Antioxidant, Physicochemical, and Cellular Secretion of Glucagon-Like Peptide-1 Properties of Oat Bran Protein Hydrolysates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
2.1. Materials and Chemicals
2.2. Protein Extraction
2.3. Gel Electrophoresis and Mass Sspectrometry Analysis of Proteins
2.4. Protein Hydrolysis and Dialysis
2.5. Ultra-Violet and Fourier Transform Infrared (FT-IR) Aanalysis of Hydrolysates
2.6. Analysis of Free Thiols and Amino Acids
2.7. Zeta Potential and Hydrophobicity Assays
2.8. Antioxidant Assays
2.9. Alpha-Amylase and Dipeptidyl Peptidase-4 Inhibition Assays
2.10. Culture and Maintenance of NCI-H716 Cells
2.11. Preparation of NCI-H716 Cells forBioassays
2.12. Effect of Hydrolysates on the Viability of Cells
2.13. Glucagon-Like Peptide-1 (GLP-1) Secretion
2.14. Statistics
3. Results and Discussion
3.1. Characterization of Isolated Proteins
3.2. Hydrolysis of Proteins and Characterization of Hydrolysates
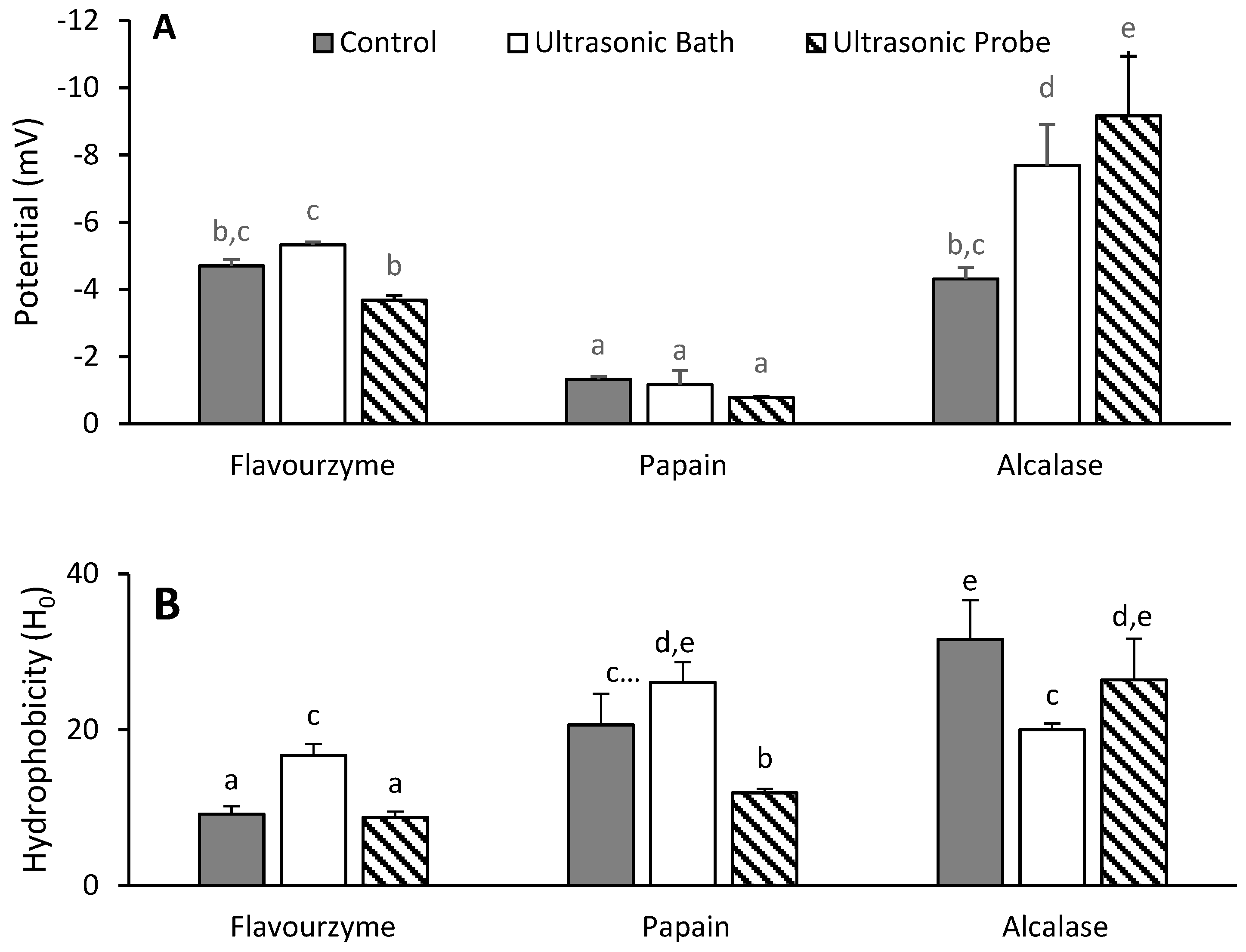
3.3. Zeta Potential and Hydrophobicity of Hydrolyzed Proteins
3.4. Spectroscopic Characterization of Hydrolyzed Proteins
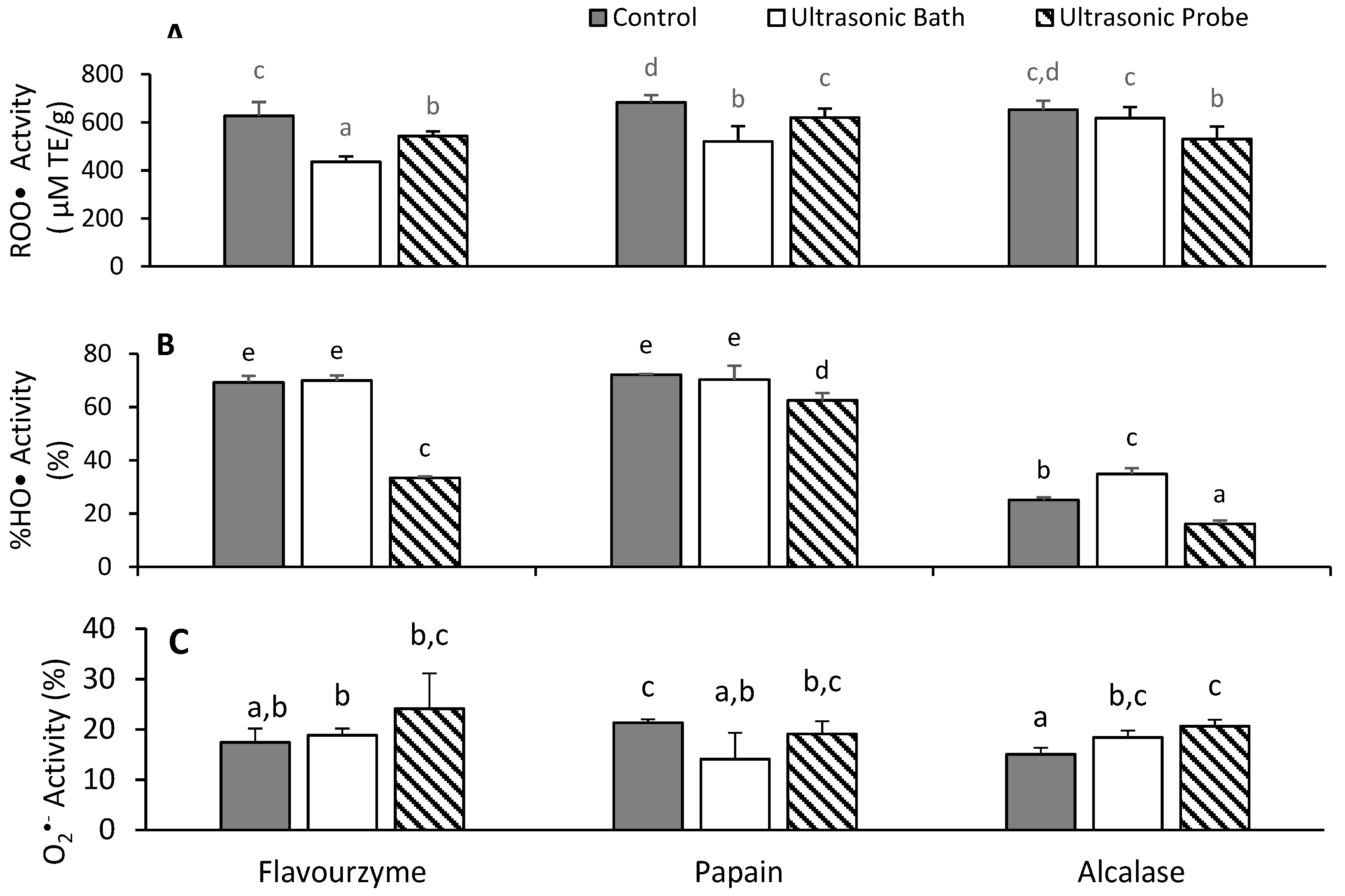
3.5. Antioxidant Activities of Hydrolyzed Proteins
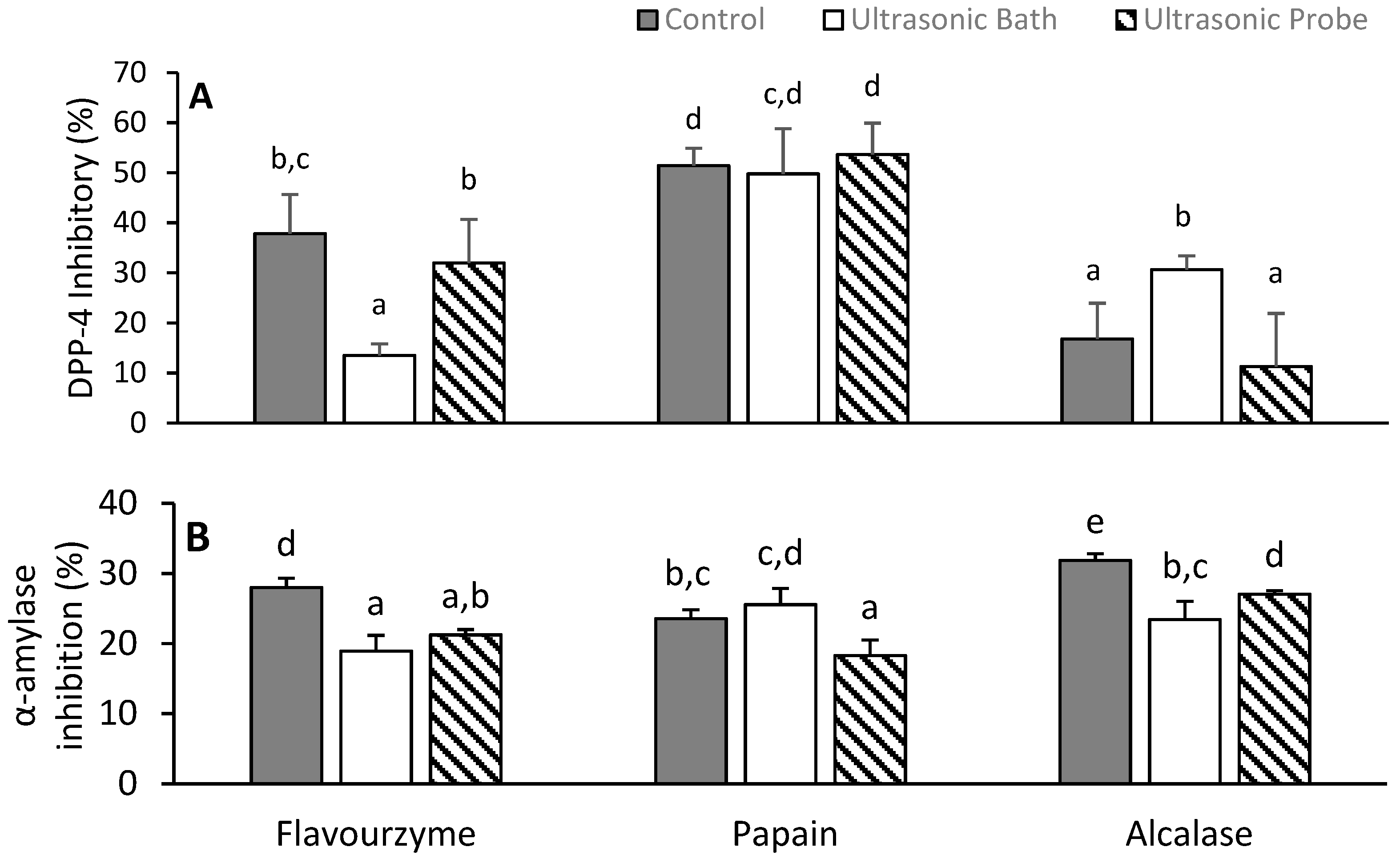
3.6. Inhibition of Alpha-Amylase and Dipeptidyl Peptidase-4
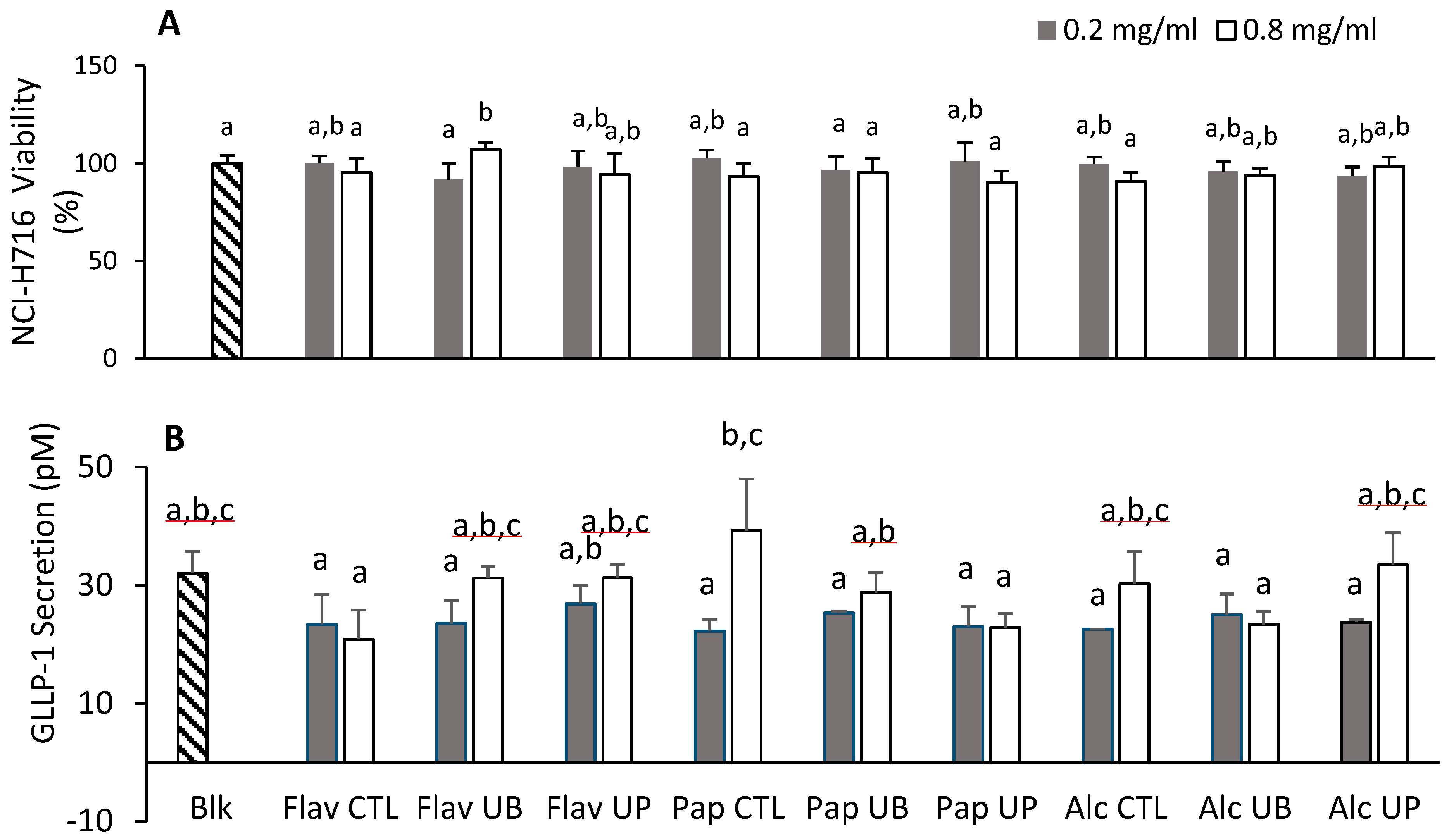
3.7. Effect of Hydrolyzed Proteins on Cellular Viability and the Secretion of Glucagon-Like Peptide in NCI-H716 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Espinosa, G.Y.C.; Walters, M.E.; Tsopmo, A. Oats. In Whole Grains: Processing, Product Development, and Nutritional Aspects; Mir, S.A., Manickavasagan, A., Shah, M.A., Eds.; CRC Press: Boca Raton, FL, USA, 2019; pp. 129–150. [Google Scholar]
