Cyanidin Chloride Induces Apoptosis by Inhibiting NF-κB Signaling through Activation of Nrf2 in Colorectal Cancer Cells


Abstract
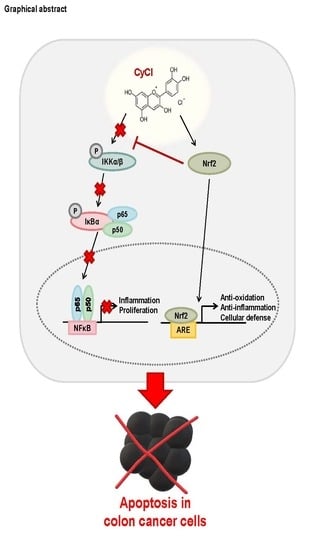
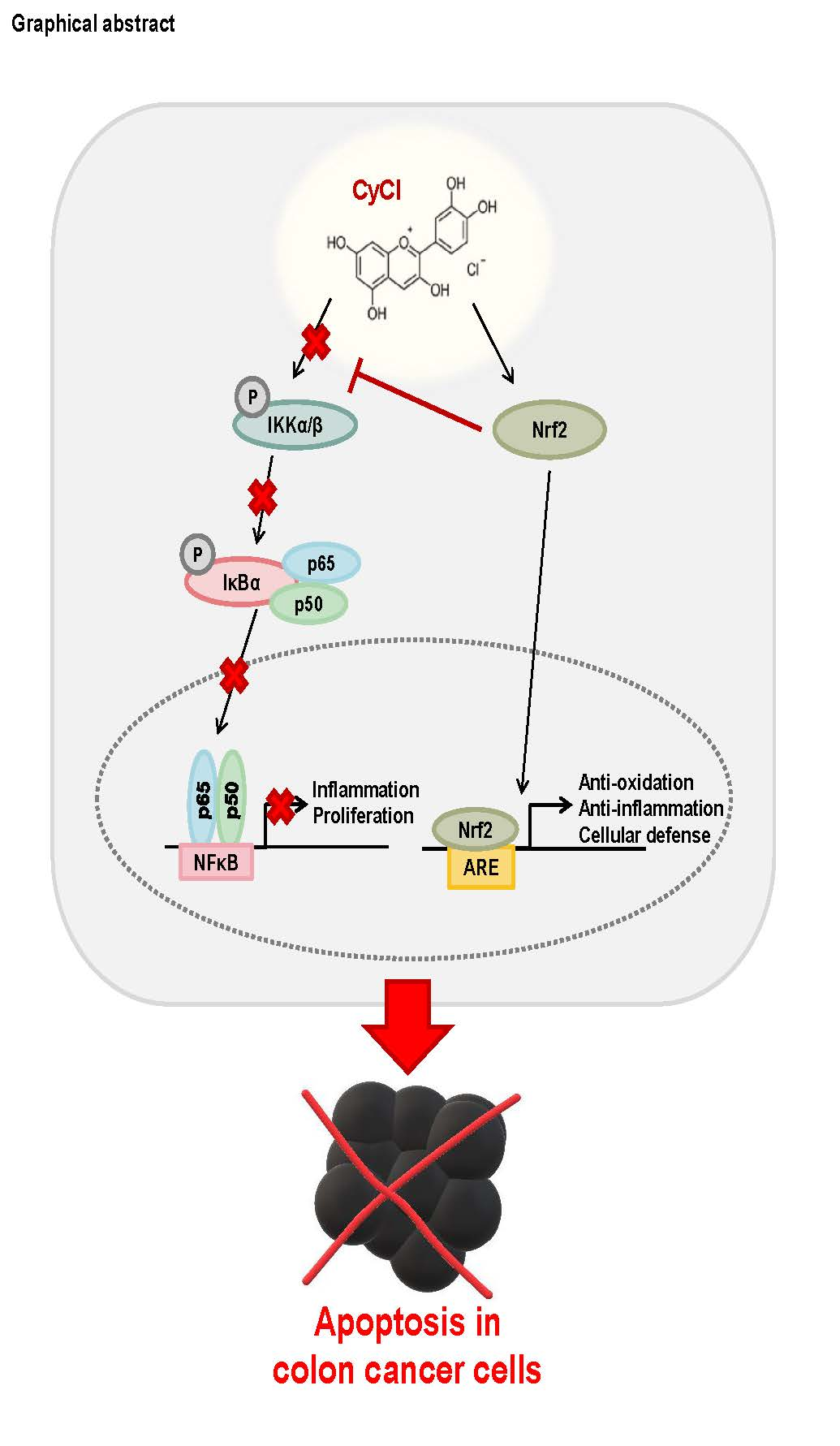
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cell Viability Assay
2.2. Flow Cytometry
2.3. MTT Assay
2.4. IKKβ Kinase Activity Measurement
2.5. Luciferase Assay
2.6. 2,2-Diphenyl-1-Picrylhydrazyl (DPPH) Free Radical Scavenging Assay
2.7. Focus-Forming Assay
2.8. Isolation of Nuclear and Cytoplasmic Fractions
2.9. Western Blot Analysis
2.10. RNA Preparation and Gene Expression Analysis
2.11. Small Interfering RNA (siRNA)
2.12. Statistical Analysis
3. Results
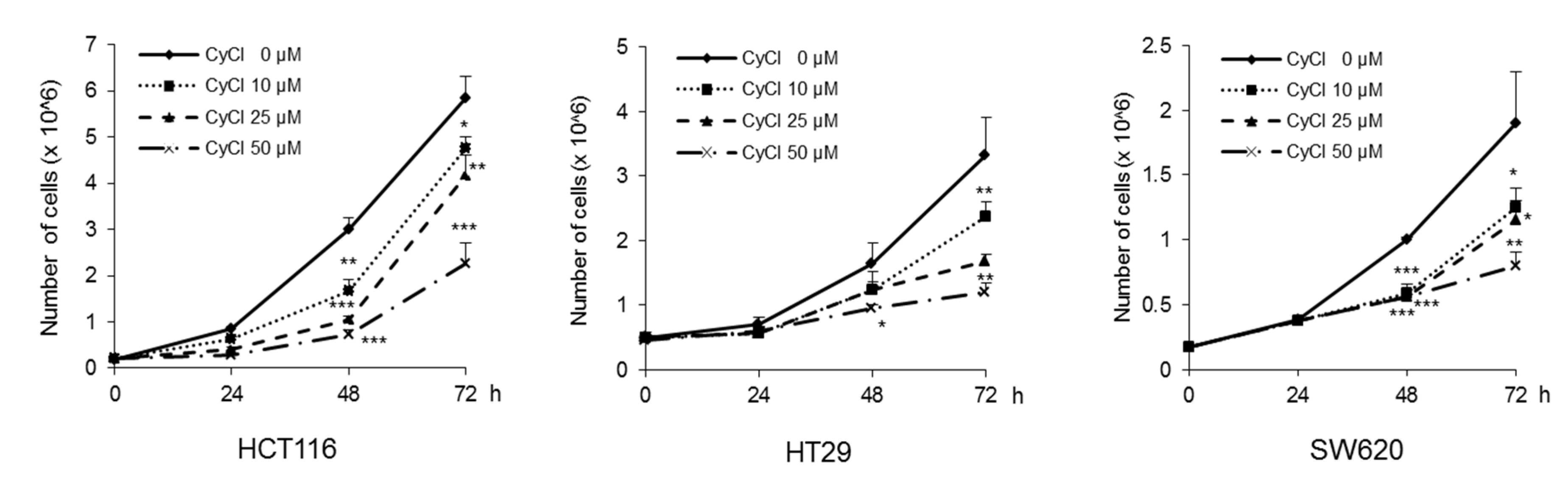
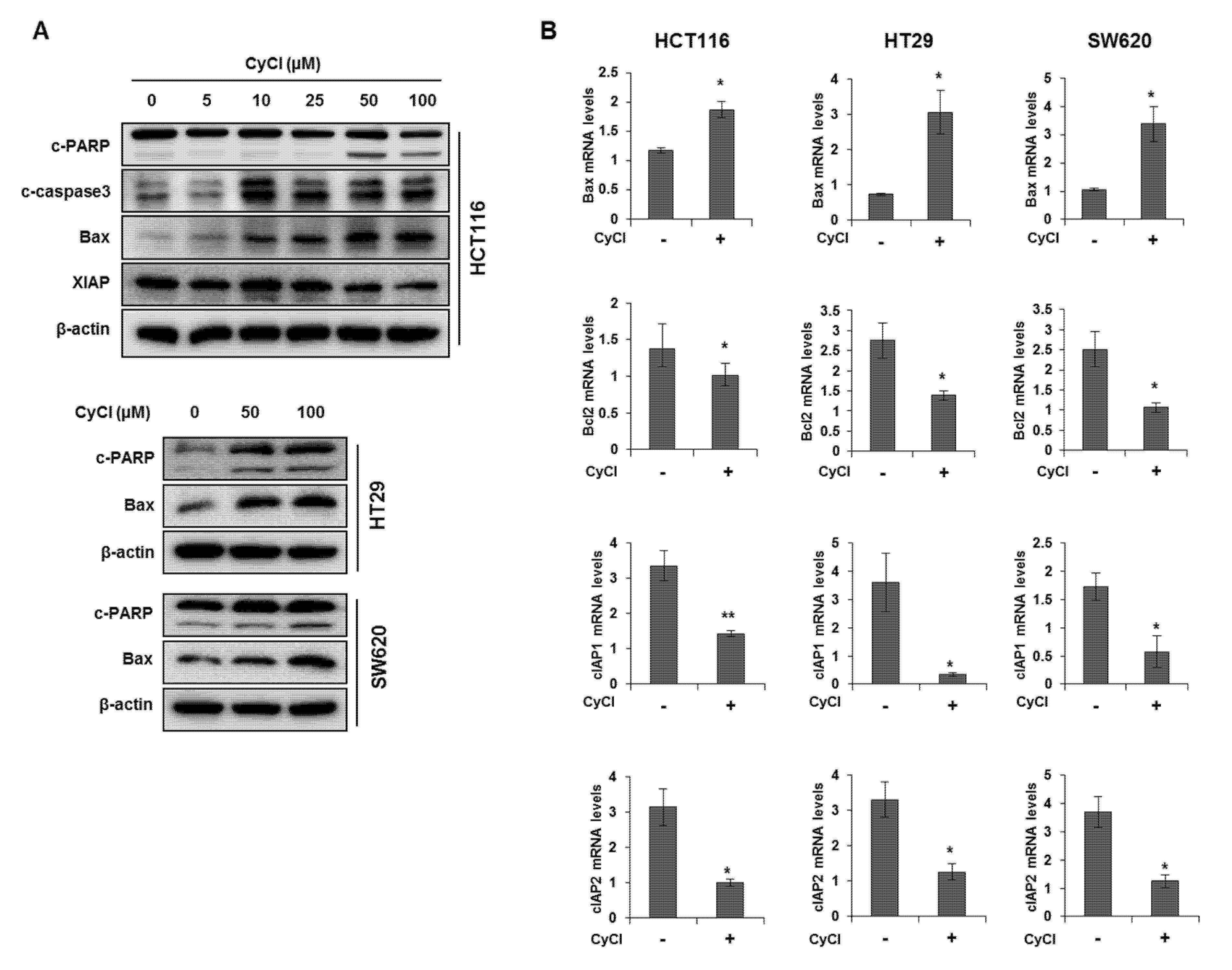
3.1. CyCl Inhibits Cell Proliferation and Induces Apoptosis in Colon Cancer Cells
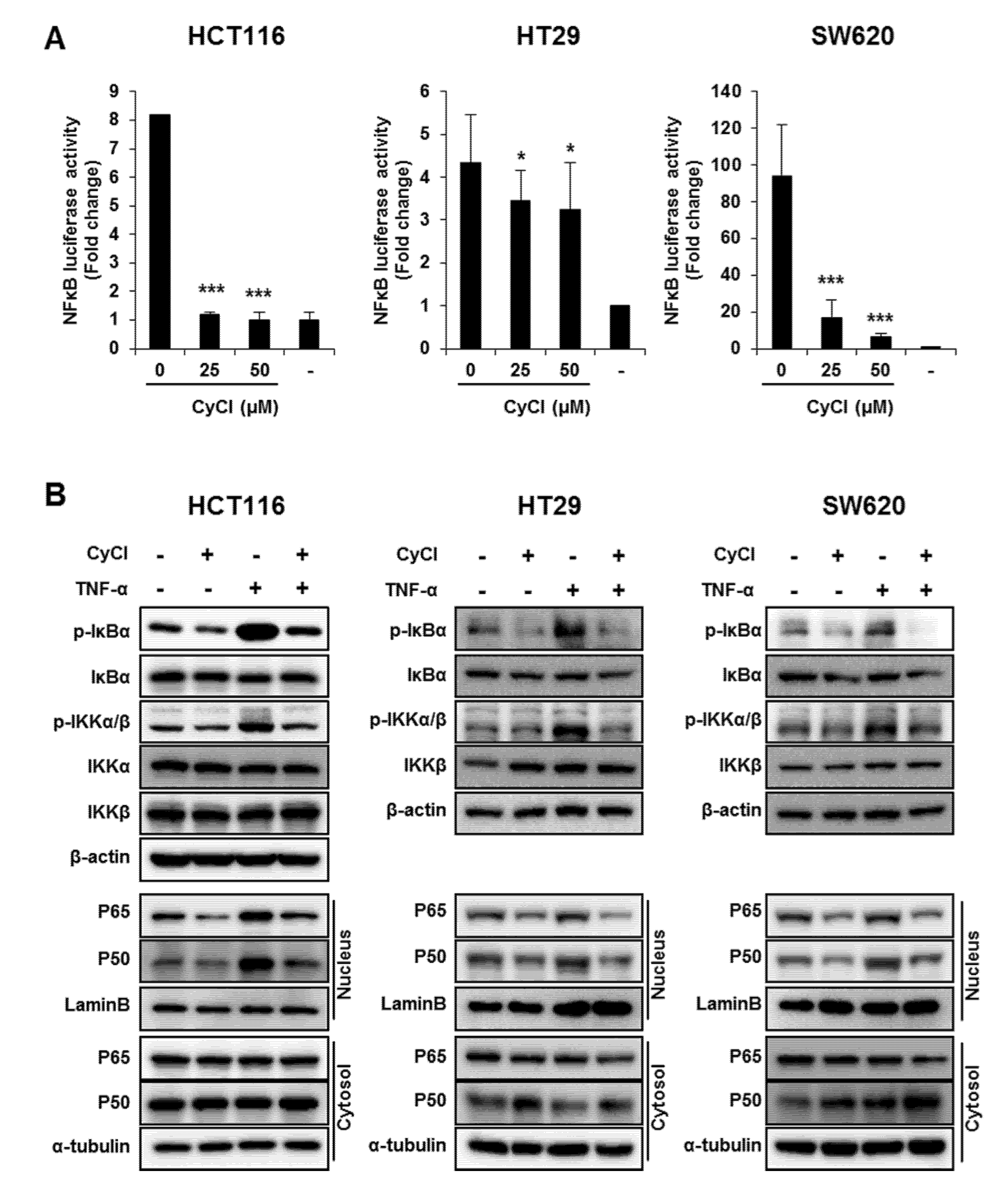
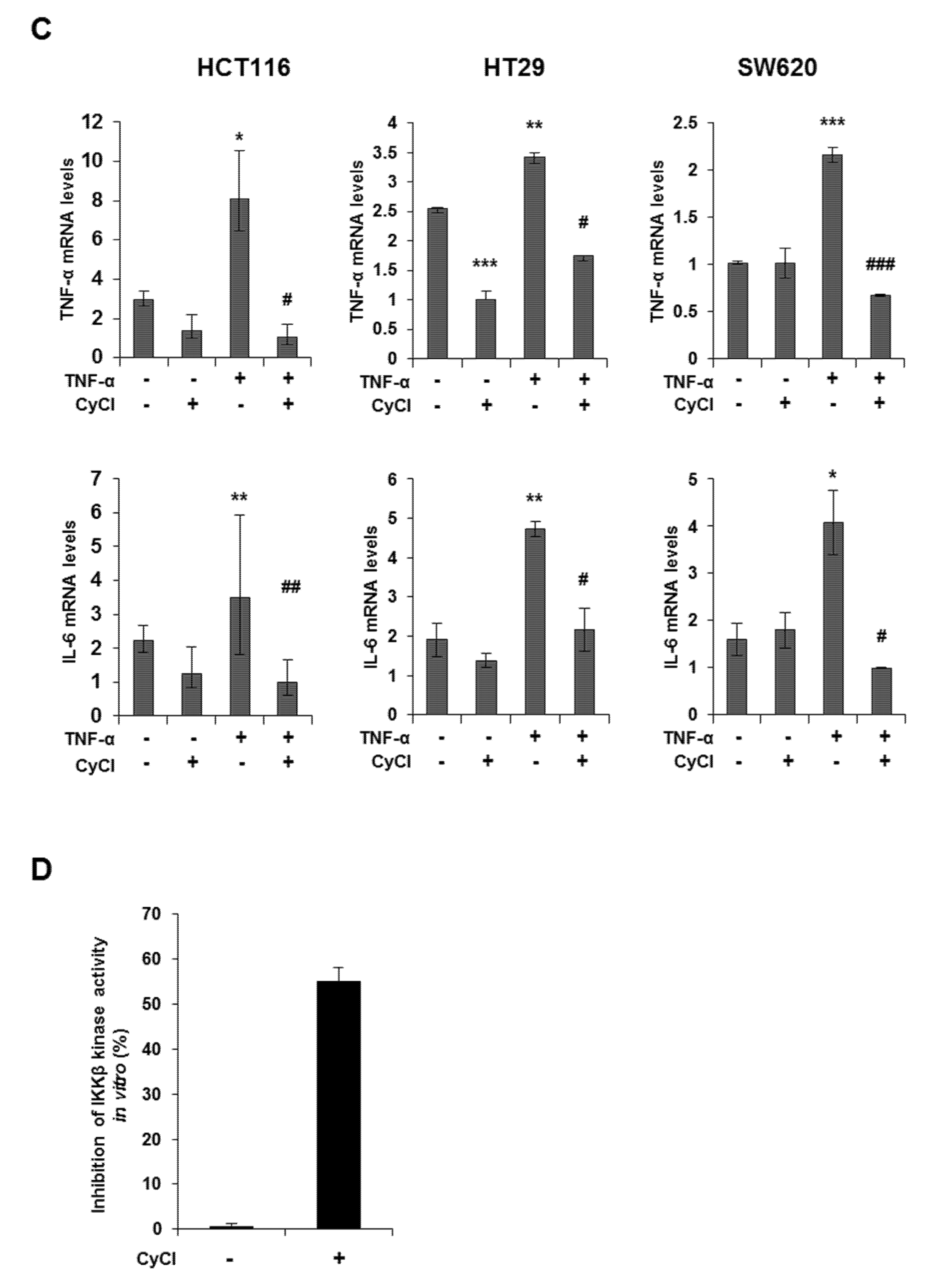
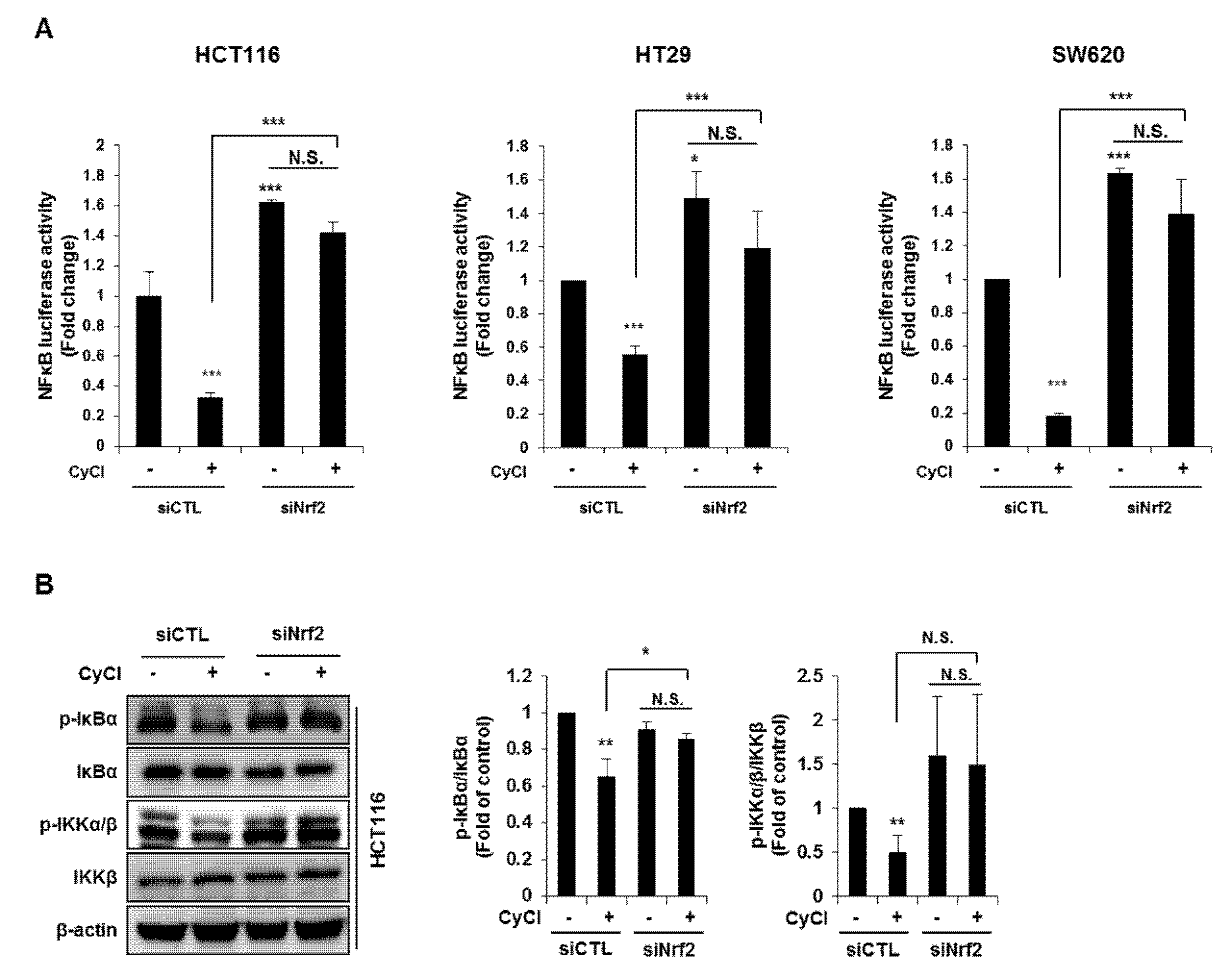
3.2. CyCl Suppresses the NF-κB Signaling Pathway in Colon Cancer Cells
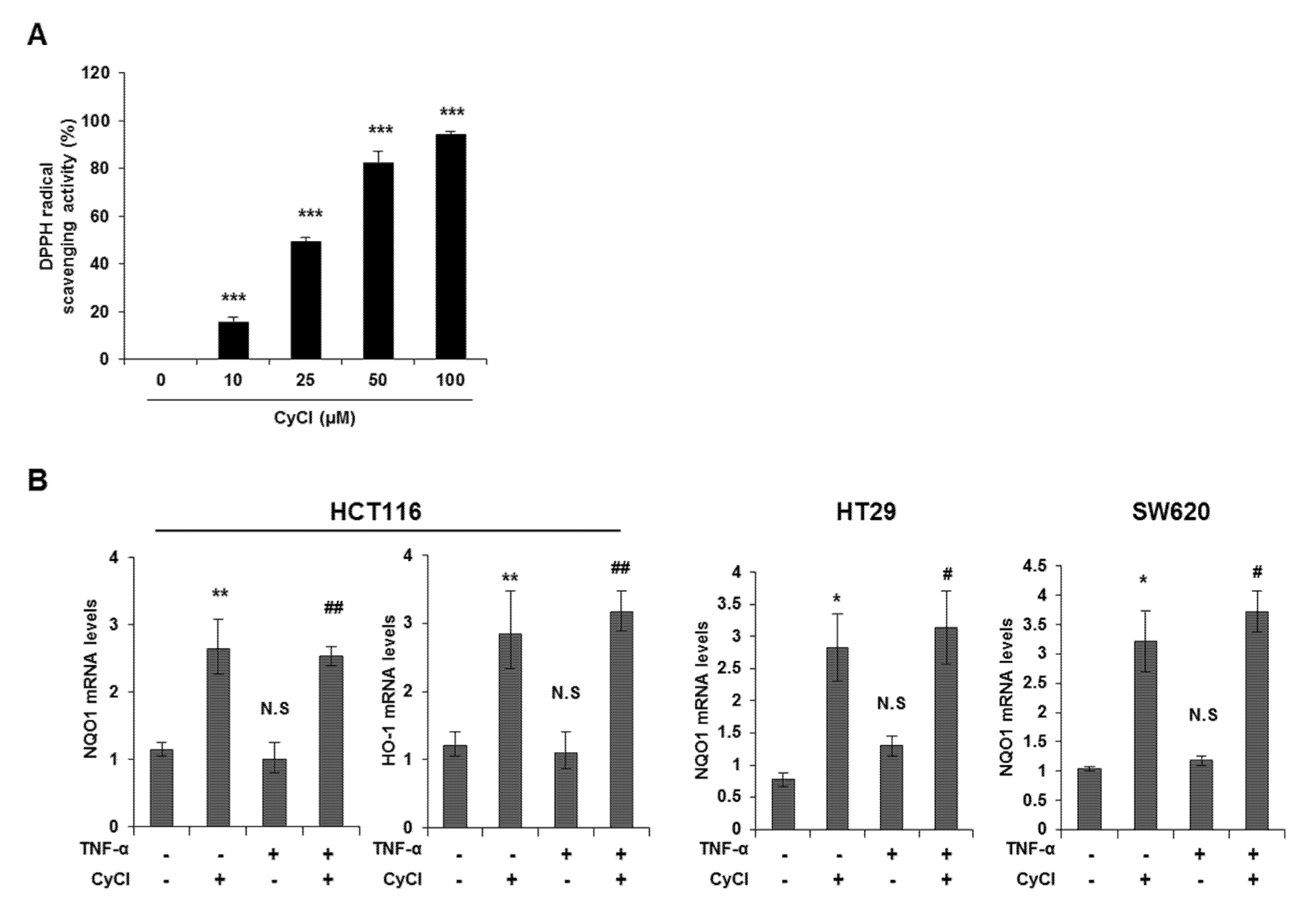
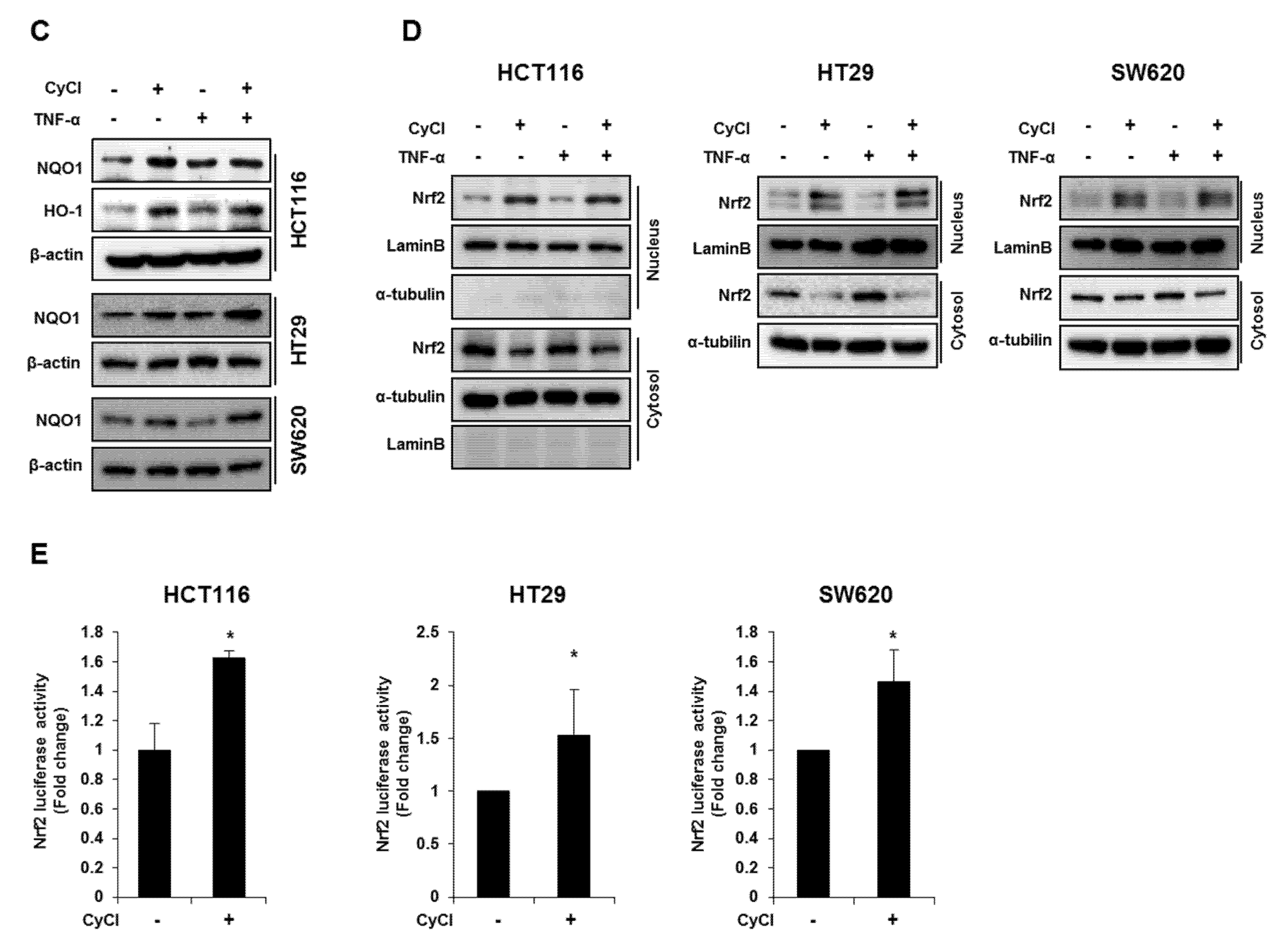
3.3. CyCl Induces Translocation of Nrf2 into the Nucleus and Activates Antioxidant Enzymes
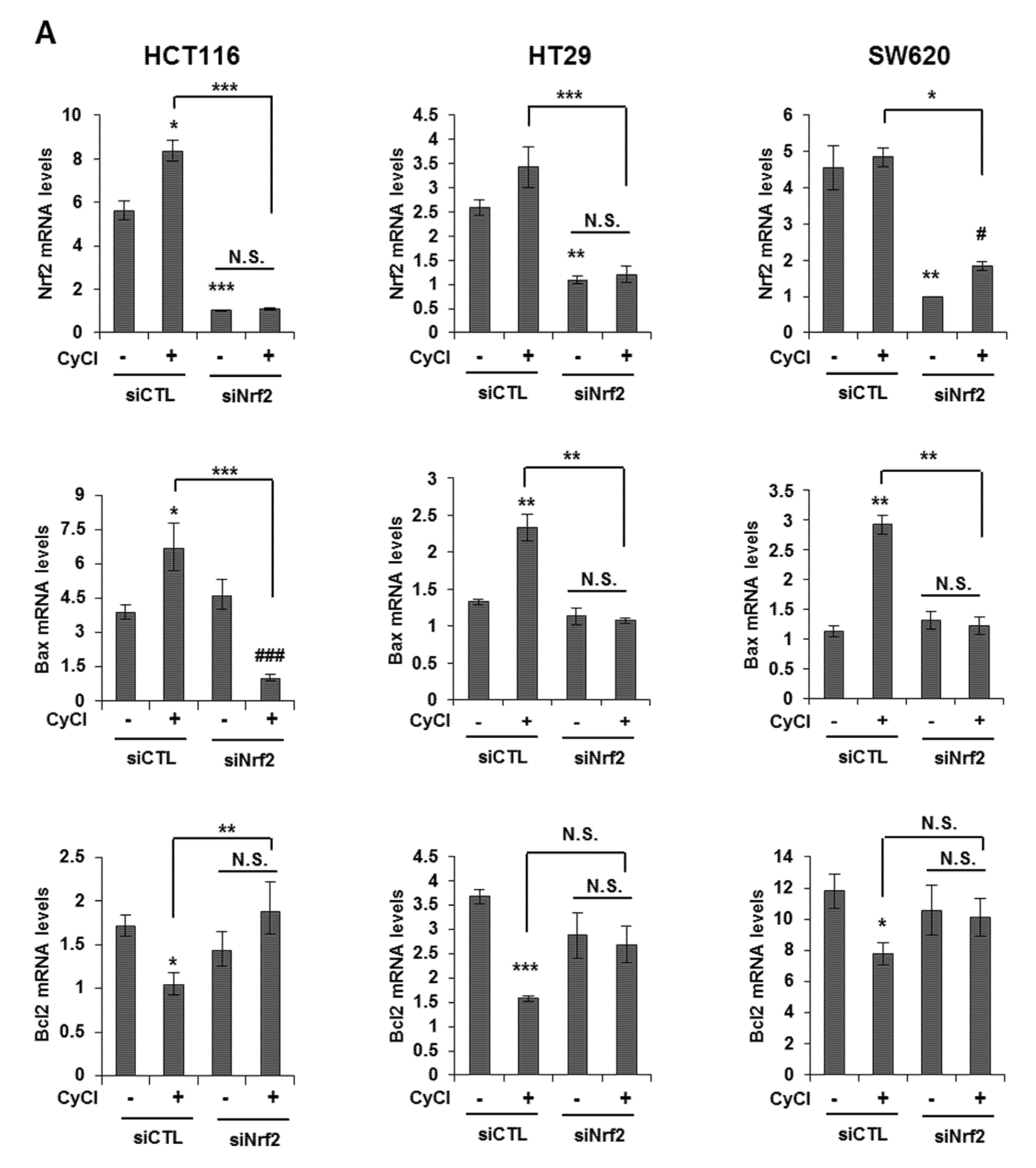
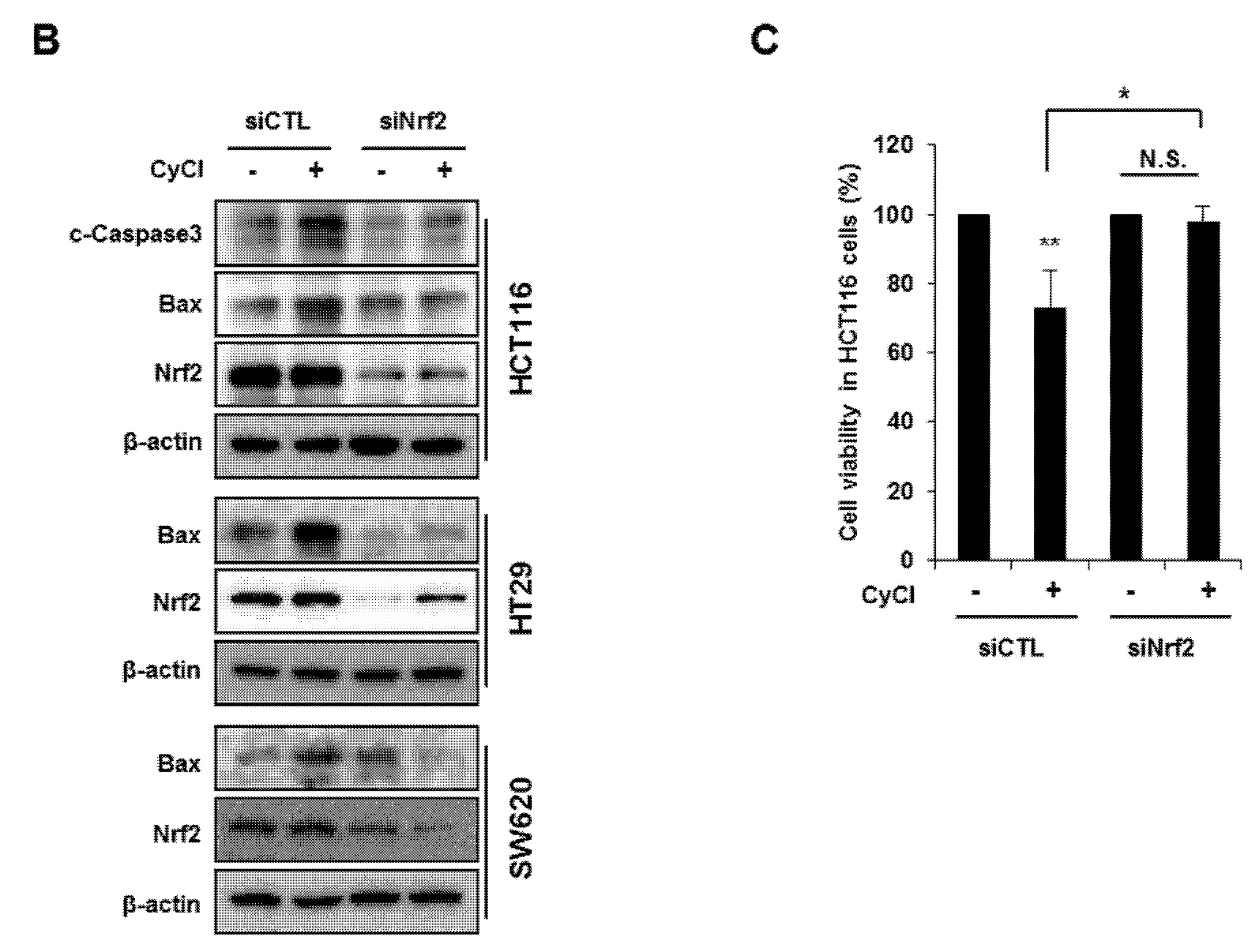
3.4. Nrf2 Activation is Crucial for Apoptosis Induced by NF-κB Suppression
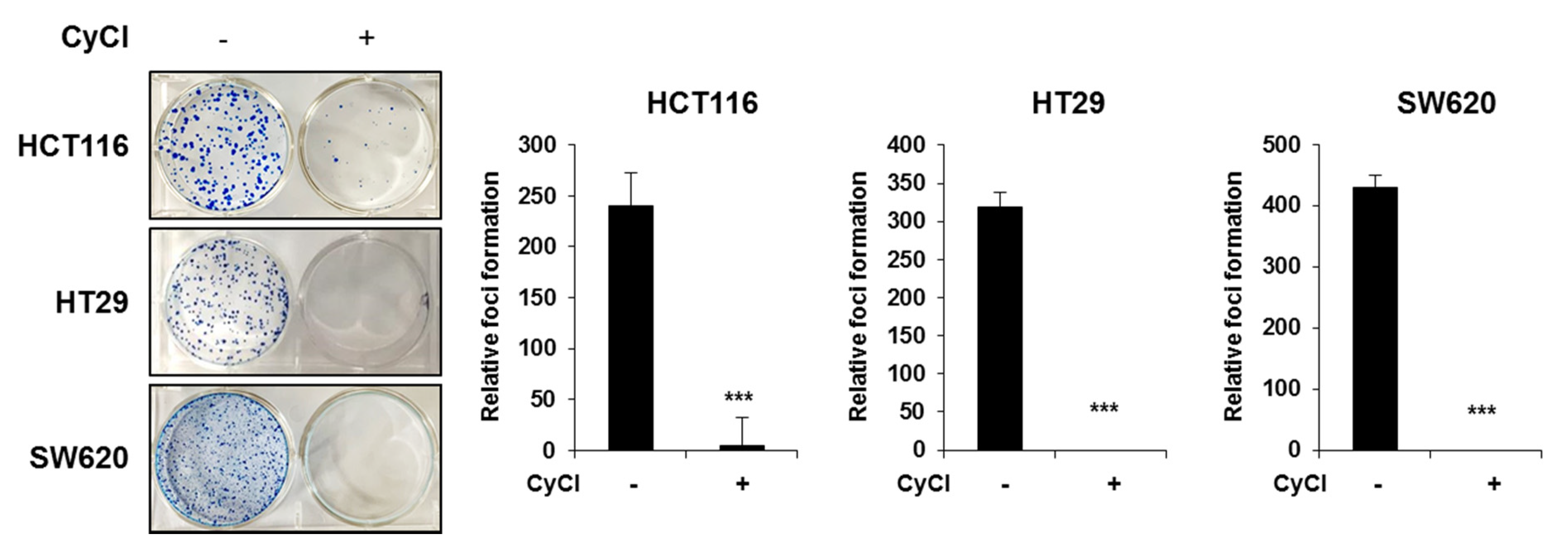
3.5. CyCl Inhibits Colon Cancer Foci Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Prev. Biomark. 2016, 25, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Triantafillidis, J.K.; Nasioulas, G.; Kosmidis, P.A. Colorectal cancer and inflammatory bowel disease: Epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009, 29, 2727–2737. [Google Scholar]
