Oxidative Stress and Photodynamic Therapy of Skin Cancers: Mechanisms, Challenges and Promising Developments

 , ,
, ,
Abstract
:1. Introduction
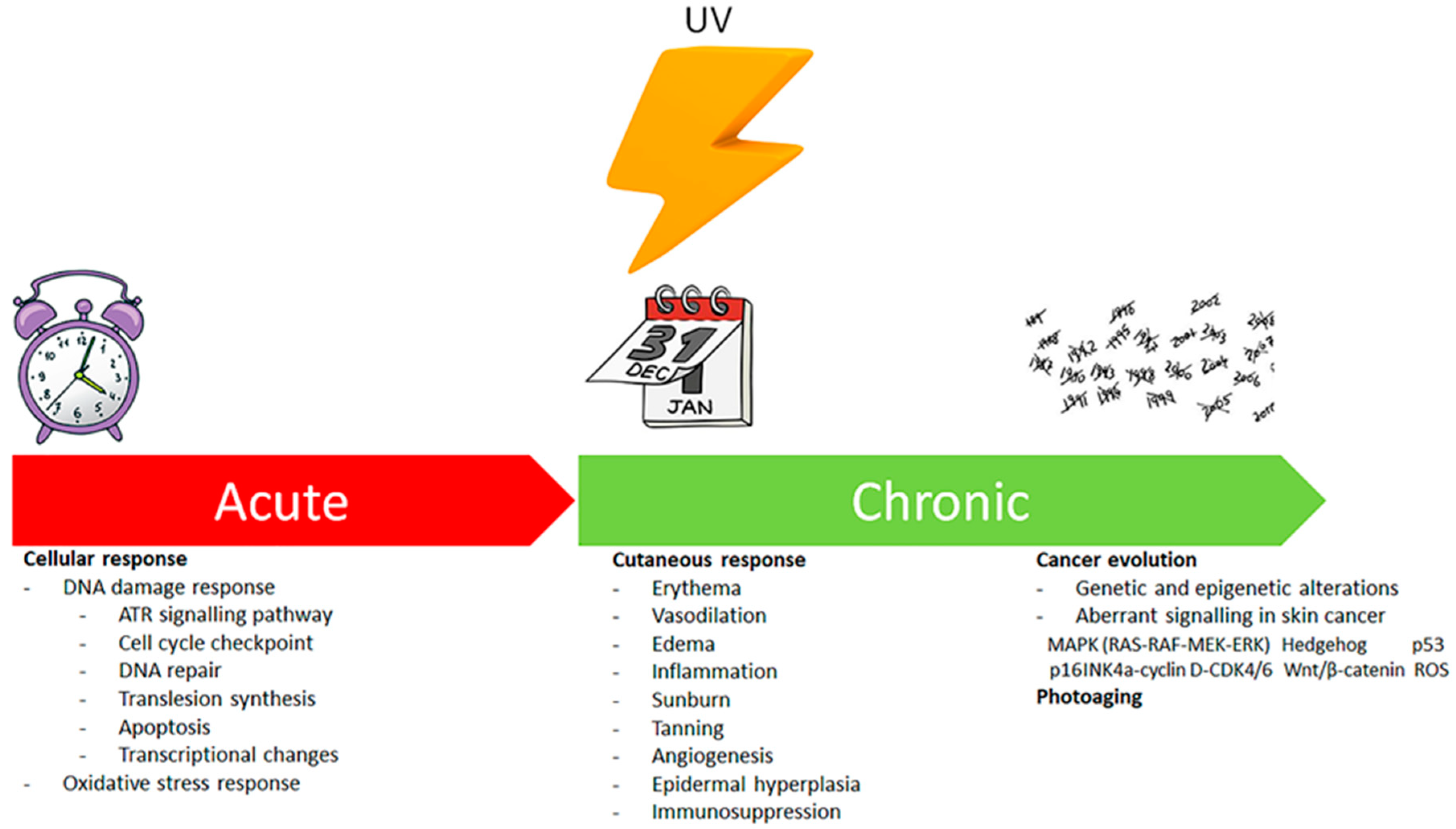
Ultraviolet Radiation, Oxidative Stress and Skin Cancer
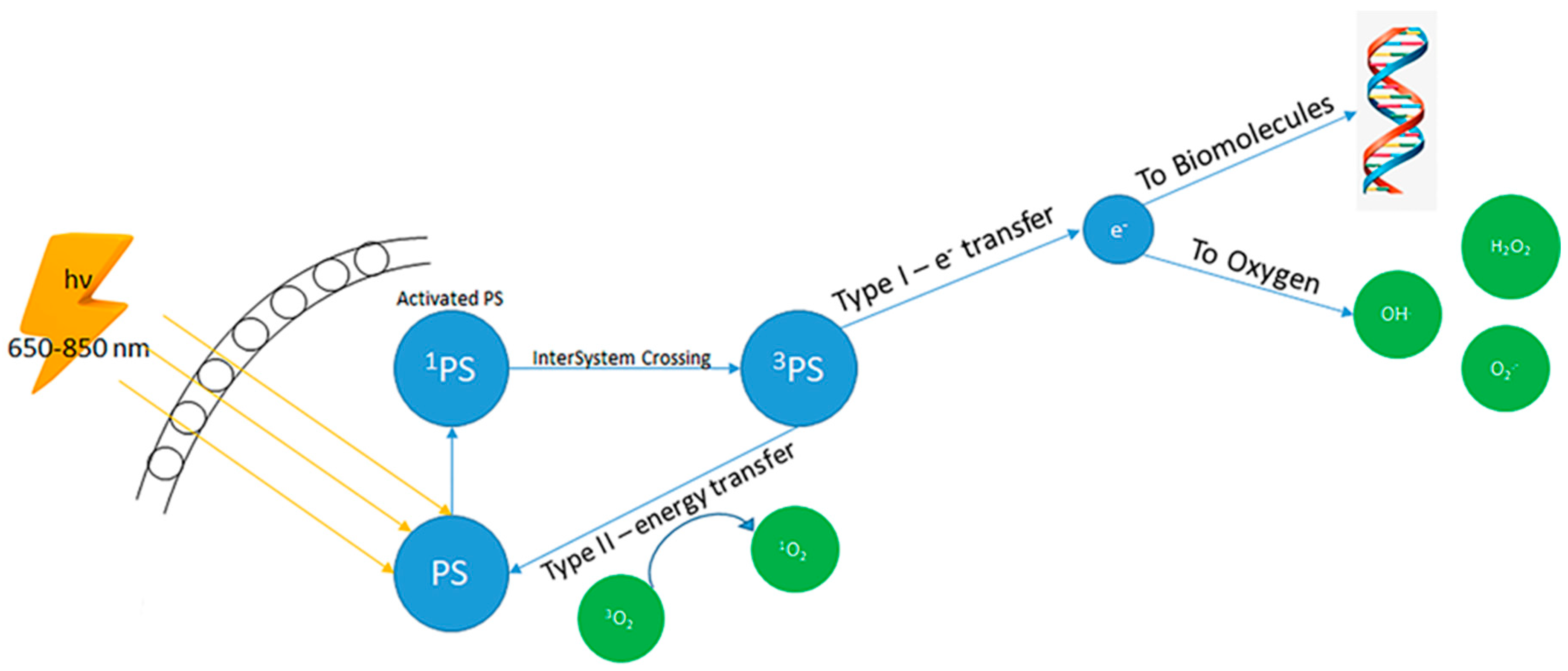
2. Photodynamic Therapy of Skin Cancer and Oxidative Stress
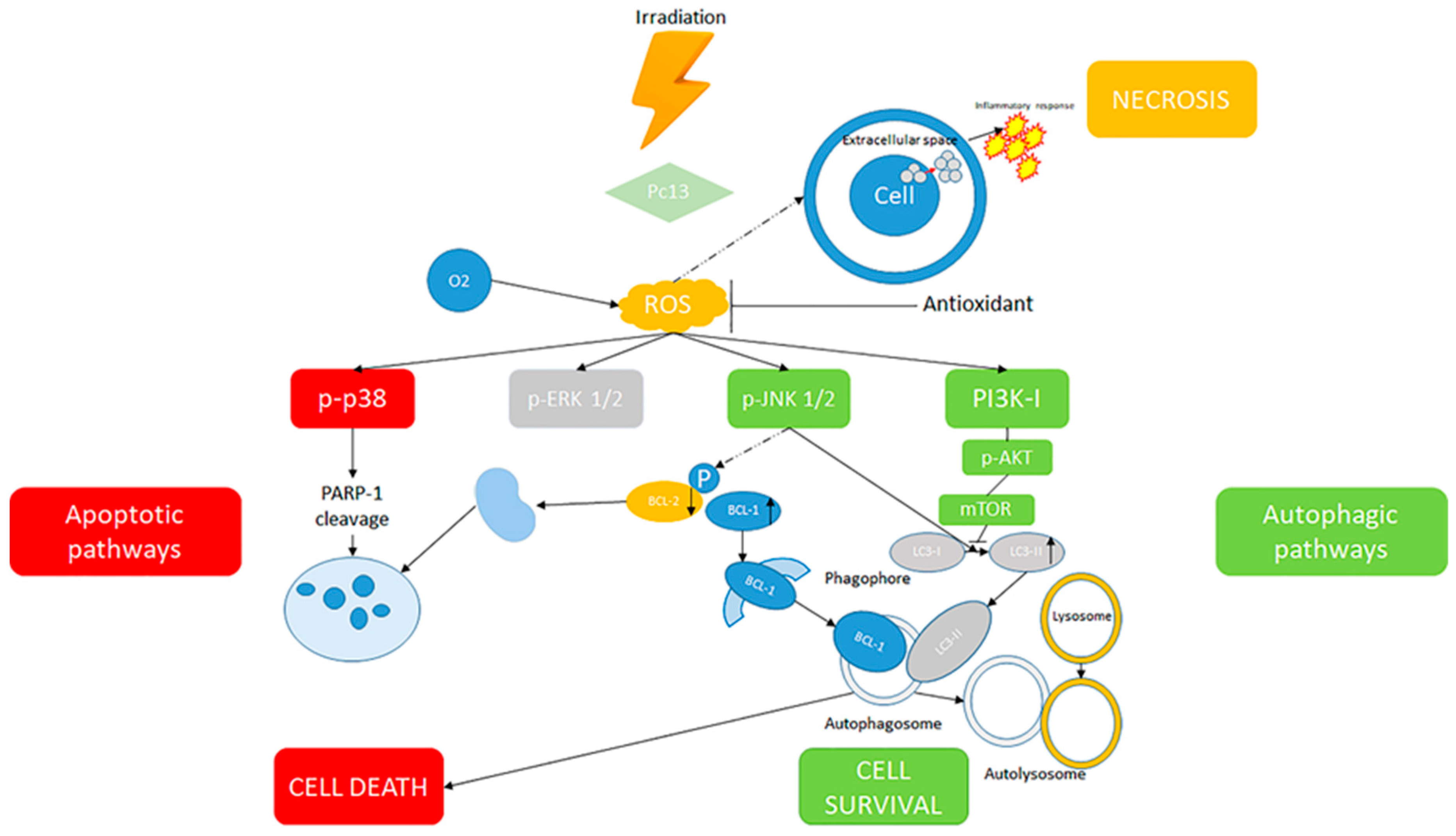
2.1. PDT, Oxidative Stress and Cellular Death
2.2. Molecular Mechanisms of Cell Death
2.3. Additional Mechanisms of Action of PDT
2.4. PSs, Lipid Peroxidation and PDT
3. Novel Photosensitisers
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Plaetzer, K.; Krammer, B.; Berlanda, J.; Berr, F.; Kiesslich, T. Photophysics and photochemistry of photodynamic therapy: Fundamental aspects. Lasers Med. Sci. 2009, 24, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Dorval, J.; Hontela, A. Role of glutathione redox cycle and catalase in defense against oxidative stress induced by endosulfan in adrenocortical cells of rainbow trout (Oncorhynchus mykiss). Toxicol. Appl. Pharmacol. 2003, 192, 191–200. [Google Scholar] [CrossRef]
- Kern, J.C.; Kehrer, J.P. Free radicals and apoptosis: Relationships with glutathione, thioredoxin and the bcl family of proteins. Front. Biosci. 2005, 10, 1727–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.I.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.O.; Schieke, S.M.; Sies, H.; Holbrook, N.J. Peroxynitrite activates the phosphoinositide 3-kinase/Akt pathway in human skin primary fibroblasts. Biochem. J. 2000, 352, 219–225. [Google Scholar] [CrossRef]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–164124. [Google Scholar] [CrossRef] [Green Version]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Suozzi, K.; Turban, J.; Girardi, M. Cutaneous Photoprotection: A Review of the Current Status and Evolving Strategies. Yale J. Biol. Med. 2020, 93, 55–67. [Google Scholar]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef]
- Tessman, I.; Liu, S.K.; Kennedy, M.A. Mechanism of SOS mutagenesis of UV-irradiated DNA: Mostly error-free processing of deaminated cytosine. Proc. Natl. Acad. Sci. USA 1992, 89, 1159–1163. [Google Scholar] [CrossRef] [Green Version]
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.H.; Halaban, R.; Douki, T.; Brash, D.E. Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- de Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Adv. Exp. Med. Biol. 2017, 996, 15–23. [Google Scholar] [PubMed]
- Liebel, F.; Kaur, S.; Ruvolo, E.; Kollias, N.; Southall, M.-D. Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J. Investig. Dermatol. 2012, 132, 1901–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venza, M.; Visalli, M.; Beninati, C.; De Gaetano, G.V.; Teti, D.; Venza, I. Cellular Mechanisms of Oxidative Stress and Action in Melanoma. Oxid. Med. Cell. Longev. 2015, 2015, 481782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wondrak, G.T.; Roberts, M.J.; Cervantes-Laurean, D.; Jacobson, M.K.; Jacobson, E.L. Proteins of the extracellular matrix are sensitizers of photo-oxidative stress in human skin cells. J. Investig. Dermatol. 2003, 121, 578–586. [Google Scholar] [CrossRef] [Green Version]
- Bergendi, L.; Benes, L.; Durackova, Z.; Ferencik, M. Chemistry, physiology and pathology of free radicals. Life Sci. 1999, 65, 1865–1874. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324 (Pt 1), 1–18. [Google Scholar]
- Hattori, Y.; Nishigori, C.; Tanaka, T.; Uchida, K.; Nikaido, O.; Osawa, H.; Hiai, T.; Imamura, S.; Toyokuni, S. 8-hydroxy-2’-deoxyguanosine is increased in epidermal cells of hairless mice after chronic ultraviolet B exposure. J. Investig. Dermatol. 1996, 107, 733–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peus, D.; Vasa, R.A.; Beyerle, A.; Meves, A.; Krautmacher, C.; Pittelkow, M.R. UVB activates ERK1/2 and p38 signaling pathways via reactive oxygen species in cultured keratinocytes. J. Investig. Dermatol. 1999, 112, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Knebel, A.; Rahmsdorf, H.J.; Ullrich, A.; Herrlich, P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996, 15, 5314–5325. [Google Scholar] [CrossRef]