- Beck, E.J.; Tosh, S.M.; Batterham, M.J.; Tapsell, L.C. Oat b-glucan increases postprandial cholecystokinin levels, decreases insulin response and extends subjective satiety in overweight subjects. Mol. Nutr. Food Res. 2009, 53, 1343–1351. [Google Scholar] [CrossRef]
- Uraipong, C.; Zhao, J. Rice bran protein hydrolysates exhibit strong in vitro α-amylase, β-glucosidase and ACE-inhibition activities. J. Sci. Food Agric. 2016, 96, 1101–1110. [Google Scholar] [CrossRef]
- Jao, C.-L.; Hung, C.-C.; Tung, Y.-S.; Lin, P.-Y.; Chen, M.-C.; Hsu, K.-C. The development of bioactive peptides from dietary proteins as a dipeptidyl peptidase IV inhibitor for the management of type 2 diabetes. BioMedicine 2015, 5, 14. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Roumie, C.L.; Min, J.Y.; McGowan, L.D.A.; Presley, C.; Grijalva, C.G.; Hackstadt, A.J.; Hung, A.M.; Greevy, R.A.; Elasy, T.; Griffin, M.R. Comparative safety of sulfonylurea and metformin monotherapy on the risk of heart failure: A cohort study. J. Am. Heart Assoc. 2017, 6, e005379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velarde-Salcedo, A.J.; Barrera-Pacheco, A.; Lara-González, S.; Montero-Morán, G.M.; Díaz-Gois, A.; Mejia, E.G.D.; De La Rosa, A.P.B. In vitro inhibition of dipeptidyl peptidase IV by peptides derived from the hydrolysis of amaranth (Amaranthus hypochondriacus L.) proteins. Food Chem. 2013, 136, 758–764. [Google Scholar] [CrossRef]
- Payan, F. Structural basis for the inhibition of mammalian and insect α-amylases by plant protein inhibitors. Biochim. Biophys. Acta-Proteins Proteom. 2004, 1696, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhu, W.; Yi, J.; Liu, N.; Cao, Y.; Lu, J.; Decker, E.A.; McClements, D.J. Effects of sonication on the physicochemical and functional properties of walnut protein isolate. Food Res. Int. 2018, 106, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Walburg, G.; Larkins, B.A. Oat Seed Globulin: Subunit characterization and demonstration of its synthesis as a precursor. Plant Physiol. 