- Martin, M.U.; Wesche, H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim. Biophys. Acta 2002, 1592, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glei, M.; Latunde-Dada, G.O.; Klinder, A.; Becker, T.W.; Hermann, U.; Voigt, K.; Pool-Zobel, B.L. Iron-overload induces oxidative DNA damage in the human colon carcinoma cell line HT29 clone 19A. Mutat. Res. 2002, 519, 151–161. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Shah, S.; MacEwan, D.J. TNF mediates the sustained activation of Nrf2 in human monocytes. J. Immunol. 2011, 187, 702–707. [Google Scholar] [CrossRef] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Surh, Y.J. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005, 224, 171–184. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef]
- Aboonabi, A.; Singh, I. Chemopreventive role of anthocyanins in atherosclerosis via activation of Nrf2-ARE as an indicator and modulator of redox. Biomed. Pharmacother. 2015, 72, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Buya, M.; Qin, W.; Sun, C.; Cai, H.; Xie, Q.; Xu, B.; Wu, Y. Anthocyanins from Chinese bayberry extract activate transcription factor Nrf2 in beta cells and negatively regulate oxidative stress-induced autophagy. J. Agric. Food Chem. 2013, 61, 8765–8772. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Giusti, M.M. Anthocyanins: Natural colorants with health-promoting properties. Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Gismondi, A.; Di Marco, G.; Canuti, L.; Canini, A. Antiradical activity of phenolic metabolites extracted from grapes of white and red Vitis vinifera L. cultivars. VITIS-J. Grapevine Res. 2017, 56, 19–26. [Google Scholar]