- Ikehata, H.; Yamamoto, M. Roles of the KEAP1-NRF2 system in mammalian skin exposed to UV radiation. Toxicol. Appl. Pharmacol. 2018, 360, 69–77. [Google Scholar] [CrossRef]
- Cannavò, S.P.; Tonacci, A.; Bertino, L.; Casciaro, M.; Borgia, F.; Gangemi, S. The role of oxidative stress in the biology of melanoma: A systematic review. Pathol. Res. Pract. 2019, 215, 21–28. [Google Scholar]
- Vaccaro, M.; Bagnato, G.; Cristani, M.; Borgia, F.; Spatari, G.; Tigano, V.; Saja, A.; Guarneri, F.; Cannavò, S.P.; Gangemi, S. Oxidation products are increased in patients affected by non-segmental generalized vitiligo. Arch. Dermatol. Res. 2017, 309, 485–490. [Google Scholar] [CrossRef]
- Imbesi, S.; Musolino, C.; Allegra, A.; Saija, A.; Morabito, F.; Calapai, G.; Gangemi, S. Oxidative stress in oncohematologic diseases: An update. Expert Rev. Hematol. 2013, 6, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, S.; Allegra, A.; Alonci, A.; Cristani, M.; Russo, S.; Speciale, A.; Penna, G.; Spatari, G.; Cannavò, A.; Bellomo, G.; et al. Increase of novel biomarkers for oxidative stress in patients with plasma cell disorders and in multiple myeloma patients with bone lesions. Inflamm. Res. 2012, 61, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Musolino, C.; Allegra, A.; Saija, A.; Alonci, A.; Russo, S.; Spatari, G.; Penna, G.; Gerace, D.; Cristani, M.; David, A.; et al. Changes in advanced oxidation protein products, advanced glycation end products, and s-nitrosylated proteins, in patients affected by polycythemia vera and essential thrombocythemia. Clin. Biochem. 2012, 45, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, S.; Allegra, A.; Aguennouz, M.; Alonci, A.; Speciale, A.; Cannavò, A.; Cristani, M.; Russo, S.; Spatari, G.; Alibrandi, A.; et al. Relationship between advanced oxidation protein products, advanced glycation end products, and S-nitrosylated proteins with biological risk and MDR-1 polymorphisms in patients affected by B-chronic lymphocytic leukemia. Cancer Investig. 2012, 30, 20–26. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Alonci, A.; Saija, A.; Russo, S.; Cannavò, A.; Cristani, M.; Centorrino, R.; Saitta, S.; Alibrandi, A.; et al. Carbonyl group serum levels are associated with CD38 expression in patients with B chronic lymphocytic leukemia. Clin. Biochem. 2011, 44, 1487–1490. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.; Johnson, A.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Bickers, D.R.; Athar, M. Oxidative stress in the pathogenesis of skin disease. J. Investig. Dermatol. 2006, 126, 2565–2575. [Google Scholar] [CrossRef] [Green Version]
- Kessel, D.; Oleinick, N.L. Cell Death Pathways Associated with Photodynamic Therapy: An Update. Photochem. Photobiol. 2018, 94, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Roguin, L.P.; Chiarante, N.; García Vior, M.C.; Marino, J. Zinc (II) phthalocyanines as photosensitizers for antitumor photodynamic therapy. Int. J. Biochem. Cell. Biol. 2019, 114, 105575. [Google Scholar] [CrossRef]
- Kwa, R.E.; Campana, K.; Moy, R.L. Biology of cutaneous squamous cell carcinoma. J. Am. Acad. Dermatol. 1992, 26, 1–26. [Google Scholar] [CrossRef]
- Albert, M.R.; Ostheimer, K.G. The evolution of current medical and popular attitudes toward ultraviolet light exposure: Part 1. J. Am. Acad. Dermatol. 2002, 47, 930–937. [Google Scholar] [CrossRef] [Green Version]
- Raab, O. Uber die Wirkung fluorischeider Stoffe auf Infusora. Z Biol. 1900, 39, 524–526. [Google Scholar]
- von Tappeiner, H.; Jesionek, A. Therapeutische Versuche mit fluorescierenden Stoffen. Munch Med Wochenschr 1903, 47, 2042–2044. [Google Scholar]
- Roelandts, R. The history of phototherapy: Something new under the sun? J. Am. Acad. Dermatol. 2002, 46, 926–930. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Grindey, G.B.; Fiel, R.; Weishaupt, K.R.; Boyle, D.G. Photoradiation therapy. II. Cure of animal tumors with hematoporphyrin and light. J. Natl Cancer Inst. 1975, 55, 115121. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Kaufman, J.E.; Goldfarb, A.; Weishaupt, K.R.; Boyle, D.; Mittleman, A. Photoradiation therapy for the treatment of malignant tumors. Cancer Res. 1978, 38, 26282635. [Google Scholar]