1983, 72, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Walters, M.E.; Udenigwe, C.C.; Tsopmo, A. Structural characterization and functional properties of proteins from oat milling fractions. J. Am. Oil Chem. Soc. 2018, 95, 991–1000. [Google Scholar] [CrossRef]
- Esfandi, R.; Willmore, W.G.; Tsopmo, A. Peptidomic analysis of hydrolyzed oat bran proteins, and their in vitro antioxidant and metal chelating properties. Food Chem. 2019, 279, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Puppo, M.C.; Calvelo, A.; Añón, M.C. Physicochemical and rheological characterization of wheat flour dough. Cereal Chem. 2005, 82, 173–181. [Google Scholar] [CrossRef]
- Panasiuk, R.; Amarowicz, R.; Kostyra, H.; Sijtsma, L. Determination of α-amino nitrogen in pea protein hydrolysates: A comparison of three analytical methods. Food Chem. 1998, 62, 363–367. [Google Scholar] [CrossRef]
- Udenigwe, C.C.; Mohan, A.; Wu, S. Peptide aggregation during plastein reaction enhanced bile acid-binding capacity of enzymatic chicken meat hydrolysates. J. Food Biochem. 2015, 39, 344–348. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Hampsch-Woodill, M.; Flanagan, J.A.; Prior, R.L. High-throughput assay of oxygen radical absorbance capacity (ORAC) using a multichannel liquid handling system coupled with a microplate fluorescence reader in 96-well format. J. Agric. Food Chem. 2002, 50, 4437–4444. [Google Scholar]
