Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance


1
Department of Pediatrics, University of Texas Medical Branch at Galveston, Galveston, TX 77555, USA
2
Department of Microbiology and Immunology, University of Texas Medical Branch at Galveston, Galveston, TX 77555, USA
*
Author to whom correspondence should be addressed.
Antioxidants 2018, 7(10), 129; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox7100129
Submission received: 21 May 2018
/
Revised: 21 September 2018
/
Accepted: 27 September 2018
/
Published: 28 September 2018
Abstract
:Hydrogen sulfide (H2S) has arisen as a critical gasotransmitter signaling molecule modulating cellular biological events related to health and diseases in heart, brain, liver, vascular systems and immune response. Three enzymes mediate the endogenous production of H2S: cystathione β-synthase (CBS), cystathione γ-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3-MST). CBS and CSE localizations are organ-specific. 3-MST is a mitochondrial and cytosolic enzyme. The generation of H2S is firmly regulated by these enzymes under normal physiological conditions. Recent studies have highlighted the role of H2S in cellular redox homeostasis, as it displays significant antioxidant properties. H2S exerts antioxidant effects through several mechanisms, such as quenching reactive oxygen species (ROS) and reactive nitrogen species (RNS), by modulating cellular levels of glutathione (GSH) and thioredoxin (Trx-1) or increasing expression of antioxidant enzymes (AOE), by activating the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2). H2S also influences the activity of the histone deacetylase protein family of sirtuins, which plays an important role in inhibiting oxidative stress in cardiomyocytes and during the aging process by modulating AOE gene expression. This review focuses on the role of H2S in NRF2 and sirtuin signaling pathways as they are related to cellular redox homeostasis.
1. Introduction
Hydrogen sulfide (H2S) is an inorganic and colorless gas, with strong odor and toxic effects at high concentrations [1]. In the last few years, H2S has been identified as the third most physiologically important gasotransmitter participating in multiple cellular signaling pathways, along with carbon monoxide (CO) and nitric oxide (NO) [2]. It plays a physiological role in a variety of cellular and organ functions and a protective role in multiple pathological conditions, displaying vasoactive, cytoprotective, anti-inflammatory and antioxidant activities (reviewed in [3]). As a gasotransmitter, it diffuses quickly through the cells, operating next to sites of biosynthesis, with a short lifetime [4]. Endogenous H2S in mammals is generated through enzymatic and non-enzymatic pathways. The former process requires the action of cytosolic and mitochondrial enzymes: cystathionine β-synthase (CBS), cystathionine γ-lsase (CSE), 3-mercaptopyruvate sulfurtransferase (3-MST) and cysteine aminotransferase (CAT), using L-cysteine or homocysteine as substrates (Figure 1). CBS and CSE are mainly expressed in the vascular, nervous, and cardiovascular systems, as well as in the liver and kidney [5,6]. These two enzymes are primarily responsible for H2S production, and they are also released in the circulatory system by hepatocytes and endothelial cells, as a part of the plasma [7]. The non-enzymatic pathway is based on the reduction of sulfur species and thiol molecules, contributing in a minor extent to H2S cellular content.
Three distinct mechanisms are implicated in the catabolism of H2S: (1) oxidation, (2) methylation, and (3) scavenging by metalloproteins. Oxidation is the most common reaction, and encompasses the rapid metabolism of H2S to sulfate and sulfite species with thiosulfate as an intermediate molecule. It takes place in the mitochondria through the sequential action of sulfide: quinone oxidoreductase (SQR), rhodanese and sulfur dioxygenase. Methylation converts endogenous H2S into dimethylsulfide and thiol S-methyltransferase (TMST) mainly in the cytoplasm, and it seems to have a lesser role than the oxidation pathway. Scavenging by metalloproteins involves the binding of H2S and hemoglobin by scavenging reaction, producing disulfide or metallo-containing products [8].
Free H2S exists in equilibrium with a pool of labile sulfur-containing molecules that can release H2S under certain physiological conditions. It has become more and more evident that part of the signaling effects attributed to H2S result from the occurrence of persulfides and polysulfides, among other sulfur-containing molecules, which have been collectively termed as “reactive sulfur species” (RSS). For more details on H2S metabolism and the formation of persulfides and polysulfides, as well as their role in cellular functions and signaling, please refer to the numerous excellent recent reviews published, such as [9,10,11,12,13].
Several studies have highlighted the role of H2S/RSS in cellular redox homeostasis, which occurs in part by modulating levels of cellular antioxidants, such as gluthatione (GSH), and increasing expression of antioxidant enzymes (AOE), and increasing activities/expressions of the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2) and the histone deacetylase protein family of sirtuins (SIRTs). This review summarizes the known role of H2S in maintaining cellular redox balance through these two mechanism(s) and its relationship with oxidative stress-related diseases.
2. Oxidative Stress and Antioxidant Effects of Hydrogen Sulfide
Reactive oxygen species (ROS) are ubiquitous, highly reactive molecules produced as a result of the reduction of molecular oxygen. Cellular sites for ROS generation include the mitochondria, and microsomes and require the involvement of various enzymes like cyclooxygenase, lipoxygenase, xanthine oxidase and membrane-bound reduced nicotinamide adenine dinucleotide phosphate NADPH-oxidase. Excessive levels of ROS can be generated by increased stimulation of the NADPH-oxidase system (mitochondrial and cell membrane-associated) or by other mechanisms, often involving mitochondrial dysfunction. Oxidative stress represents an imbalance between the ROS generation and the cellular antioxidant defensive system, which includes scavenging and repairing molecules. The first include a number of AOEs, such as superoxide dismutase (SOD) (three isoforms of SOD have been identified in mammals: the cytoplasmic Cu/ZnSOD or SOD1, the mitochondrial MnSOD or SOD2, and the extracellular ECSOD or SOD3), catalase and glutathione peroxidase (GPx). The latter include glutathione (GSH) and thioredoxin (Trx-1), which are the predominant antioxidants acting as a defense net during the oxidative stress process [15,16]. GSH is a tripeptide made of cysteine, glycine and glutamate, existing often as a reduced form, and it is synthesized from cysteine. GSH reduces disulfide bonds formed within cytoplasmic proteins to cysteines by serving as an electron donor. In the process, GSH is converted to its oxidized form, glutathione disulfide (GSSG). Trx-1 is a 12-kD oxidoreductase enzyme containing a dithiol–disulfide active site, which acts as an antioxidant by facilitating the reduction of other proteins by cysteine thiol-disulfide [17]. H2S has been shown to exert antioxidant effects through several mechanisms including direct quenching of ROS, modulation of cellular levels of GSH and Trx-1, or increased expression of AOE, by activating the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2), as described below and summarized in Figure 2.