- de Albuquerque, I.O.; Nunes, J.; Figueiro Longo, J.P.; Muehlmann, L.A.; Azevedo, R.B. Photodynamic therapy in superficial basal cell carcinoma treatment. Photodiagnosis Photodyn. Ther. 2019, 27, 428–432. [Google Scholar] [CrossRef]
- Tampa, M.; Sarbu, M.I.; Matei, C.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Constantin, C.; Neagu, M.; Georgescu, S.R. Photodynamic therapy: A hot topic in dermato-oncology. Oncol. Lett. 2019, 17, 4085–4093. [Google Scholar] [CrossRef] [Green Version]
- Keyal, U.; Bhatta, A.K.; Zhang, G.; Wang, X.L. Present and future perspectives of photodynamic therapy for cutaneous squamous cell carcinoma. J. Am. Acad. Dermatol. 2019, 80, 765773. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Yanovsky, R.L.; Bartenstein, D.W.; Rogers, G.S.; Isakoff, S.J.; Chen, S.T. Photodynamic therapy for solid tumors: A review of the literature. Photodermatol. Photoimmunol. Photomed. 2019, 35, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Ion, R.M. Derivative UV-Vis spectrophotometry for porphyrins interactions in photodynamic therapy. Anal. Lett. 2010, 43, 1277–1286. [Google Scholar] [CrossRef]
- Garland, M.J.; Cassidy, C.M.; Woolfson, D.; Donnelly, R.F. Designing photosensitizers for photodynamic therapy: Strategies, challenges and promising developments. Future Med. Chem. 2009, 1, 667–691. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumor immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, B.K.; Kricker, A.; English, D.R. Sun exposure and skin cancer. Australas. J. Dermatol. 1997, 38 (Suppl. 1), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.; Holick, M.F. Sunlight and Vitamin D: A global perspective for health. Dermatoendocrinology 2013, 5, 51–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clydesdale, G.J.; Dandie, G.W.; Muller, H.K. Ultraviolet light induced injury: Immunological and inflammatory effects. Immunol. Cell. Biol. 2001, 79, 547–568. [Google Scholar] [CrossRef]
- Warren, J.B. Nitric oxide and human skin blood flow responses to acetylcholine and ultraviolet light. FASEB J. 1994, 8, 247–251. [Google Scholar] [CrossRef]
- Johnson, K.E.; Wulff, B.C.; Oberyszyn, T.M.; Wilgus, T.A. Ultraviolet light exposure stimulates HMGB1 release by keratinocytes. Arch. Dermatol. Res. 2013, 305, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Homig-Holzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.T.; Fisher, D.E. MITF and UV responses in skin: From pigmentation to addiction. Pigment Cell Melanoma Res. 2019, 32, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Abaseri, T.B.; Putta, S.; Hansen, L.A. Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis 2006, 27, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Imokawa, G.; Ishida, K. Biological mechanisms underlying the ultraviolet radiation induced formation of skin wrinkling and sagging I: Reduced skin elasticity, highly associated with enhanced dermal elastase activity, triggers wrinkling and sagging. Int. J. Mol. Sci. 2015, 16, 7753–7775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, J.R.; Brieva, J.C. Images in clinical medicine. Unilateral dermatoheliosis. N. Engl. J. Med. 2012, 366, e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, T. Mechanisms of UV-induced immunosuppression. Keio J. Med. 2005, 54, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chapman, N.M.; Zhang, B.; Li, M.; Fan, M.; Laribee, R.N.; Zaidi, M.R.; Pfeffer, L.M.; Chi, J.; Wu, Z.H. Upregulation of PD-L1 via HMGB1-Activated IRF3 and NF-kappaB Contributes to UV Radiation-Induced Immune Suppression. Cancer Res. 2019, 79, 2909–2922. [Google Scholar] [CrossRef]
- Fell, G.L.; Robinson, K.C.; Mao, J.; Woolf, C.J.; Fisher, D.E. Skin beta-endorphin mediates addiction to UV light. Cell 2014, 157, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Moan, J.; Berg, K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem. Photobiol. 1991, 53, 549–553. [Google Scholar] [CrossRef]