- Wallace, T.C.; Giusti, M.M. Anthocyanins. Adv. Nutr. 2015, 6, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.M.; Chia, L.S.; Goh, N.K.; Chia, T.F.; Brouillard, R. Analysis and biological activities of anthocyanins. Phytochemistry 2003, 64, 923–933. [Google Scholar] [CrossRef]
- Galvano, F.; La Fauci, L.; Lazzarino, G.; Fogliano, V.; Ritieni, A.; Ciappellano, S.; Battistini, N.C.; Tavazzi, B.; Galvano, G. Cyanidins: Metabolism and biological properties. J. Nutr. Biochem. 2004, 15, 2–11. [Google Scholar] [CrossRef]
- Renis, M.; Calandra, L.; Scifo, C.; Tomasello, B.; Cardile, V.; Vanella, L.; Bei, R.; La Fauci, L.; Galvano, F. Response of cell cycle/stress-related protein expression and DNA damage upon treatment of CaCo2 cells with anthocyanins. Br. J. Nutr. 2008, 100, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Shi, Y.; Ren, Y.; Wu, H.; Yao, F.; Wei, J.; Wu, M.; Hou, Y.; Duan, H. Anthocyanins inhibit high-glucose-induced cholesterol accumulation and inflammation by activating LXRα pathway in HK-2 cells. Drug Des. Dev. Ther. 2015, 9, 5099. [Google Scholar]
- Sun, G.Y.; Chen, Z.; Jasmer, K.J.; Chuang, D.Y.; Gu, Z.; Hannink, M.; Simonyi, A. Quercetin Attenuates Inflammatory Responses in BV-2 Microglial Cells: Role of MAPKs on the Nrf2 Pathway and Induction of Heme Oxygenase-1. PLoS ONE 2015, 10, e0141509. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Han, Y.M.; Kangwan, N.; Lee, S.Y.; Jung, M.K.; Kim, E.H.; Hahm, K.B. S-allyl cysteine alleviates nonsteroidal anti-inflammatory drug-induced gastric mucosal damages by increasing cyclooxygenase-2 inhibition, heme oxygenase-1 induction, and histone deacetylation inhibition. J. Gastroenterol. Hepatol. 