- Ratnasari, N.; Walters, M.; Tsopmo, A. Antioxidant and lipoxygenase activities of polyphenol extracts from oat brans treated with polysaccharide degrading enzymes. Heliyon 2017, 3, e00351. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Yin, Y.; Zhao, W.; Liu, J.; Chen, F. Anti-diabetic activity peptides from albumin against α-glucosidase and α-amylase. Food Chem. 2012, 135, 2078–2085. [Google Scholar]
- Kuhre, R.E.; Albrechtsen, N.J.W.; Deacon, C.F.; Balk-Møller, E.; Rehfeld, J.F.; Reimann, F.; Gribble, F.M.; Holst, J.J. Peptide production and secretion in GLUTag, NCI-H716, and STC-1 cells: A comparison to native L-cells. J. Mol. Endocrinol. 2016, 56, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Park, M.; Lee, Y.M.; Rhyu, M.R.; Kim, H.Y. Ginsenoside metabolite compound K stimulates glucagon-like peptide-1 secretion in NCI-H716 cells via bile acid receptor activation. Arch. Pharm. Res. 2014, 37, 1193–1200. [Google Scholar] [CrossRef]
- Esfandi, R.; Willmore, W.G.; Tsopmo, A. Antioxidant and anti-apoptotic properties of oat bran protein hydrolysates in stressed hepatic cells. Foods 2019, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Inguglia, E.S.; Tiwari, B.K.; Kerry, J.P.; Burgess, C.M. Effects of high intensity ultrasound on the inactivation profiles of Escherichia coli K12 and Listeria innocua with salt and salt replacers. Ultrason. Sonochem. 2018, 48, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Gazza, L.; Taddei, F.; Conti, S.; Gazzelloni, G.; Muccilli, V.; Janni, M.; Ovidio, R.D.; Alfieri, M.; Redaelli, R.; Pogna, N.E. Biochemical and molecular characterization of Avena indolines and their role in kernel texture. Mol. Genet. Genom. 2015, 290, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, O.; Sontag-Strohm, T.; Salovaara, H.; Lampi, A.M.; Sibakov, J.; Loponen, J. Foaming of differently processed oats: Role of nonpolar lipids and tryptophanin proteins. Cereal Chem. 2011, 88, 239–244. [Google Scholar] [CrossRef]