- Chen, B.; Pogue, B.W.; Hoopes, P.J.; Hasan, T. Vascular and cellular targeting for photodynamic therapy. Crit. Rev. Eukaryot. Gene Expr. 2006, 16, 279–305. [Google Scholar] [CrossRef]
- Davids, L.M.; Kleemann, B. Combating melanoma: The use of photodynamic therapy as a novel, adjuvant therapeutic tool. Cancer Treat. Rev. 2011, 37, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Golab, J.; Nowis, D.; Skrzycki, M.; Czeczot, H.; Baranczyk-Kuzma, A.; Wilczynski, G.M.; Makowski, M.; Mroz, P.; Kozar, K.; Kaminski, R.; et al. Antitumor effects of photodynamic therapy are potentiated by 2methoxyestradiol. Asuperoxide dismutase inhibitor. J. Biol. Chem. 2003, 278, 407–414. [Google Scholar] [PubMed] [Green Version]
- Clichici, S.; Filip, A.; Daicoviciu, D.; Ion, R.M.; Mocan, T.; Tatomir, C.; Rogojan, L.; Olteanu, D.; Muresan, A. The dynamics of reactive oxygen species in photodynamic therapy with tetra sulfophenyl-porphyrin. Acta Physiol. Hung. 2010, 97, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Daicoviciu, D.; Filip, A.; Ion, R.M.; Clichici, S.; Decea, N.; Muresan, A. Oxidative photodamage induced by photodynamic therapy with methoxyphenyl porphyrin derivatives in tumour-bearing rats. Folia Biol Prague 2010, 56, 12–19. [Google Scholar]
- Woods, J.A.; Traynor, N.J.; Brancaleon, L.; Moseley, H. The effect of photofrin on DNAstrand breaks and base oxidation in HaCaT keratinocytes: A comet assay study. Photochem. Photobiol. 2004, 79, 105–113. [Google Scholar] [CrossRef]
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid Redox Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef] [Green Version]
- Morton, C.A.; Szeimies, R.M.; Sidoroff, A.; Braathen, L.R. European guidelines for topical photodynamic therapy part 1: Treatment delivery and current indications—Actinic keratoses, Bowen’s disease, basal cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 536–544. [Google Scholar] [CrossRef]
- Kiesslich, T.; Plaetzer, K.; Oberdanner, C.B.; Berlanda, J.; Obermair, F.J.; Krammer, B. Differential effects of glucose deprivation on the cellular sensitivity towards photodynamic treatment-based production of reactive oxygen species and apoptosis-induction. FEBS Lett. 2005, 579, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Barberi-Heyob, M.; Vedrine, P.O.; Merlin, J.L.; Millon, R.; Abecassis, J.; Poupon, M.-F.; Guillemin, F. Wild type p53 gene transfer into mutated p53 HT29 cells improves sensitivity to photodynamic therapy via induction of apoptosis. Int. J. Oncol. 2004, 24, 951–958. [Google Scholar] [CrossRef]
- Gupta, S.; Dwarakanath, B.S.; Muralidhar, K.; Jain, V. Role of apoptosis in photodynamic sensitivity of human tumour cell lines. Ind. J. Exp. Biol. 2003, 41, 33–40. [Google Scholar]
- Lam, M.; Oleinick, N.L.; Nieminen, A.L. Photodynamic therapyi nduced apoptosis in epidermoid carcinoma cells. Reactive oxygen species and mitochondrial inner membrane permeabilization. J. Biol. Chem. 2001, 276, 47379–47386. [Google Scholar] [CrossRef] [Green Version]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar]
- Susan, M.; Baldea, I.; Senila, S.; Macovei, V.; Dreve, S.; Ion, R.M.; Cosgarea, R. Photodamaging effects of porphyrins and chitosan on primary human keratinocytes and carcinoma cell cultures. Int. J. Dermatol. 2011, 50, 280–286. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Carvalho, A.P.; Duarte, C.B. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar] [CrossRef] [Green Version]
- Filip, A.G.; Clichici, S.; Daicoviciu, D.; Ion, R.M.; Tatomir, C.; Rogojan, L.; Opris, I.; Mocan, T.; Olteanu, D.; Muresan, A. Possible in vivo mechanisms involved in photodynamic therapy using tetrapyrrolic macrocycles. Braz. J. Med. Biol. Res. 2011, 44, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.-H. Photodynamic treatment induces an apoptotic pathway involving calcium, nitric oxide, p53, p21-activated kinase 2, and c-Jun N-terminal kinase and inactivates survival signal in human umbilical vein endothelial cells. Int. J. Mol. Sci. 2011, 12, 1041–1059. [Google Scholar] [CrossRef] [Green Version]