2014, 29, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Bae, J.S.; Hahm, K.B.; Cha, J.Y. Endogenously synthesized n-3 polyunsaturated fatty acids in fat-1 mice ameliorate high-fat diet-induced non-alcoholic fatty liver disease. Biochem. Pharmacol. 2012, 84, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.M.; Hahm, K.B.; Park, J.M.; Hong, S.P.; Kim, E.H. Paradoxically augmented anti-tumorigenic action of proton pump inhibitor and GastrininAPCMin/+ intestinal polyposis model. Neoplasia 2014, 16, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einbond, L.S.; Reynertson, K.A.; Luo, X.-D.; Basile, M.J.; Kennelly, E.J. Anthocyanin antioxidants from edible fruits. Food Chem. 2004, 84, 23–28. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.L.; Lin, L.; Lou, P.J.; Chang, T.C.; Young, T.H. Detection of cell carcinogenic transformation by a quadruplex DNA binding fluorescent probe. PLoS ONE 2014, 9, e86143. [Google Scholar] [CrossRef]
- Van Deventer, S.J. Tumour necrosis factor and Crohn’s disease. Gut 1997, 40, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Kronke, M.; Schutze, S.; Scheurich, P.; Pfizenmaier, K. TNF signal transduction and TNF-responsive genes. Immunol. Ser. 1992, 56, 189–216. [Google Scholar]
- Yan, B.; Wang, H.; Rabbani, Z.N.; Zhao, Y.; Li, W.; Yuan, Y.; Li, F.; Dewhirst, M.W.; Li, C.-Y. Tumor necrosis factor-α is a potent endogenous mutagen that promotes cellular transformation. Cancer Res. 2006, 66, 11565–11570. [Google Scholar] [CrossRef] [Green Version]