- Cumby, N.; Zhong, Y.; Naczk, M.; Shahidi, F. Antioxidant activity and water-holding capacity of canola protein hydrolysates. Food Chem. 2008, 109, 144–148. [Google Scholar] [CrossRef]
- Vioque, J.; Sánchez-Vioque, R.; Clemente, A.; Pedroche, J.; Bautista, J.; Millan, F. Production and characterization of an extensive rapeseed protein hydrolysate. J. Am. Oil Chem. Soc. 1999, 76, 819–823. [Google Scholar] [CrossRef]
- Malik, M.A.; Sharma, H.K.; Saini, C.S. High intensity ultrasound treatment of protein isolate extracted from dephenolized sunflower meal: Effect on physicochemical and functional properties. Ultrason. Sonochem. 2017, 39, 511–519. [Google Scholar]
- Kang, D.C.; Zou, Y.H.; Cheng, Y.P.; Xing, L.J.; Zhou, G.H.; Zhang, W.G. Effects of power ultrasound on oxidation and structure of beef proteins during curing processing. Ultrason. Sonochem. 2016, 33, 47–53. [Google Scholar] [CrossRef]
- Zhang, Z.; Regenstein, J.M.; Zhou, P.; Yang, Y. Effects of high intensity ultrasound modification on physicochemical property and water in myofibrillar protein gel. Ultrason. Sonochem. 2017, 34, 960–967. [Google Scholar] [CrossRef]
- Udechukwu, M.C.; Downey, B.; Udenigwe, C.C. Influence of structural and surface properties of whey-derived peptides on zinc-chelating capacity, and in vitro gastric stability and bioaccessibility of the zinc-peptide complexes. Food Chem. 2018, 240, 1227–1232. [Google Scholar] [CrossRef]
- Ambigaipalan, P.; Al-Khalifa, A.S.; Shahidi, F. Antioxidant and angiotensin I converting enzyme (ACE) inhibitory activities of date seed protein hydrolysates prepared using Alcalase, Flavourzyme and Thermolysin. J. Funct. Foods 2015, 18, 1125–1137. [Google Scholar] [CrossRef]