- Cory, S.; Huang, D.C.; Adams, J.M. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 85908607. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.E.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef] [Green Version]
- Tong, Z.; Singh, G.; Rainbow, A.J. The role of the p53 tumor suppressor in the response of human cells to photofrin-mediated phphotodynamic therapy. Photochem. Photobiol. 2000, 71, 201–210. [Google Scholar] [CrossRef]
- Davids, L.M.; Kleemann, B.; Kacerovska, D.; Pizinger, K.; Kidson, S.H. Hypericin phototoxicity induces different modes of cell death in melanoma and human skin cells. J. Photochem. Photobiol. 2008, 91, 67–76. [Google Scholar] [CrossRef]
- Wyld, L.; Reed, M.W.; Brown, N.J. Differential cell death response to photodynamic therapy is dependent on dose and cell type. Br. J. Cancer 2001, 84, 1384–1386. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Casares, N.; Pequignot, M.O.; Chaput, N.; Albert, M.L.; Kroemer, G. Immune response against dying tumor cells. Adv. Immunol. 2004, 84, 131–179. [Google Scholar]
- Oble, D.A.; Loewe, R.; Yu, P.; Mihm, M.C., Jr. Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immun. 2009, 9, 3. [Google Scholar]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta 2007, 1776, 86–107. [Google Scholar] [CrossRef]
- Valli, F.; García Vior, M.C.; Roguin, L.P.; Marino, J. Crosstalk between oxidative stress-induced apoptotic and autophagic signaling pathways in Zn(II) phthalocyanine photodynamic therapy of melanoma. Free Radic Biol. Med 2020, S0891-5849(19), 32561–32565. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Lomaestro, B.M.; Malone, M. Glutathione in health and disease: Pharmacotherapeutic issues. Ann. Pharmacother. 1995, 29, 1263–1273. [Google Scholar] [CrossRef]
- Calzavara-Pinton, P.G.; Venturini, M.; Sala, R. Photodynamic therapy: Update 2006. Part 1: Photochemistry and photobiology. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 293–302. [Google Scholar] [CrossRef]
- Champeau, M.; Vignoud, S.; Mortier, L.; Mordon, S. Photodynamic therapy for skin cancer: How to enhance drug penetration? J. Photochem. Photobiol. B 2019, 197, 111544. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Kimura, M.; Nishioka, A.; Kato, H.; Morita, A. Dual wavelength 5aminolevulinic acid photodynamic therapy using a novel flexible light-emitting diode unit. J. Dermatol. Sci. 2019, 93, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanesio, M.E.; Alvarez, M.G.; Yslas, E.I.; Borsarelli, C.D.; Silber, J.J.; Rivarola, V.; Durantini, E.N. Photodynamic studies of metallo 5,10,15,20-tetrakis(4-methoxyphenyl) porphyrin: Photochemical characterization and biological consequences in a human carcinoma cell line. Photochem. Photobiol. 2001, 74, 14–21. [Google Scholar] [CrossRef]
- Alvarez, M.G.; Yslas, E.I.; Rivarola, V.; Mori, G.; La Penna, M.; Silber, J.J.; Durantini, E.N. Photodynamic effect of 5,10,15,20-tetrakis(4-methoxyphenyl) porphirine (TMP) on Hep-2 cell lines. Molecules 2000, 5, 379–380. [Google Scholar] [CrossRef] [Green Version]
- Daicoviciu, D.; Filip, A.; Clichici, S.; Suciu, S.; Muresan, A.; Decea, N.; Dreve, S. Oxidative effects after photodynamic therapy in rats. Bull. UASVM Vet. Med. Cluj-Napoca 2008, 65, 364–369. [Google Scholar]
- Saczko, J.; Skrzypek, W.; Chwilkowska, A.; Choromanska, A.; Pola, A.; Gamian, A.; Kulbacka, J. Photo-oxidative action in cervix carcinoma cells induced by HPD-mediated photodynamic therapy. Exp. Oncol. 2009, 31, 195–199. [Google Scholar]
- Zhang, D.; Song, H.; Qin, Y. Total synthesis of indoline alkaloids: A cyclopropanation strategy. Acc. Chem. Res. 2011, 44, 447457. [Google Scholar] [CrossRef]
- Hu, W.P.; Kuo, K.K.; Senadi, G.C.; Chang, L.S.; Wang, J.J. Photodynamic Therapy Using Indolines-Fused-Triazoles Induces Mitochondrial Apoptosis in Human Non-Melanoma BCC Cells. Anticancer Res. 2017, 37, 5499–5505. [Google Scholar] [CrossRef]