- Beutler, B. TNF, immunity and inflammatory disease: Lessons of the past decade. J. Investig. Med. 1995, 43, 227–235. [Google Scholar] [PubMed]
- Barkett, M.; Gilmore, T.D. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6910–6924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugina, D.; Hanganu, D.; Diaconeasa, Z.; Tabaran, F.; Coman, C.; Leopold, L.; Bunea, A.; Pintea, A. Antiproliferative and apoptotic potential of cyanidin-based anthocyanins on melanoma cells. Int. J. Mol. Sci. 2017, 18, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, D.; Speciale, A.; Cristani, M.; Fratantonio, D.; Molonia, M.S.; Ranaldi, G.; Saija, A.; Cimino, F. Cyanidin-3-O-glucoside inhibits NF-kB signalling in intestinal epithelial cells exposed to TNF-alpha and exerts protective effects via Nrf2 pathway activation. Toxicol. Lett. 2016, 264, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S. NF-κB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Valassiadou, K.E.; Stefanaki, K.; Tzardi, M.; Datseris, G.; Georgoulias, V.; Melissas, J.; Tsiftsis, D.D.; Delides, G.; Kanavaros, P. Immunohistochemical expression of p53, bcl-2, mdm2 and waf1/p21 proteins in colorectal adenocarcinomas. Anticancer Res. 1997, 17, 2571–2576. [Google Scholar] [PubMed]
- Cherbonnel-Lasserre, C.; Dosanjh, M.K. Suppression of apoptosis by overexpression of Bcl-2 or Bcl-xL promotes survival and mutagenesis after oxidative damage. Biochimie 1997, 79, 613–617. [Google Scholar] [CrossRef]