2.1. H2S and Repairing Antioxidant Defenses
H2S has been shown to be able to scavenge ROS and reactive nitrogen species (RNS), including hypochlorous acid, hydrogen peroxide, lipid hydroperoxides, superoxide and peroxynitrite (reviewed in [18,19]). Molecules containing an SH group such as H2S, HS–, HS–SH, and HSS– can reverse the damage due to ROS/RNS by donating a hydrogen atom to carbon-centered radicals; however, the very low concentrations of H2S and related molecules in blood and tissues limit their efficacy of repairing free radical cellular damage.
Cysteine, in addition to be a precursor for H2S, is also the source of GSH production. Cysteine exists as two unstable redox forms in the body: the oxidized form—cystine and the reduced form—cysteine. The extracellular cystine form is carried into cells through the cystine/glutamate antiporter system, after which cysteine is reduced and ready for GSH synthesis. The release of H2S into the extracellular space has been shown to induce a reduction of cystine into cysteine, increasing the amount of cysteine available as a substrate for GSH synthesis, and to enhance cystine transport [20]. GSH is synthesized by the consecutive catalysis of two enzymes, γ-glutamyl cysteine synthetase (γ-GCS) and glutathione synthetase (GS). H2S administration has been shown to enhance γ-GCS activity, without changing its expression [20]. H2S administration is also associated with augmented levels of GSH in the mitochondria. As cytoplasmic GSH is transported into mitochondria, because mitochondria cannot synthesize GSH, the enhanced mitochondrial GSH concentration following H2S administration is suggested to depend on the increased cytoplasmic GSH levels and enhanced transport into the mitochondria [20].
As mentioned above, thioredoxins are small thiol-oxidoreductase enzymes that control cellular redox homeostasis. In a mouse model of ischemia-induced heart failure, H2S treatment increased the Trx-1 gene and protein levels, as well as basal Trx-1 activity. H2S-dependent cardioprotection was dependent on an intact Trx-1 protein [21]. H2S has been found to up-regulate Trx-1 in part through an NRF2-independent, unidentified mechanism [22].
2.2. H2S-Mediated NRF2 Activation
NRF2 is a basic leucine-zipper protein, belonging to the Cap’n’Collar family of transcription factors, that mediates expression of cytoprotective genes that defend cells from oxidative stress and cellular damage, including AOEs. NRF2-driven gene expression occurs through NRF2 binding to promoters’ antioxidant responsive element (ARE) sequences. Under normal physiologic conditions, this transcription factor is confined to the cytoplasm by binding to Kelch-like ECH-associated protein 1 (Keap1) dimer forming an inactive complex. Whenever a change in redox status occurs by increased cellular ROS levels, Keap1 dimer changes conformation due to the breaking of disulfide bonds between cysteine residues, and releases NRF2, which translocates to the nucleus and induces the transcription of AOE genes to attain redox homestastis [23].
At pH 7.4 under normal conditions, H2S is present mainly as dissociated anion (HS−, S2−) and 20% as not dissociated species. S-sulfhydration or persulfidation is a post-translational modification in which a sulfhydryl group (R-SH) attaches to the cysteine residues of target proteins in order to regulate the protein function. A variety of key proteins acting as a switch/sensor of different cellular pathways in mammals are sulfhydrated by H2S, leading to modulation of cell signaling that relates to oxidative stress, cell survival/death, metabolism, cell proliferation, and inflammation [24,25]. Various studies have shown that S-sulfhydration is one mechanism where H2S interacts directly with the NRF2 pathway. H2S has been shown to S-sulfhydrate Keap1 at the cysteine-151 residue, leading to NRF2 dissociation, increased nuclear translocation and expression of antioxidant genes through binding to promoters’ ARE sites [26]. Furthermore, H2S can S-sulfhydrate Keap1 at the cysteine-226 and cysteine-613 residues, leading to Keap1 inactivation, NRF2 release and promotion of NRF2-dependent gene expression [27].
3. H2S and Sirtuin Interaction during Oxidative Stress
SIRTs are enzymes that catalyze post-translational modifications of both histone and nonhistone proteins. There are seven members in the mammalian family with different cellular localizations, enzymatic activities and targets (reviewed in [28]). SIRT1 and SIRT6 are present in the nucleus; SIRT2 is in the cytoplasm; SIRT3, SIRT4, SIRT5 are localized in the mitochondria. Originally identified as deacetylases, SIRTs have more recently been found to catalyze a variety of other reactions, including desuccinylation, demalonylation, and deglutarylation. SIRTs are classified as class III histone deacetylases (HDACs) and they use β-Nicotinamide adenine dinucleotide (NAD+) as cofactor, different from HDAC classes I and II, which use zinc instead. They are involved in a variety of cellular functions and are regulated in response to a wide range of stimuli, including nutritional and metabolic changes, inflammatory signals and oxidative stress. Disruption of redox cellular homeostasis affects SIRTs at different levels, including inducing or repressing their expression, and leading to post-translational modifications such as cysteine oxidation and nitrosylation, which can lead to loss of their function (reviewed in [29]).