- Valencia, A.; Kochevar, I.E. Ultraviolet A induces apoptosis via reactive oxygen species in a model for Smith-Lemli-Opitz syndrome. Free Radic Biol. Med. 2006, 40, 641–650. [Google Scholar] [CrossRef]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef]
- Choromańska, A.; Saczko, J.; Kulbacka, J.; Kamińska, I.; Skołucka, N.; Majkowski, M. Comparison of the influence of photodynamic reaction on the Me45 and MEWO cell lines in vitro. Contemp.Oncol. (Pozn). 2012, 16, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.; Tschopp, J. Clusterin: The intriguing guises of a widely expressed glycoprotein. Trends Biochem. Sci. 1992, 17, 154–159. [Google Scholar] [CrossRef]
- French, L.E.; Sappino, A.-P.; Tschopp, J.; Schifferli, J.A. Distinct sites of production and deposition of the putative cell death marker clusterin in the human thymus. J. Clin. Investig. 1992, 90, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Kyprianou, N.; Alexander, R.B.; Isaacs, J.T. Activation of programmed cell death by recombinant human tumor necrosis factor plus topoisomerase II-targeted drugs in L929 tumor cells. J. Natl. Cancer Inst. 1991, 83, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.R.; Yeh, S.; Leskov, K.; Odegaard, E.; Hsu, H.L.; Chang, C.; Kinsella, T.J.; Chen, D.J.; Boothman, D.A. Isolation of Ku-70-binding proteins (KUBs). Nucleic Acids Res. 1999, 27, 2165–2174. [Google Scholar] [CrossRef] [Green Version]
- Viard, I.; Wehrli, P.; Jornot, L.; Bullani, R.; Vechietti, J.-L.; Schifferli, J.A.; Tschopp, J.; French, L.E. Clusterin gene expression mediates resistance to apoptotic cell death by heat shock and oxidative stress. J. Investig. Dermatol. 1999, 112, 290–296. [Google Scholar] [CrossRef]
- Kalka, K.; Ahmad, N.; Criswell, T.; Boothman, D.; Hasan Mukhta, H. Up-Regulation of Clusterin during Phthalocyanine 4 Photodynamic Therapy mediated Apoptosis of Tumor Cells and Ablation of Mouse Skin Tumors. Cancer Res. 2000, 60, 5984–5987. [Google Scholar]
- Allison, R.R.; Moghissi, K. Oncologic photodynamic therapy: Clinical strategies that modulate mechanisms of action. Photodiagn. Photodyn. Ther. 2013, 10, 331–341. [Google Scholar] [CrossRef]
- Vaidya, A.; Sun, Y.; Ke, T.; Jeong, E.K.; Lu, Z.R. Contrast enhanced MRI-guided photodynamic therapy for site-specific cancer treatment. Magn. Reson. Med. 2006, 56, 761–767. [Google Scholar] [CrossRef]
- Kercher, E.M.; Zhang, K.; Waguespack, M.; Lang, R.T.; Olmos, A.; Spring, B.Q. High-power light-emitting diode array design and assembly for practical photodynamic therapy research. J. Biomed. Opt. 2020, 25, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Attili, S.K.; Lesar, A.; McNeill, A.; Camacho-Lopez, M.; Moseley, H.; Ibbotson, S.; Samuel, I.D.W.; Ferguson, J. An open pilot study of ambulatory photodynamic therapy using a wearable low-irradiance organic light-emitting diode light source in the treatment of nonmelanoma skin cancer. Br. J. Dermatol. 2009, 161, 170–173. [Google Scholar] [CrossRef]
- Firczuk, M.; Winiarska, M.; Szokalska, A.; Jodlowska, M.; Swiech, M.; Bojarczuk, K.; Salwa, P.; Nowis, D. Approaches to improve photodynamic therapy of cancer. Front. Biosci. 2011, 16, 208–224. [Google Scholar] [CrossRef] [Green Version]
- Serra-Guillen, C.; Hueso, L.; Nagore, E.; Vila, M.; Llombart, B.; Requena Caballero, C.; Botell-Estrada, R.; Sanmartin, O.; Alfaro-Rubio, A.; Guillen, C. Comparative study between cold air analgesia and supraorbital and supratrochlear nerve block for the management of pain during photodynamic therapy for actinic keratoses of the frontotemporal zone. Br. J. Dermatol. 2009, 161, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Halldin, C.B.; Paoli, J.; Sandberg, C.; Gonzalez, H.; Wennberg, A.M. Nerve blocks enable adequate pain relief during topical photodynamic therapy of field cancerization on the forehead and scalp. Br. J. Dermatol. 2009, 160, 795–800. [Google Scholar] [CrossRef]