- Bonnotte, B.; Favre, N.; Moutet, M.; Fromentin, A.; Solary, E.; Martin, M.; Martin, F. Bcl-2-mediated inhibition of apoptosis prevents immunogenicity and restores tumorigenicity of spontaneously regressive tumors. J. Immunol. 1998, 161, 1433–1438. [Google Scholar]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Tong, L.; Chuang, C.C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef]
- Perse, M. Oxidative stress in the pathogenesis of colorectal cancer: Cause or consequence? Biomed. Res. Int. 2013, 2013, 725710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N. Nrf2: Friend or foe for chemoprevention? Carcinogenesis 2010, 31, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Khor, T.O.; Huang, M.-T.; Prawan, A.; Liu, Y.; Hao, X.; Yu, S.; Cheung, W.K.L.; Chan, J.Y.; Reddy, B.S.; Yang, C.S. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 2008, 1, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, L.; Kwong, M.; Hou, S.; Lee, C.; Chan, J.Y. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 2003, 278, 48021–48029. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Han, X.-D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The absence of Nrf2 enhances NF-kappaB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediat. Inflamm. 2012, 2012, 217580. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Dilution | Product No. | Species of Origin and Supplier |
|---|---|---|---|
| β-actin | 1:4000 | sc-47778 | Mouse polyclonal, Santa Cruz Biotechnology, Inc. |
| α-tubulin | 1:1000 | sc-5286 | Rabbit polyclonal, Santa Cruz Biotechnology, Inc. |
| LaminB | 1:1000 | sc-6217 | Goat polyclonal, Santa Cruz Biotechnology, Inc. |
| PARP | 1:1000 | 9542 | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| c-caspase3 | 1:1000 | 9661L | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| Bax | 1:1000 | sc-7480 | Mouse polyclonal, Santa Cruz Biotechnology, Inc. |