- Caer, D.; Baniel, A.; Subirade, M.; Gueguen, J.; Colas, B. Preparation and physicochemical properties of glycosylated derivatives of pea legumin. J. Agric. Food Chem. 1990, 38, 1700–1706. [Google Scholar] [CrossRef]
- Wang, K.; Sun, D.-W.; Pu, H.; Wei, Q. Principles and applications of spectroscopic techniques for evaluating food protein conformational changes: A review. Trends Food Sci. Technol. 2017, 67, 207–219. [Google Scholar] [CrossRef]
- Chang, C.; Li, X.; Li, J.; Niu, F.; Zhang, M.; Zhou, B.; Su, Y.; Yang, Y. Effect of enzymatic hydrolysis on characteristics and synergistic efficiency of pectin on emulsifying properties of egg white protein. Food Hydrocoll. 2017, 65, 87–95. [Google Scholar] [CrossRef]
- Liu, G.; Li, J.; Shi, K.; Wang, S.; Chen, J.; Liu, Y.; Huang, Q. Composition, secondary structure, and self-assembly of oat protein isolate. J. Agric. Food Chem. 2009, 57, 4552–4558. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, H.; Ali, A.; Xin, H.; Valizadeh, R. Detecting carbohydrate molecular structural makeup in different types of cereal grains and different cultivars within each type of grain grown in semi-arid area using FTIR spectroscopy with uni- and multi-variate molecular spectral analyses. Anim. Feed Sci. Technol. 2014, 194, 136–144. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Handa, C.L.; Xu, J. Effects of ultrasound pre-treatment on the structure of β-conglycinin and glycinin and the antioxidant activity of their hydrolysates. Food Chem. 2017, 218, 165–172. [Google Scholar] [CrossRef]
- Ngoh, Y.Y.; Gan, C.Y. Enzyme-assisted extraction and identification of antioxidative and α-amylase inhibitory peptides from Pinto beans (Phaseolus vulgaris cv. Pinto). Food Chem. 2016, 190, 331–337. [Google Scholar] [CrossRef]