- Heckl, C.; Aumiller, M.; Rühm, A.; Sroka, R.; Stepp, H. Fluorescence and Treatment Light Monitoring for Interstitial Photodynamic Therapy. Photochem. Photobiol. 2020, 96, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Gollnick, S.O.; Musser, D.A.; Oseroff, A.R.; Vaughan, L.; Owczarczak, B.; Henderson, B.W. IL-10 does not play a role in cutaneous Photofrin photodynamic therapy-induced suppression of the contact hypersensitivity response. Photochem. Photobiol. 2001, 74, 811–816. [Google Scholar] [CrossRef]
- Evangelou, G.; Farrar, M.D.; Cotterell, L.; Andrew, S.; Tosca, A.D.; Watson, R.E.B.; Rhodes, L.E. Topical photodynamic therapy significantly reduces epidermal Langerhans cells during clinical treatment of basal cell carcinoma. Br. J. Dermatol. 2012, 166, 1112–1115. [Google Scholar] [CrossRef]
- Frost, G.A.; Halliday, G.M.; Damian, D.L. Photodynamic therapy-induced immunosuppression in humans is prevented by reducing the rate of light delivery. J. Investig. Dermatol. 2011, 131, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Kripke, M.L. Reflections on the field of photoimmunology. J. Investig. Dermatol. 2013, 133, 27–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelou, G.; Farrar, M.D.; White, R.D.; Sorefan, N.B.; Wright, K.P.; McLean, K.; Andrew, S.; Watson, R.E.B.; Rhodes, L.E. Topical aminolaevulinic acid-photodynamic therapy produces an inflammatory infiltrate but reduces Langerhans cells in healthy human skin in vivo. Br. J. Dermatol. 2011, 165, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Powers, J.G.; Bhawan, J.; Polyak, I.; Gilchrest, B.A. Effective Blue Light Photodynamic Therapy Does Not Affect Cutaneous Langerhans Cell Number or Oxidatively Damage DNA. Dermatol. Surg. 2014, 40, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S.K.; Porter, S.L.; Rizk, N.; Sheng, Y.; McKaig, T.; Burnett, K.; White, B.; Nesbitt, H.; Matin, R.N.; McHale, A.P.; et al. Rose Bengal-Amphiphilic Peptide Conjugate for Enhanced Photodynamic Therapy of Malignant Melanoma. J. Med. Chem. 2020, 63, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R. Fluorinated fullerenes. Chemistry 2001, 7, 4074–4083. [Google Scholar] [CrossRef]
- Krusic, P.J.; Wasserman, E.; Keizer, P.N.; Morton, J.R.; Preston, K.F. Radical Reactions of c60. Science 1991, 254, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.; Bettreich, M. Radical Additions. In Fullerenes: Chemistry and Reactions; Weinheim FRG: Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 213–230. [Google Scholar]
- Rondags, A.; Yuen, W.Y.; Jonkman, M.F.; Horváth, B. Fullerene C60 with cytoprotective and cytotoxic potential: Prospects as a novel treatment agent in Dermatology? Exp. Dermatol. 2017, 26, 220–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Huang, Y.; Wu, P.; Tsai, Y. Topical delivery of 5-aminolevulinic acid-encapsulated ethosomes in a hyperproliferative skin animal model using the CLSM technique to evaluate the penetration behavior. Eur. J. Pharm. Biopharm. 2009, 73, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Penna, G.; Alonci, A.; Rizzo, V.; Russo, S.; Musolino, C. Nanoparticles in oncology: The new theragnostic molecules. Anticancer Agents Med. Chem. 2011, 11, 669–686. [Google Scholar] [CrossRef]
- Tham, H.P.; Xu, K.; Lim, W.Q.; Chen, H.; Zheng, M.; Thng, T.G.S.; Venkatraman, S.S.; Xu, C.; Zhao, Y. Microneedle-Assisted Topical Delivery of Photodynamically Active Mesoporous Formulation for Combination Therapy of Deep-Seated Melanoma. ACS Nano 2018, 12, 11936–11948. [Google Scholar] [CrossRef]
- Lv, R.C.; Yang, D.; Yang, P.P.; Xu, J.T.; He, F.; Gai, S.L.; Li, C.X.; Dai, Y.L.; Yang, G.X.; Lin, J. Integration of Upconversion Nanoparticles and Ultrathin Black Phosphorus for Efficient Photodynamic Theranostics under 808 nm Near-Infrared Light Irradiation. Chem. Mater. 2016, 28, 4724–4734. [Google Scholar] [CrossRef]
- Pucelik, B.; Arnaut, L.G.; Stoche, G.; Dąbrowski, J.M. Design of Pluronic-Based Formulation for Enhanced Redaporfin-Photodynamic Therapy Against Pigmented Melanoma. ACS Appl. Mater. Interfaces 2016, 8, 22039–22055. [Google Scholar] [CrossRef]
- Samia, A.; Chen, X.; Burda, C. Semiconductor Quantum Dots for Photodynamic Therapy. J. Am. Chem. Soc. 2003, 125, 15736–15737. [Google Scholar] [CrossRef] [PubMed]