| XIAP | 1:1000 | sc-11426 | Rabbit polyclonal, Santa Cruz Biotechnology, Inc. |
| p-IκBα | 1:1000 | 2856 | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| IκBα | 1:1000 | 9242 | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| p-IKKα/β | 1:1000 | 2697 | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| IKKα | 1:1000 | 2682 | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| IKKβ | 1:1000 | 2678 | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| P65 | 1:1000 | 8242 | Rabbit polyclonal, Cell Signaling Technology, Inc. |
| P50 | 1:1000 | sc-1190 | Goat polyclonal, Santa Cruz Biotechnology, Inc. |
| NQO1 | 1:1000 | ab34173 | Mouse polyclonal, Abcam, Inc. |
| HO-1 | 1:2000 | ADI-SPA-895 | Rabbit polyclonal, Enzo, Inc. |
| Nrf2 | 1:1000 | ab62352 | Rabbit polyclonal, Abcam, Inc. |
| Accession Number | Gene | Forward | Reverse | bp | |
|---|---|---|---|---|---|
| qRT PCR | NM138763 | Bax | CCT GTG CAC CAA GGT GCC GGA ACT | CCA CCC TGG TCT TGG ATC CAG CCC | 99 |
| NM000657 | Bcl2 | TTG TGG CCT TCT TTG AGT TCG GTG | GGT GCC GGT TCA GGT ACT CAG TCA | 114 | |
| NM001166 | cIAP1 | GCTTCCGTGTTTGCTGCG | ACTGCAGGGGGACAAAATAGG | 78 | |
| NM001165 | cIAP2 | TCTGGGCAGCAGGTTTACAA | CCCGAGATTAGACTAAGTCCCTT | 127 | |
| NM020529 | NFκbia | TGT GCT TCG AGT GAC TGA CC | TCA CCC CAC ATC ACT GAA CG | 81 | |
| NM032721 | NFκbid | ACC CCA TCT GTG AAT GAG GC | GTC CAG AAC CCA CAG TCT CC | 73 | |
| NM000584 | IL-8 | TCC TTG TTC CAC TGT GCC TTG | TGC TTC CAC ATG TCC TCA CAA | 101 | |