SIRT1 is localized predominantly in the nucleus, but is also present in the cytosol. Among its numerous known targets are the tumor-suppressor protein p53, nuclear nactor kappa B (NF-κB), peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α), forkhead box protein O (FOXO), and many other transcription factors and nuclear receptors participating in the regulation of multiple cellular functions, including mitochondrial biogenesis, glucose, and lipid metabolism, DNA repair, apoptosis, inflammation and oxidative stress resistance. In a model of atherosclerosis, lack of H2S-generating enzyme CSE, or H2S donor administration, have been shown to induce SIRT1 expression and increase deacetylation activity by sulfhydration of two CXXC domains, which caused SIRT1 to bind more zinc, therefore promoting its activity, and decreasing its ubiquitin-dependent degradation [30]. In a cardiomyocytes culture model of oxidative damage induced by hydrogen peroxide (H2O2), H9c2 cells treated with the H2S donor sodium hydrosulfide (NaHS) displayed a lower oxidants level and higher expression of the AOE SOD, GPx and GST, as well as increased SIRT1 expression. Treatment of cells with the SIRT1 inhibitor Ex 527 reverted the NaHS effect, indicating that H2S antioxidant effect was mediated through the SIRT pathway [31]. In an endothelial cell model of senescence induced by H2O2, treatment with NaHS resulted in increased SIRT1 activity, although not expression, and inhibition of endothelial cell dysfunction in a SIRT-dependent manner [32]. Changes in SIRT1 activity in endothelial cells after exogenous administration of NaHS have been linked to regulation of intracellular levels of NAD+ [33]. Diallyl trisulfide (DATS), an organosulfur compound of garlic, is a natural H2S donor. In a mouse model of ischemia-reperfusion injury, DATS treatment up-regulated cardiac SIRT1 expression and nuclear distribution, leading to reduced oxidative stress and endoplasmic reticulum stress-dependent apoptosis [34].
SIRT3 is a major regulator of mitochondrial function. SIRT3 catalyzes deacetylation of mitochondrial proteins, which in turn affects mitochondrial energy metabolism. SIRT3 is regulated by nutritional status and metabolic stress. To investigate the ability of H2S to modulate oxidative stress in endothelial cells via SIRT3 activation, the endothelial cell line EA.hy926 was pretreated with the H2S slow-releasing donor GYY4137, and then exposed to H2O2. GYY4137-treated cells exhibited decreased ROS formation and increased levels of total SOD activity, compared to the cells treated only with H2O2 [35]. GYY4137 treatment was able to restore the SIRT3 expression level, which was decreased by H2O2 exposure, through increased activator protein (AP)-1 binding to the SIRT3 promoter—effects abolished by treatment of endothelial cells with the AP-1 inhibitor SR11302 [35]. To investigate the mechanism by which H2S protects against cardiac hypertrophy, neonatal rat cardiomyocytes were pretreated with NaHS and treated with angiotensin II. H2S treatment was associated with increased SIRT3 expression and was able to reverse angiotensin-induced mitochondrial dysfunction and SOD2 expression (the latter was due to reduced FOXO3a activation—effects abolished by SIRT3 silencing in cells [36]. In a mouse model of transverse aortic constriction (TAC) of myocardial hypertrophy, the NaHS treatment was able to reduce hypertrophy, inhibit oxidative stress, and restore mitochondria structure, volume and number only in wild-type but not SIRT3 knockout mice [36]. The summary of the relationship between H2S and SIRT is presented in Figure 3.
4. H2S Treatment in Animal Models of Diseases Associated with Oxidative Damage
H2S has been recognized as playing a protective role in a variety of diseases. Reduced endogenous H2S levels, redox imbalance, and oxidative damage are associated with disease severity and progression in cardiac, neurological, pulmonary, gastric, nephrological, hepatic diseases, as well as in aging. H2S donor administration has proven beneficial in a variety of diseases associated with oxidative damage. Table 1 summarizes findings in animal models, where H2S donor administration results in changes in oxidative stress and/or NRF2 activation and AOE expression/activity.
A H2S donor—Na2S—provided profound protection against myocardial ischemic injury in mice as evidenced by significant decreases in infarct size, and oxidative damage. H2S increased S-sulfhydration of Keap1, induced NRF2 dissociation from Keap1, enhanced NRF2 nuclear translocation, and expression of antioxidant enzymes to neutralize ROS [22]. Treatment with slow-releasing H2S donor GYY4137 protected rats against myocardial ischemia and reperfusion injury by suppressing superoxide anion levels, oxidative damage, and extracellular signal–regulated kinase ERK pathway in the myocardium [37]. H2S donor (NaHS) treatment in rats decreased NADPH oxidase 4-ROS-ERK1/2 signaling axis and increased heme oxygenase-1 (HO-1) expression and attenuated myocardial fibrotic response [38]. A novel H2S-donor-4-carboxyphenyl isothiocyanate (4CPI) treatment significantly decreased ROS levels, oxidative damage and ischemia/reperfusion-induced tissue injury in an in vivo model of acute myocardial infarction in rats [39]. Treatment with H2S donor (NaHS)-reduced NADPH oxidase 4 (NOX4) and ROS levels and cellular oxidative stress, ameliorating cardiac dysfunction in Takotsubo cardiomyopathy (TCM) in rats [40]. The organosulfur compound diallyl trisulfide (DATS) treatment in mice attenuated cardiac dysfunction after heart failure via induction of angiogenesis. DATS treatment provided a proangiogenic environment for the growth of new vessels by inducing expression of the proliferation marker, Ki67, as well as GPx-1 and HO-1 [41]. Treatment with NaHS significantly attenuated angiotensin II-induced hypertension and oxidative stress in mice by decreasing superoxide radical, resulting in lowered blood pressure and endothelial dysfunction [42].
H2S treatment offers beneficial roles in neurodegenerative disorders. Parkinson’s disease (PD) is characterized by a progressive loss of dopaminergic neurons in the substantia nigra that leads to movement dysfunction. Treatment with NaHS protected rats from 6-hydroxydopamine (6-OHDA)-induced PD by suppressing NADPH oxidase activation, ROS levels, oxidative damage, and inflammation [43]. Progressive losses of neurons and memory are hallmarks of Alzheimer’s disease (AD), and beta-amyloid plaques and oxidative stress play a crucial role in the pathogenesis. NaHS treatment in AD mice exerted antioxidant and neuroprotective effects by inducing NRF2, HO-1, GST, and ameliorating learning memory impairment [44]. Huntington’s disease is a fatal genetic disorder associated with accumulation of expanded polyglutamine repeats in huntingtin protein, leading to oxidative stress, neurotoxicity, and motor and behavioral changes. Recently, the researchers observed a significant depletion of CSE, the biosynthetic enzyme for cysteine, in Huntington’s disease tissues, and supplementation with cysteine-reverted abnormalities in a mouse model of Huntington’s disease [45].