- Tsay, J.; Trzoss, M.; Shi, L.; Kong, X.; Selke, M.; Jung, M.; Weiss, S. Singlet oxygen production by Peptide-coated quantum dot-photosensitizer conjugates. J. Am. Chem. Soc. 2007, 129, 6865–6871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nun, Y.; Merquiol, E.; Brandis, A.; Turk, B.; Scherz, A.; Blum, G. Photodynamic quenched cathepsin activity based probes for cancer detection and macrophage targeted therapy. Theranostics 2015, 5, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Shon, S.M.; Choi, Y.; Kim, J.Y.; Lee, D.K.; Park, J.-Y.; Schellingerhout, D.; Kim, D.-F. Photodynamic therapy using a protease-mediated theranostic agent reduces cathepsin-B activity in mouse atheromata in vivo. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1360–1365. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV light touches the brain and endocrine system through skin, and why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef] [Green Version]
- Chaiprasongsuk, A.; Janjetovic, Z.; Kim, T.K.; Jarrett, S.G.; D’Orazio, J.A.; Holick, M.F.; Tang, E.K.Y.; Tuckey, R.C.; Panich, U.; Li, W.; et al. Protective effects of novel derivatives of vitamin D3 and lumisterol against UVB-induced damage in human keratinocytes involve activation of Nrf2 and p53 defense mechanisms. Redox Biol. 2019, 24, 101206. [Google Scholar] [CrossRef]
- Slominski, A.T.; Hardeland, R.; Zmijewski, M.A.; Slominski, R.M.; Reiter, R.J.; Paus, R. Melatonin: A Cutaneous Perspective on its Production, Metabolism, and Functions. J. Investig. Dermatol. 2018, 138, 490–499. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Lesions | |
| Skin erythema | |
| Vasodilation | |
| Inflammation | |
| Immunosuppression | |
| Dermatoheliosis | |
| Epidermal hyperplasia | |
| Skin carcinogenesis | |
| Biological Response | |
| Response | Mechanism(s) |
| ROS production | UV radiation causes extreme quantities of ROS that overcome antioxidant systems; ECM proteins act as photosensitisers producing ROS after UV irradiation |
| Tumorigenesis | ROS promote altered cell growth, DNA damage and epigenetic modifications, and cause the onset of tumours |
| Cell proliferation block | MAPK and PI3K pathway activation |
| Cell death | MAPK and PI3K pathway activation |
| Necrosis | Cytosolic constituents spill into the extracellular space through the damaged plasma membrane and provoke a robust inflammatory response, in turn potentiating immunity by attracting host leukocytes into the tumour and increasing antigen presentation |
| Apoptosis | Minor oxidative stress induces apoptosis |
| Autophagy | Selective destruction of cellular elements via ROS |
| Innate immune system stimulation | Higher concentration of TNF-α and MDA, related to histological modifications due to ROS production |
| Lipid peroxidation | Modification of the ratio in the stimulation of cell death by apoptosis or by necrosis. 5,10,15,20-tetrakis(4-methoxyphenyl)-porphyrin (TMPP PDT) and zinc complex TMPP PDT provoked an augmented concentration of thiobarbituric reactive substances in tumour tissue and in blood plasma at 24 h after the PDT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Pioggia, G.; Tonacci, A.; Musolino, C.; Gangemi, S. Oxidative Stress and Photodynamic Therapy of Skin Cancers: Mechanisms, Challenges and Promising Developments. Antioxidants 2020, 9, 448. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9050448
Allegra A, Pioggia G, Tonacci A, Musolino C, Gangemi S. Oxidative Stress and Photodynamic Therapy of Skin Cancers: Mechanisms, Challenges and Promising Developments. Antioxidants. 2020; 9(5):448. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9050448
Chicago/Turabian StyleAllegra, Alessandro, Giovanni Pioggia, Alessandro Tonacci, Caterina Musolino, and Sebastiano Gangemi. 2020. "Oxidative Stress and Photodynamic Therapy of Skin Cancers: Mechanisms, Challenges and Promising Developments" Antioxidants 9, no. 5: 448. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9050448