| NM000600 | IL-6 | AGG GCT CTT CGG CAA ATG TA | GAA GGA ATG CCC ATT AAC AAC AA | 62 | |
| NM000594 | TNF-α | GAA AGT CCC GTG GAA ATC CC | CTG CGC AGT CTG CTT TGC | 70 | |
| NM006164 | Nrf2 | AGGTTGCCCACATTCCCAAA | AGTGACTGAAACGTAGCCGA | 118 | |
| NM000903 | NQO1 | AAG AGC ACT GAT CGT ACT GG | CTT CAG TTT ACC TGT GAT GTC C | 172 | |
| NM002133 | HO-1 | CAA CAT CCA GCT CTT TGA GG | AGA AAG CTG AGT GTA AGG AC | 204 | |
| NR003286 | 18s rRNA | GCAATTATTCCCCATGAACG | GGCCTCACTAAACCATCCAA | 111 | |
| RT PCR | NM001166 | c-IAP1 | GCCTTTCTCCAAACCCTCTT | ATTCGAGCTGCATGTGTCTG | 355 |
| NM182962 | c-IAP2 | GCAGGCAAAGGCTAGTCATC | GAATGCTGCACAGAGACCAA | 370 | |
| NM006164 | Nrf2 | CGG TATGCA ACAGGACAT TG | ACTGGTTGGGGTCTTCTGTG | 280 | |
| NR003286 | 18s rRNA | CCCAACTTCTTAGAGGGACAAGT | TAGTCAAGTTCGACCGTCTTCTC | 350 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-Y.; Yun, S.-M.; Song, M.-Y.; Jung, K.; Kim, E.-H. Cyanidin Chloride Induces Apoptosis by Inhibiting NF-κB Signaling through Activation of Nrf2 in Colorectal Cancer Cells. Antioxidants 2020, 9, 285. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9040285
Lee D-Y, Yun S-M, Song M-Y, Jung K, Kim E-H. Cyanidin Chloride Induces Apoptosis by Inhibiting NF-κB Signaling through Activation of Nrf2 in Colorectal Cancer Cells. Antioxidants. 2020; 9(4):285. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9040285
Chicago/Turabian StyleLee, Da-Young, Sun-Mi Yun, Moon-Young Song, Kiwon Jung, and Eun-Hee Kim. 2020. "Cyanidin Chloride Induces Apoptosis by Inhibiting NF-κB Signaling through Activation of Nrf2 in Colorectal Cancer Cells" Antioxidants 9, no. 4: 285. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9040285