Treatment with NaHS or GYY4137 or supplementation with L-cysteine in rats protected against gastric ischemia/reperfusion (I/R) lesions. H2S exerted antioxidative properties by inducing expression of SOD2 and GPx-1, leading to an increase in gastric microcirculation and prevention of further progression of I/R injury into deeper gastric ulcers [46,47]. NaHS treatment in rats attenuated pulmonary I/R injury by inducing SOD and catalase activities, quenching superoxide production and reducing lipid damage [48]. Administration of NaHS gave protection against pulmonary fibrosis in smoking rats by attenuating oxidative stress and inflammation. H2S induced NRF2 activity and up-regulated antioxidant genes HO-1 and Trx-1 and inhibited NF-κB activity in the smoking rat lungs [49]. H2S protected the murine liver against I/R injury through up-regulation of GSH, and Trx-1 activity, attenuated lipid damage, and inhibited inflammatory factors and the progression of apoptosis [50,51]. NaHS protected rat kidneys against diabetic nephropathy and uranium-induced toxicities and murine kidneys against I/R injury through activation of the NRF2-antioxidant defense pathway and suppression of the inflammatory response [52,53,54].
5. Conclusions
An increased number of studies have confirmed the beneficial use of H2S donors in neuronal, cardiovascular and other oxidative stress-dependent diseases [3,4]. The role of H2S in modulating redox signaling has still not been fully understood, as H2S explicates an antioxidant effect through multiple mechanisms and interactions with different targets. Additionally, low or high cellular levels of H2S are linked to different outcomes of the cellular responses. The review goal was to discuss the connection between H2S and modulation of redox signaling and summarize the studies elucidating the role of H2S administration as a potential therapeutic approach for diseases due to altered redox cellular balance.
Author Contributions
N.K. and T.C. wrote the paper, A.C. conceived and edited the paper.
Funding
This work was partially supported by NIH grants AI122142and AI25434.
Acknowledgments
The authors would like to thank Cynthia Tribble for assistance in editing and submitting paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mancardi, D.; Penna, C.; Merlino, A.; Del, S.P.; Wink, D.A.; Pagliaro, P. Physiological and pharmacological features of the novel gasotransmitter: Hydrogen sulfide. Biochim. Biophys. Acta 2009, 1787, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Luciano, M.G.; Ingrosso, D.; Raiola, I.; Pulzella, P.; Sepe, I.; Lanza, D.; Violetti, E.; Capasso, R.; Lombardi, C.; et al. Hydrogen sulfide, the third gaseous signaling molecule with cardiovascular properties, is decreased in hemodialysis patients. J. Ren. Nutr. 2010, 20, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Production and physiological effects of hydrogen sulfide. Antioxid. Redox Signal. 2014, 20, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Olas, B. Hydrogen sulfide in signaling pathways. Clin. Chim. Acta 2015, 439, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Wong, P.T.; Bian, J.S. Hydrogen sulfide: A novel signaling molecule in the central nervous system. Neurochem. Int. 2010, 56, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 6904327. [Google Scholar] [CrossRef] [PubMed]
- Bearden, S.E.; Beard, R.S.; Pfau, J.C., Jr. Extracellular transsulfuration generates hydrogen sulfide from homocysteine and protects endothelium from redox stress. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1568–H1576. [Google Scholar] [CrossRef] [PubMed]
- Lowicka, E.; Beltowski, J. Hydrogen sulfide (H2S)—The third gas of interest for pharmacologists. Pharmacol. Rep. 2007, 59, 4–24. [Google Scholar] [PubMed]
- Giuffre, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxid. Med. Cell. Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed]
