, K.R. Antioxidant status and lipid peroxidation in type II diabetes mellitus with and without complications. Clin. Sci. 1996, 90, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Memisogullari, R.; Taysi, S.; Bakan, E.; Capoglu, I. Antioxidant status and lipid peroxidation in type II diabetes mellitus. Cell Biochem. Funct. 2003, 21, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Reisman, S.A.; Yeager, R.L.; Goedken, M.J.; Klaassen, C.D. Nuclear factor erythroid 2-related factor 2 deletion impairs glucose tolerance and exacerbates hyperglycemia in type 1 diabetic mice. J. Pharmacol. Exp. Ther. 2010, 333, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef]
- Zhao, S.; Song, T.; Gu, Y.; Zhang, Y.; Cao, S.; Miao, Q.; Zhang, X.; Chen, H.; Gao, Y.; Zhang, L.; et al. Hydrogen Sulfide Alleviates Liver Injury Through the S-Sulfhydrated-Kelch-Like ECH-Associated Protein 1/Nuclear Erythroid 2-Related Factor 2/Low-Density Lipoprotein Receptor-Related Protein 1 Pathway. Hepatology 2021, 73, 282–302. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, J.; Kocan, L.; Vasko, L.; Perjesi, P. Glutathione-Related Enzymes and Proteins: A Review. Molecules 2023, 28, 1447. [Google Scholar] [CrossRef] [PubMed]
- Tuell, D.; Ford, G.; Los, E.; Stone, W. The Role of Glutathione and Its Precursors in Type 2 Diabetes. Antioxidants 2024, 13, 184. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) | Accession No. |
|---|---|---|---|
| Ho-1 | CCCAGAGGCTGTGAACTCTG | GGGGAAAGCAGTCATGGTCA | NM_012580.2 |
| Nqo1 | GAAAGGATGGGAGGTGGTCG | GCTCCCCTGTGATGTCGTTT | NM_017000.3 |
| Gclc | TGATTGAAGGGACACCTGGC | TGTGCTCTGGCAGTGTGAAT | NM_012815.2 |
| Grx1 | CGTGGTCTCCTGGAATTTGTG | AAGACCCGAGGAACTGTTCTTG | NM_022278.1 |
| Nrf2 | TAGATCTTGGGGTAAGTCGAGA | CTCTTGTCTCTCCTTTTCGAGT | NM_001399173.1 |
| Ins2 | TGGAAGCTCTCTACCTGGTGT | GTGCCAAGGTCTGAAGGTCAC | NM_019130.2 |
| Ins1 | CTACACACCCAAGTCCCGTC | CCAAGGTCTGAAGATCCCCG | NM_019129.3 |
| Ngn3 | GTCAGAGACTGTCACACCCC | TGGAACTGAGCACTTCGTGG | NM_021700.1 |
| MafA | TTCTGGAGAGCGAGAAGTGC | CGCGCTCACAGAAAGAAGTC | XM_017603453.1 |
| Pdx1 | CGGACATCTCCCCATACG | AAAGGGAGATGAACGCGG | NM_022852.3 |
| β-actin | TTTAATGTCACGCACGATTTCCC | CCCATCTATGAGGGTTACGC | NM_031144.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Zhou, Q.; Chen, H.; Bai, J.; An, R.; Zhang, K.; Zhang, X.; An, H.; Zhang, J.; Wang, Y.; et al. Glutathione Induces Keap1 S-Glutathionylation and Mitigates Oscillating Glucose-Induced β-Cell Dysfunction by Activating Nrf2. Antioxidants 2024, 13, 400. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox13040400
Chen X, Zhou Q, Chen H, Bai J, An R, Zhang K, Zhang X, An H, Zhang J, Wang Y, et al. Glutathione Induces Keap1 S-Glutathionylation and Mitigates Oscillating Glucose-Induced β-Cell Dysfunction by Activating Nrf2. Antioxidants. 2024; 13(4):400. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox13040400
Chicago/Turabian StyleChen, Xiufang, Qian Zhou, Huamin Chen, Juan Bai, Ruike An, Keyi Zhang, Xinyue Zhang, Hui An, Jitai Zhang, Yongyu Wang, and et al. 2024. "Glutathione Induces Keap1 S-Glutathionylation and Mitigates Oscillating Glucose-Induced β-Cell Dysfunction by Activating Nrf2" Antioxidants 13, no. 4: 400. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox13040400