- Wang, B.; Atungulu, G.G.; Khir, R.; Geng, J.; Ma, H.; Li, Y.; Wu, B. Ultrasonic Treatment Effect on Enzymolysis Kinetics and Activities of ACE-Inhibitory Peptides from Oat-Isolated Protein. Food Biophys. 2015, 10, 244–252. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; Maux, S.L.; Dubrulle, C.; Barre, C.; FitzGerald, R.J. Quinoa (Chenopodium quinoa Willd.) protein hydrolysates with in vitro dipeptidyl peptidase IV (DPP-IV) inhibitory and antioxidant properties. J. Cereal Sci. 2015, 65, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Yu, G.; Zhang, Y.; Zhang, B.; Fan, J. Dipeptidyl peptidase IV inhibitory peptides derived from oat (Avena sativa L.), buckwheat (Fagopyrum esculentum), and highland barley (Hordeum vulgare trifurcatum (L.) Trofim) proteins. J. Agric. Food Chem. 2015, 63, 9543–9549. [Google Scholar] [CrossRef]
- Chen, Q.; Reimer, R.A. Dairy protein and leucine alter GLP-1 release and mRNA of genes involved in intestinal lipid metabolism in vitro. Nutrition 2009, 25, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walters, M.E.; Willmore, W.G.; Tsopmo, A. Antioxidant, Physicochemical, and Cellular Secretion of Glucagon-Like Peptide-1 Properties of Oat Bran Protein Hydrolysates. Antioxidants 2020, 9, 557. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9060557
Walters ME, Willmore WG, Tsopmo A. Antioxidant, Physicochemical, and Cellular Secretion of Glucagon-Like Peptide-1 Properties of Oat Bran Protein Hydrolysates. Antioxidants. 2020; 9(6):557. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9060557
Chicago/Turabian StyleWalters, Mallory E., William G. Willmore, and Apollinaire Tsopmo. 2020. "Antioxidant, Physicochemical, and Cellular Secretion of Glucagon-Like Peptide-1 Properties of Oat Bran Protein Hydrolysates" Antioxidants 9, no. 6: 557. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9060557