- Olson, KR. H2S and polysulfide metabolism: Conventional and unconventional pathways. Biochem. Pharmacol. 2018, 149, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, L.; Bianco, C.L.; Toscano, J.P.; Lin, J.; Akaike, T.; Fukuto, J.M. Chemical Biology of Hydropersulfides and Related Species: Possible Roles in Cellular Protection and Redox Signaling. Antioxid. Redox Signal. 2017, 27, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Bazhanov, N.; Ansar, M.; Ivanciuc, T.; Garofal, R.P.; Casola, A. Hydrogen sulfide: A novel player in airway development, pathophysiology of respiratory diseases, and antiviral defenses. Am. J. Respir. Cell Mol. Biol. 2017, 57, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Dominko, K.; Dikic, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arch. Ind. Hyg. Toxicol. 2018, 69, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, J.; Arner, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen sulfide in biochemistry and medicine. Antioxid. Redox Signal. 2012, 17, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Winterbourn, C.C. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 2010, 23, 1541–1543. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, C.K.; Lambert, J.P.; Molkentin, J.D.; Sadoshima, J.; Calvert, J.W. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Du, J.; Tang, C.; Huang, Y.; Jin, H. H2S-Induced Sulfhydration: Biological Function and Detection Methodology. Front. Pharmacol. 2017, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The Gasotransmitter Hydrogen Sulfide Induces Nrf2-Target Genes by Inactivating the Keap1 Ubiquitin Ligase Substrate Adaptor Through Formation of a Disulfide Bond between Cys-226 and Cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Andersson, U.; Hakkola, J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. 2016, 30, 3942–3960. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Escande, C.; Denicola, A. Potential Modulation of Sirtuins by Oxidative Stress. Oxid. Med. Cell. Longev. 2016, 2016, 9831825. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Lin, X.; Xu, W.; Zheng, F.; Cai, J.; Yang, J.; Cui, Q.; Tang, C.; Cai, J.; Xu, G.; et al. Sulfhydrated sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, Q.; Liu, X.; Pan, L.; Xiong, Q.; Zhu, Y.Z. Hydrogen sulfide protects against apoptosis under oxidative stress through SIRT1 pathway in H9c2 cardiomyocytes. Nitric Oxide 2015, 46, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Suo, R.; Zhao, Z.Z.; Tang, Z.H.; Ren, Z.; Liu, X.; Liu, L.S.; Wang, Z.; Tang, C.K.; Wei, D.H.; Jiang, Z.S. Hydrogen sulfide prevents H2O2-induced senescence in human umbilical vein endothelial cells through SIRT1 activation. Mol. Med. Rep. 2013, 7, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an endothelial NAD(+)-H2S signaling network is a reversible cause of vascular aging. Cell 2018, 173, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, S.; Tang, X.; Li, Z.; Zhang, J.; Xue, X.; Han, J.; Liu, Y.; Zhang, Y.; Zhang, Y.; et al. Diallyl trisulfide ameliorates myocardial ischemia-reperfusion injury by reducing oxidative stress and endoplasmic reticulum stress-mediated apoptosis in type 1 diabetic rats: Role of SIRT1 activation. Apoptosis 2017, 22, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Feng, H.; Li, S.; Meng, G.; Liu, S.; Tang, X.; Ma, Y.; Han, Y.; Xiao, Y.; Gu, Y.; et al. SIRT3 mediates the antioxidant effect of hydrogen sulfide in endothelial cells. Antioxid. Redox Signal. 2016, 24, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Liu, J.; Liu, S.; Song, Q.; Liu, L.; Xie, L.; Han, Y.; Ji, Y. Hydrogen sulfide pretreatment improves mitochondrial function in myocardial hypertrophy via a SIRT3-dependent manner. Br. J. Pharmacol. 2018, 175, 1126–1145. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Wang, J.; Xiao, Y.; Bai, W.; Xie, L.; Shan, L.; Moore, P.K.; Ji, Y. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 2015, 29, 203–213. [Google Scholar] [PubMed] [Green Version]
- Pan, L.L.; Liu, X.H.; Shen, Y.Q.; Wang, N.Z.; Xu, J.; Wu, D.; Xiong, Q.H.; Deng, H.Y.; Huang, G.Y.; Zhu, Y.Z. Inhibition of NADPH oxidase 4-related signaling by sodium hydrosulfide attenuates myocardial fibrotic response. Int. J. Cardiol. 2013, 168, 3770–3778. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Marino, A.; Piano, I.; Brancaleone, V.; Tomita, K.; Di Cesare, M.L.; Martelli, A.; Citi, V.; Breschi, M.C.; Levi, R.; et al. The novel H2S-donor 4-carboxyphenyl isothiocyanate promotes cardioprotective effects against ischemia/reperfusion injury through activation of mitoKATP channels and reduction of oxidative stress. Pharmacol. Res. 2016, 113, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jin, S.; Teng, X.; Duan, X.; Chen, Y.; Wu, Y. Hydrogen sulfide attenuates cardiac injury in takotsubo cardiomyopathy by alleviating oxidative stress. Nitric Oxide 2017, 67, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Al-Magableh, M.R.; Kemp-Harper, B.K.; Hart, J.L. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens. Res. 2015, 38, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.F.; Lu, M.; Tiong, C.X.; Dawe, G.S.; Hu, G.; Bian, J.S. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 2010, 9, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Deng, Y.; Liu, H.; Yin, C.; Li, X.; Gong, Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 2016, 150–151, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine gamma-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 509, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Magierowski, M.; Magierowska, K.; Hubalewska-Mazgaj, M.; Sliwowski, Z.; Pajdo, R.; Ginter, G.; Kwiecien, S.; Brzozowski, T. Exogenous and endogenous hydrogen sulfide protects gastric mucosa against the formation and time-dependent development of ischemia/reperfusion-induced acute lesions progressing into deeper ulcerations. Molecules 2017, 22, 295. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, L.; Zou, J.; Qiao, W.; Liu, H.; Qi, Y.; Yan, C. Protective effect of endogenous hydrogen sulfide against oxidative stress in gastric ischemia-reperfusion injury. Exp. Ther. Med. 2013, 5, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Liu, X.; Geng, B.; Fang, L.; Tang, C. Hydrogen sulfide protects rat lung from ischemia-reperfusion injury. Life Sci. 2008, 82, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; An, G.; Chen, J. Inhibitory effects of hydrogen sulphide on pulmonary fibrosis in smoking rats via attenuation of oxidative stress and inflammation. J. Cell. Mol. Med. 2014, 18, 1098–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, S.; Calvert, J.W.; Duranski, M.R.; Ramachandran, A.; Lefer, D.J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H801–H806. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Zhao, M.; Jiang, H.; Tan, G.; Pan, S.; Sun, X. Role of hydrogen sulfide in hepatic ischemia-reperfusion-induced injury in rats. Liver Transpl. 2009, 15, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhao, T.; Yuan, Y.; Hu, N.; Tang, X. Hydrogen sulfide (H2S) attenuates uranium-induced acute nephrotoxicity through oxidative stress and inflammatory response via Nrf2-NF-kappaB pathways. Chem. Biol. Interact. 2015, 242, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Feng, Y.; Zhan, Z.; Chen, J. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J. Biol. Chem. 2014, 289, 28827–28834. [Google Scholar] [CrossRef] [PubMed]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.L.; Wang, M.J.; Sun, C.; Huang, Y.; Jin, S.; Mu, X.P.; Chen, Y.; Zhu, Y.C. Protective Effects of Hydrogen Sulfide in the Ageing Kidney. Oxid. Med. Cell. Longev. 2016, 2016, 7570489. [Google Scholar] [CrossRef] [PubMed]
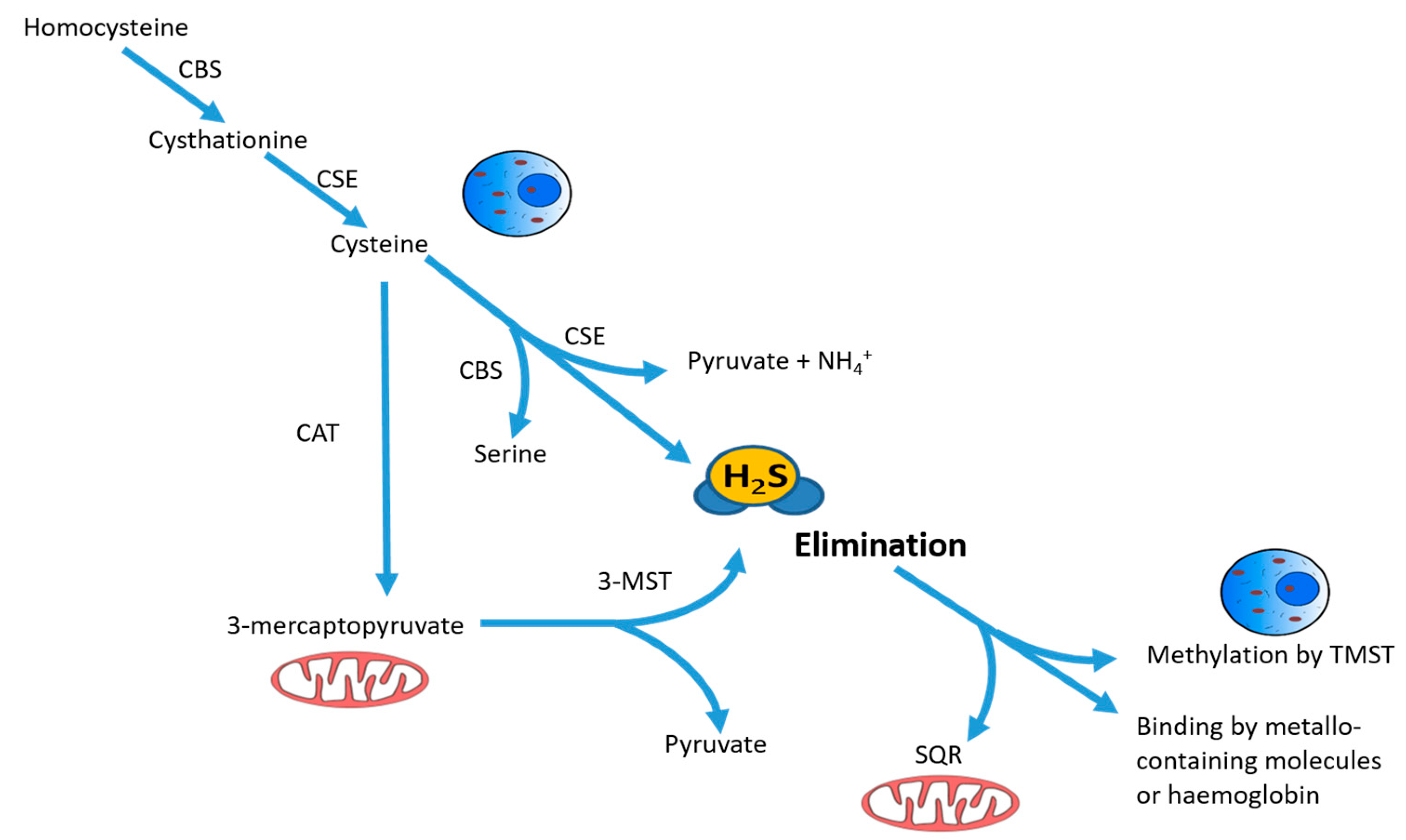
Figure 1.
Schematic description of intracellular synthesis and degradation of hydrogen sulfide H2S. H2S is produced by cytoplasmic and mitochondrial enzymes cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), 3-mercaptopyruvate sulfurtransferase (3-MST) and cysteine aminotransferase (CAT) using cysteine or homocysteine as substrates. The intracellular non-toxic H2S level is being actively maintained by oxidation in mitochondria by the enzyme sulfide:quinone reductase (SQR), together with rhodanese and sulfur dioxygenase, or by methylation in the cytoplasm using thiol S-methyltransferase (TMST). Free H2S can also be bound by methemoglobin and by molecules with metallic or disulfide bonds and excreted with biological fluids. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society [14].
Figure 1.
Schematic description of intracellular synthesis and degradation of hydrogen sulfide H2S. H2S is produced by cytoplasmic and mitochondrial enzymes cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), 3-mercaptopyruvate sulfurtransferase (3-MST) and cysteine aminotransferase (CAT) using cysteine or homocysteine as substrates. The intracellular non-toxic H2S level is being actively maintained by oxidation in mitochondria by the enzyme sulfide:quinone reductase (SQR), together with rhodanese and sulfur dioxygenase, or by methylation in the cytoplasm using thiol S-methyltransferase (TMST). Free H2S can also be bound by methemoglobin and by molecules with metallic or disulfide bonds and excreted with biological fluids. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society [14].

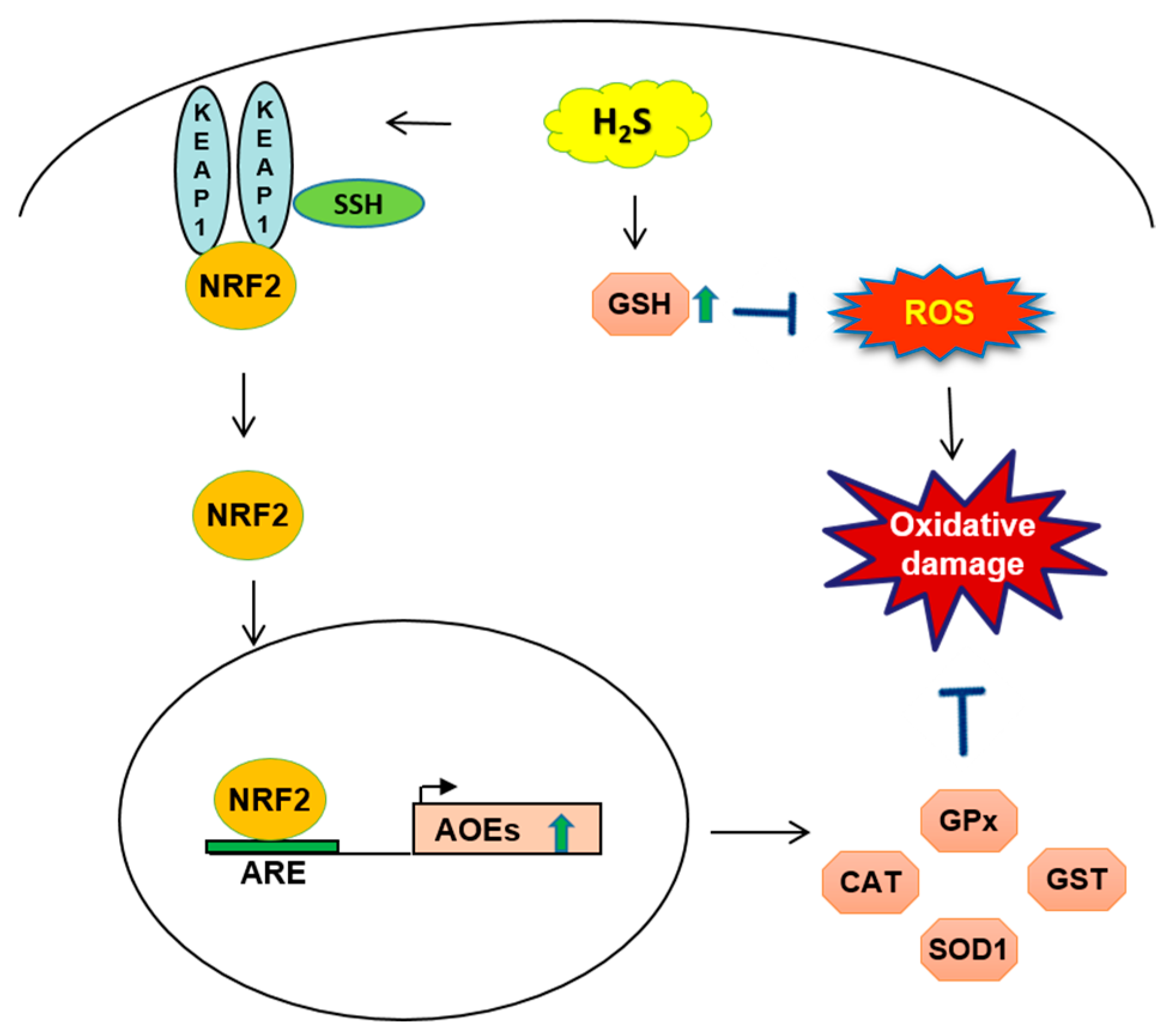
Figure 2.
Schematic of H2S mechanism related to glutathione GSH and nuclear factor (erythroid-derived 2)-like 2 NRF2 targets in oxidative cell-damage. The endogenous release of H2S increases GSH synthesis and blocks reactive oxygen species ROS production. When the cellular level of H2S is increased, Kelch-like ECH-associated protein 1 Keap1 protein is S-sulfhydrated SSH: which brings a conformational change of the protein and NRF2 release from Keap1. NRF2 translocates to the nucleus, binding to the promoter containing antioxidant response element (ARE) sequences and increased transcription of antioxidant genes as catalase CAT, superoxide dismutase SOD1, glutathione-S-transferase GST, glutathione peroxidase GPx. AOE: antioxidant enzyme.
Figure 2.
Schematic of H2S mechanism related to glutathione GSH and nuclear factor (erythroid-derived 2)-like 2 NRF2 targets in oxidative cell-damage. The endogenous release of H2S increases GSH synthesis and blocks reactive oxygen species ROS production. When the cellular level of H2S is increased, Kelch-like ECH-associated protein 1 Keap1 protein is S-sulfhydrated SSH: which brings a conformational change of the protein and NRF2 release from Keap1. NRF2 translocates to the nucleus, binding to the promoter containing antioxidant response element (ARE) sequences and increased transcription of antioxidant genes as catalase CAT, superoxide dismutase SOD1, glutathione-S-transferase GST, glutathione peroxidase GPx. AOE: antioxidant enzyme.

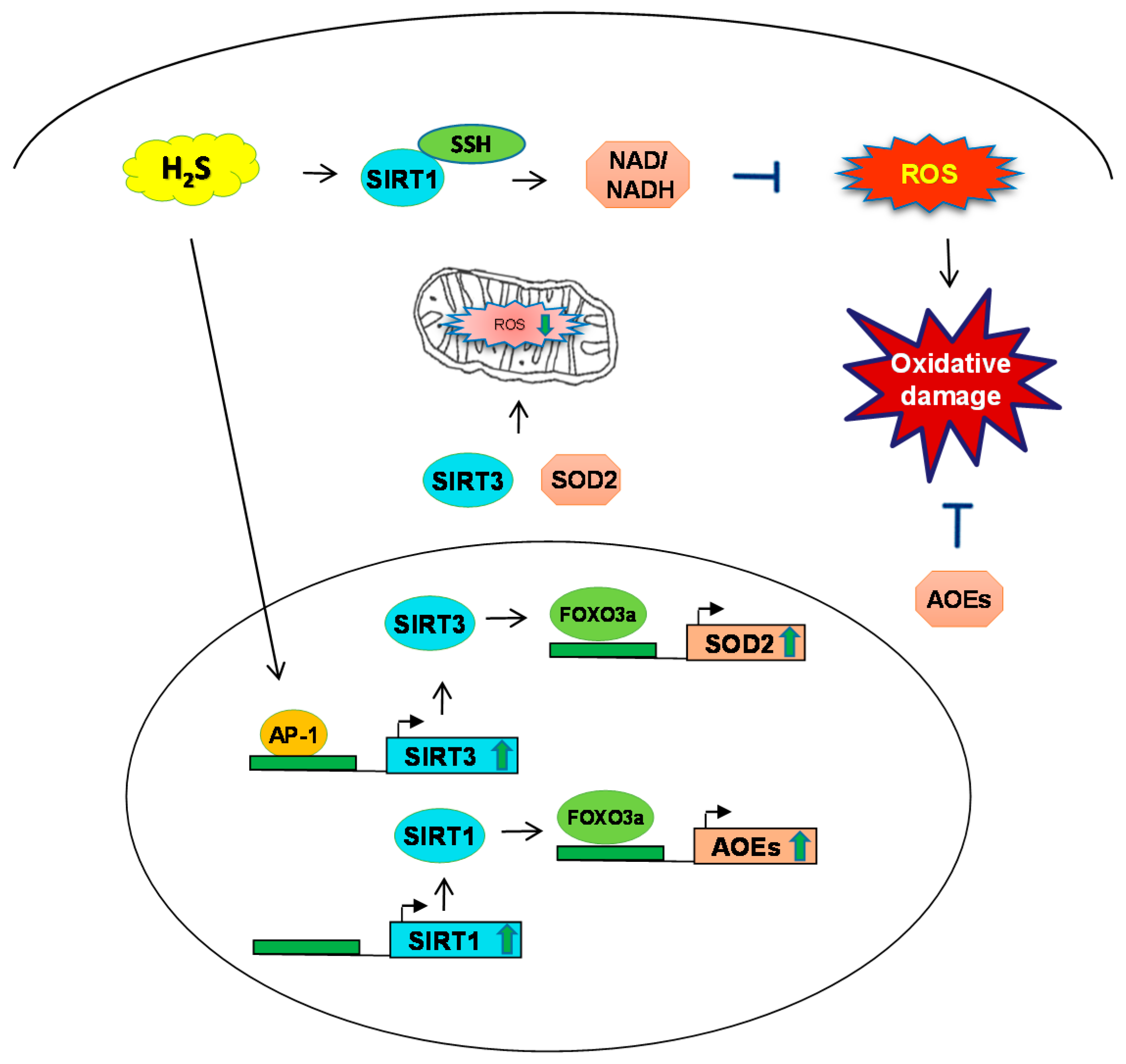
Figure 3.
Schematic of H2S mechanism and sirtuins SIRT-1, SIRT-3 during oxidative stress. H2S induces SIRT1 to regulate the levels of nicotinamide adenine dinucleotide and nicotinamide adenine dinucleotide phosphate NAD/NADH to prevent ROS generation. SIRT-3 induces the expression of transcription factor FOXO3 and consequent ROS production. Additionally, H2S has been shown to induce SOD2 through SIRT3 in mitochondria and regulate oxidative stress. SSH: S-sulfhydration; AP-1: activator protein-1.
Figure 3.
Schematic of H2S mechanism and sirtuins SIRT-1, SIRT-3 during oxidative stress. H2S induces SIRT1 to regulate the levels of nicotinamide adenine dinucleotide and nicotinamide adenine dinucleotide phosphate NAD/NADH to prevent ROS generation. SIRT-3 induces the expression of transcription factor FOXO3 and consequent ROS production. Additionally, H2S has been shown to induce SOD2 through SIRT3 in mitochondria and regulate oxidative stress. SSH: S-sulfhydration; AP-1: activator protein-1.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Beneficial role of H2S donors in animal models of oxidative stress-dependent diseases.
| Model | Mechanism | H2S donors | Reference |
|---|---|---|---|
| Heart | |||
| (Mouse) | |||
| Ischemic heart disease | NRF2 activation and up-regulation of AOE expression | Na2S | [22] |
| Angiogenesis | Up-regulation of AOE | DATS | [41] |
| Hypertension | Decrease of NADPH-dependent superoxide | NaHS | [42] |
| (Rat) | |||
| Fibrosis | Decrease in ROS generation | NaHS | [38] |
| Myocardial ischemia | Decrease of NADPH-dependent superoxide generation | 4CPI and GYY4137 | [37,39] |
| Myocardial dysfunction | Decrease of cellular oxidative stress | NaHS | [40] |
| Nervous system | |||
| (Mouse) | |||
| Alzheimer’s disease | NRF2 activation | NaHS | [44] |
| Huntington’s disease | Decreased oxidative stress | cysteine | [45] |
| (Rat) | |||
| Parkinson’s disease | Inhibition of NADPH oxidase activity and production of ROS | NaHS | [43] |
| Intestine | |||
| (Rat) | |||
| Gastric ischemia-reperfusion | Up-regulation of SOD and GSH-Px activity | NaHS and GYY4137 | [46] |
| Decrease of free radical production | L-cysteine | [47] | |
| Lungs | |||
| (Rat) | |||
| Ischemia–reperfusion injury | Reduction of lipid peroxidation and up-regulation of catalase, SOD activity | H2S | [48] |
| Pulmonary fibrosis | NRF2 activation and up-regulation of Trx-1 | NaHS | [49] |
| Liver | |||
| (Mouse and Rat) | |||
| Ischemia–reperfusion injury | Reduction of lipid peroxidation and up-regulation of | Na2S | [50] |
| GSH and Trx-1 activity | NaHS | [51] | |
| Aging | |||
| (Mouse) | NRF2 activation, enhanced SIRT1 and decreased ROS | NaHS | [26,55] |
| Kidney | |||
| (Mouse) | |||
| Renal Ischemia | Reduction of ROS, modulation of oxidative stress via NRF2 | NaHS | [54] |
| (Rat) | |||
| Uranium-induced toxicity | NRF2 activation | NaHS | [52] |
| Diabetic nephropathy | [53] |
NRF2: nuclear factor (erythroid-derived 2)-like 2; AOE: antioxidant enzyme; DATS: diallyl trisulfide; NADPH: reduced nicotinamide adenine dinucleotide phosphate; ROS: reactive oxygen species; SOD; superoxide dismutase; GSH-Px: glutathione peroxidase.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox7100129
AMA Style
Corsello T, Komaravelli N, Casola A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants. 2018; 7(10):129. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox7100129
Chicago/Turabian StyleCorsello, Tiziana, Narayana Komaravelli, and Antonella Casola. 2018. "Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance" Antioxidants 7, no. 10: 129. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox7100129
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.